

Журнал физической химии, 2022, T. 96, № 1, стр. 18-38
Образование атома позитрония в молекулярных средах. Выявление канцерогенных и антиканцерогенных свойств химических соединений
В. М. Бяков a, b, С. В. Степанов a, *
a ФГБУ “Институт теоретической и экспериментальной физики имени А.И. Алиханов
а Национального исследовательского центра ”Курчатовский институт”
117218 Москва, Россия
b Российский химико-технологический университет им. Д.И. Менделеева
125047 Москва, Россия
* E-mail: stepanov@itep.ru
Поступила в редакцию 13.05.2021
После доработки 16.06.2021
Принята к публикации 16.06.2021
- EDN: NGUTKB
- DOI: 10.31857/S0044453722010058
Аннотация
Признано, что одной из основных причин онкологических заболеваний являются химические канцерогены. В обзоре рассмотрены как биологические, так и физико-химические методы определения канцерогенной активности веществ. Физико-химические методы основываются на том, что большинство канцерогенов являются, в частности, эффективными акцепторами электронов, порождаемых ионизирующими частицами при их прохождении через вещество, моделирующее внутриклеточную среду. Показано, что полное ингибирование образования атома позитрония испытуемым химическим соединением может служить указанием на наличие у него канцерогенных свойств. Отмечено сходство позитронного метода с предложенным Дж. Бакалом радиационно-химическим тестом, использующим установки наносекундного импульсного радиолиза. Преимущества позитронного подхода по сравнению с методом Бакала сводятся к простоте, быстроте и экономической выгоде.
1. ВВЕДЕНИЕ
Наша жизнь в среде обитания и технический прогресс постоянно вынуждают нас соприкасаться с большим количеством новых химических соединений, часто обладающих высокой биологической активностью. С водой и пищей, через кожу или просто вдыхая воздух, эти вещества поступают в организм человека, и данный процесс остановить невозможно.
В настоящее время основным источником онкологических заболеваний являются химические канцерогены [1, 2]. Это вещества, которые при попадании в организм приводят к возникновению злокачественных опухолей. Последние характеризуются появлением бесконтрольно делящихся клеток, способных как к проникновению в прилежащие ткани, так и в отдаленные органы (метастазирование). Обычно под определение канцерогенов подпадают и мутагены – вещества, которые вызывают появление наследственных изменений (мутаций) в генотипе клетки: изменения, потерю хромосом или их частей. Сильные канцерогены являются одновременно и мутагенами [3]. Этот факт объясняется тем, что генетические повреждения являются необходимой стадией возникновения онкологического процесса.
По механизму воздействия химические канцерогены обычно подразделяют на две группы: генотоксические и негенотоксические [4]. Генотоксические канцерогены, попадая в водную среду, диссоциируют с образованием высокоактивных производных, содержащих электрофильную группу, несущую избыточный положительный заряд [5]. Эта группа образует с молекулами ДНК прочные ковалентные химические связи. Тем самым нарушается точность воспроизведения генома клетки, что и приводит к мутациям и образованию опухолевых клеток.
Некоторые вещества приобретают канцерогенные свойства испытав биохимические превращения внутри организма, после так называемой метаболической активации, происходящей под действием ферментов, “биологических” катализаторов. К этому заключению пришли более 60-ти(!) лет назад супруги Джеймс и Элизабет Миллеры [6–9]. Такие вещества получили название протоканцерогенов или негенотоксичных канцерогенов. Главным признаком негенотосичных канцерогенов является их неспособность непосредственно ковалентно связываться с ДНК и вызывать мутации [10].
Таким образом, протоканцерогены в результате метаболической активации превращаются в более реакционноспособные электрофильные соединения (ultimate carcinogens), которые уже взаимодействуют с нуклеофильными группами ДНК, образуют с ними ковалентные связи и затем модифицируют геном. Была обнаружена также способность негенотоксичных канцерогенов стимулировать процесс бесконтрольного размножения клеток и предотвращать апоптоз (“нормальный” регулируемый процесс гибели клеток) [11].
Большинство химических канцерогенов являются органическими соединениями, лишь небольшое число неорганических веществ обладают канцерогенными свойствами.
Существует также понятие физических канцерогенов. Сюда относят различные виды излучений (прежде всего ионизирующих). Несмотря на то, что они применяются для лечения онкологических заболеваний, эти излучения способны вызывать образование опухолей как тканей, так и органов организма, а также метастаз [12, 13].
Большое число организаций по всему миру (World Health Organization (WHO), European Food Safety Authority (EFSA), Food and Agriculture Organization (FAO), The International Agency for Research on Cancer (IARC) осуществляют мониторинг канцерогенной опасности химических веществ, с которыми возможен контакт, осуществляют связь научных исследований с политикой в области здравоохранения, выработкой рекомендаций по минимизации возможных рисков [14]. Тем не менее, ожидается, что в 2025 г. смертность на Земле по причине рака превысит 20 млн людей [2].
2. МЕТОДЫ ИССЛЕДОВАНИЯ КАНЦЕРОГЕННЫХ И МУТАГЕННЫХ СВОЙСТВ ХИМИЧЕСКИХ СОЕДИНЕНИЙ
В настоящее время существует довольно много подходов к диагностике канцерогенов [10]. Рассмотрим их кратко.
1) Без сомнения наиболее надежным и достоверным способом оценки канцерогенной опасности в отношении людей является эпидемиологический метод, когда отслеживаются последствия воздействия конкретного вещества на большое число людей (assays in humans) [15]. Эти весьма ценные данные, собираются в течение длительного времени и имеют ретроспективный характер. Однако такой подход не позволяет судить о канцерогенной опасности данного веще- ства до его контакта с человеком, тогда как суть диагностики канцерогенов заключается именно в том, чтобы получить априорную, а не апостериорную оценку. Неточность интерпретации эпидемиологических данных бывает связана с трудностью раздельной оценки влияния различных факторов (химические канцерогены, курение, профессия, болезни). В организме часто реализуется ситуация, когда вредные воздействия происходят одновременно. В этих случаях оценка рисков и последствий особенно сложна [16].
2) Диагностировать развитие заболевания, вызванное действием химических канцерогенов еще до появления явных клинических признаков, позволяет метод биомаркеров [4]. Биомаркером может быть почти любой измеряемый показатель (measurement), отражающий взаимодействие организма и потенциальной опасности. Параметры крови, мочи, кала – вот примеры наиболее распространенных биомаркеров. Однако конкретно в исследованиях по оценке канцерогенности решающее значение имеет правильный подбор биомаркеров. Именно это определяет точность выводов. К сожалению, таких маркеров довольно много, и они реагируют на нарушения в работе различных систем организма (респираторной, репродуктивной, иммунной и т.д.), либо на наличие в организме конкретных химических соединений. В случае детектирования канцерогенов в качестве биомаркеров часто выступают аддукты ДНК, белков, хромосомные аберрации. Так или иначе, этот подход также предполагает непосредственный контакт человека с канцерогенным веществом, и проведение опытов непосредственно на людях, что опять-таки существенно ограничивает область его использования.
3) Опыты на животных (animal carcinogenicity bioassay) [17]. В них выбирается какой-то биологический вид животных (или какая-либо клеточная культура), на который воздействуют испытуемым веществом. Затем производится биопсия, отбор живых клеток организма и их исследование. На основе этого делается вывод о степени канцерогенной опасности вещества. Наиболее распространены опыты на грызунах (мышах и крысах, дающих наиболее надежные результаты) и аквариумных рыбках. Преимуществом данного подхода является, то, что в подобных опытах можно воспроизвести основные фармокинетические факторы, в которых принимает участие исследуемое вещество (метаболизм, внутриклеточная миграция, рост опухоли и т.д.). Действительно, большинство канцерогенов в отношении человека являются канцерогенами и в отношении животных. Более 3000 опасных в отношении человека канцерогенов (1/3 от общего числа известных к настоящему времени) были изначально идентифицированы в опытах на животных. Вещество считается канцерогенным, если оно приводит к значимому повышению числа опухолевых заболеваний у опытной группы по сравнению с контрольной [17].
Однако следует учитывать ряд обстоятельств, осложняющих проведение опытов на животных и ограничивающих количество веществ, которые могут быть в них исследованы: 1) подобные опыты достаточно дороги (до 1 млн $ на одно вещество) и длительность каждого исследования составляет около пяти лет (на крысах); 2) процесс метаболизма у крыс и людей имеет ряд существенных отличий; 3) в Евросоюзе и США опыты на животных подпадают под прямые запреты с этической точки зрения [18, 19].
Необходимо также учитывать, что в результате биологических испытаний канцерогенные вещества могут быть не идентифицированы как канцерогены по причине того, что одновременно они являются сильно токсичными соединениями. В ходе их испытаний гибель организма наступает из-за интоксикации организма, пока рак просто не успел развиться. Поэтому разработка быстрых и относительно дешевых физико-химических методов детектирования канцерогенов является важной задачей.
4) Тесты на мутации бактерий, дрозофил и растений. Их главное достоинство – быстрота (несколько дней) и дешевизна. Наиболее популярен экспресс-тест Эймса (Ames) на обратную мутацию в генах бактерий сальмонелл, а также на мутации кишечной палочки Е. Coli для оценки канцерогенной опасности химических соединений. Недостаток теста Эймса связан с большим различием процессов метаболизма бактерий и млекопитающих. Однако его преодолевают, имитируя процесс обмена веществ млекопитающих в тесте Эймса посредством использования вытяжки из печени крыс. Это позволяет детектировать протоканцерогены (например, бензопирены), не обладающие канцерогенной активностью, но метаболиты которых уже проявляют генотоксичность.
Поскольку сальмонеллы являются прокариотами (не имеют ядер в клетках), экстраполяция результатов теста Эймса на человека не является однозначной. Тем не менее, предсказательная сила данного теста велика: 80–90% положительных по Эймсу результатов, оказались канцерогенами в экспериментах, проведенных на животных [17]. Считается, что тест Эймса хорошо выявляет электрофильные канцерогены.
В отличие от бактерий, многие насекомые способны активировать в ходе метаболизма широкий спектр генотоксичных химических веществ. С этой точки зрения изучение генетических свойств в химических опытах по исследованию летальных мутаций у дрозофил представляет собой более полезный тест: примерно 2/3 генов, ответственных за болезни человека, гомологичны генам дрозофил [20].
Опыты с растениями (фасоль, лук, кукуруза, ячмень) в генетической токсикологии из-за большого прогресса в использовании бактериальных культур и микробов производятся реже [21]. Тем не менее, возможность вести исследования изменений как на генетическом, так и на хромосомном уровне без сложного лабораторного оборудования, что необходимо для проведения других тестов, в ряде случаев дает большое преимущество.
5) Поиск количественных закономерностей структура–свойство (Quantitative Structure-Activity Relationship; QSAR) [22]. Этот подход основан на поиске корреляций между строением и свойствами молекул конкретного вещества. С одной стороны – наличие различных функциональных групп, геометрические размеры молекул, образование водородных связей, растворимость в воде и углеводородах, с другой – биологическая активность, степень канцерогенности данного вещества и не только [18].
Разработано много компьютерных программ, использующих обширные базы данных, позволяющих предсказывать возможную канцерогенность химических соединений. Наиболее известной из такого рода программ является система TOPKAT (TOxicity Prediction by C(K)omputer Assisted Technology), предназначенная для анализа органических соединений [23]. Программа является коммерческим продуктом, принадлежащим компании BIOVIA (ранее Accelrys Inc). Цель разработчиков этого продукта состояла в замене практически всех тестов на животных (причем не только на канцерогенность) компьютерными. Программа позволяет прогнозировать биологическое воздействие конкретного вещества (например, предсказывать результаты теста Эймса). Граница области “опасных” веществ определяется “тренировкой” по известным соединениям (“машинное обучение” или machine learning). Однако, стоимость такого компьютерного прогноза биологического действия одного вещества оценивается примерно в 1000$ (использование суперкомпьютеров и платного программного обеспечения).
6) Физико-химические подходы к определению канцерогенной опасности веществ опираются на их физико-химические свойства и представления о молекулярных механизмах, лежащих в основе канцерогенеза. Как уже отмечалось, согласно многолетним исследованиям, проведенным четой Миллеров [6–9], большинство канцерогенов являются сильными электрофилами. Они легко взаимодействуют с нуклеофильными группами нуклеиновых кислот, в частности, с ДНК, образуя с ними прочные донорно-акцепторные ковалентные связи. Именно вследствие этого (из-за появления ДНК-аддуктов) нарушается процесс правильной репликации ДНК. Связанные с канцерогеном нуклеотиды неправильно считываются ферментом (ДНК-полимеразой), осуществляющим репликацию ДНК. Вследствие этого и возникают мутации. Накопление большого числа мутаций в геноме превращает нормальную клетку в опухолевую и вызывает канцерогенез.
Данное обстоятельство побудило группу американских радиационных химиков под руководством Джорджа Бакала из Case Western Reserve University (Огайо, США), которая занималась изучением электропроводимости жидкостей, наводимой воздействием наносекундных импульсов быстрых e–, применить свою установку импульсного радиолиза для исследования связи между электрофильностью и канцерогенностью.
Идея Бакала состояла в следующем. Согласно Миллерам канцерогенно-опасная молекула является сильным электрофилом. Это значит, что находясь в химически инертном растворителе в окружении электронов, порожденных ионизирующим излучением, молекула захватывает одного из них при первой же встрече, причем так быстро, что длительность контакта реагентов гораздо короче, нежели предварительная стадия сближения, лимитируемая их взаимной диффузией. Само химическое превращение в таких реакциях протекает безактивационно и мгновенно, так что скорость реакции в целом контролируется только диффузией. Именно это свойство по Бакалу выделяет канцерогенно-опасные вещества среди прочих. Итак, согласно Бакалу наличие или отсутствие диффузионного контроля над константой скорости реакции захвата электрона тестируемым веществом, является критерием степени его канцерогенности.
Схема экспериментов Бакала представлена на рис. 1. Электронный ускоритель Ван де Граафа формирует на выходе импульсы (банчи) длительностью ≈15 нс электронов, ускоренных до энергий в несколько МэВ. Электроны порождают в измерительной ячейке многочисленные ион-электронные пары. В ячейке, заполненной циклогексаном с растворенным в нем испытуемым веществом S (Solute; Scavenger), находятся два электрода, расстояние между которыми 0.6 мм. К ним приложена разность потенциалов в 300 В. После каждого стартового импульса избыточные электроны исчезают вследствие 1) ион-электронной рекомбинации, 2) захвата электронов растворенным веществом S, 3) достижения анода за счет дрейфа в постоянном электрическом поле (5 кВ/см) в исследуемом растворе.
Рис. 1.
Схема экспериментов Дж. Бакала по определению констант скорости ke захвата избыточных электронов молекулами различных испытуемых веществ, растворенных в циклогексане (pulse-conductivity measurements).

Удобным растворителем для проведения подобных измерений оказался циклогексан. В нем характерное время убыли вторичных e– составляет ≈400 нс (легко измерять). Примеси, введенные в малых количествах (типичные концентрации S – несколько микромолей/литр), уменьшают это время в несколько раз. Экспериментально наблюдается укорочение времени спада тока электронов, которое по сути есть произведение константы скорости захвата ${{k}_{e}} = k({{e}^{ - }} + {\text{S}})$ избыточных электронов акцептором S на известную концентрацию [S] акцептора. Однако основной причиной для выбора циклогексана в качестве растворителя, моделирующего внутриклеточную среду, послужила наилучшая корреляция значений констант ke, полученных при его использовании с биологическими данными по канцерогенам [24]. Естественно, что процесс захвата электрона акцептором зависит от природы растворителя (полярный или неполярный), его агрегатного состояния и температурных условий [24].
На первый взгляд может показаться, что обычная вода должна быть наиболее подходящим растворителем, моделирующим внутриклеточную среду. Однако результаты экспериментов Бакала и компьютерного моделирования взаимодействия биомолекул с окружающей водой указывают, что это не так. На пространственно-временн${\text{ы}}'$х масштабах, относящихся к процессам переноса e–, вода, “окаймляющая” нуклеофильные биомолекулы, является “льдоподобной” (iceliked), структурированной (structured water). В такой воде существенно удлиняются времена ее диэлектрической релаксации, гидратации предсольватированных электронов и расстояния, на которые они мигрируют [25].
С начала 80-х годов прошлого века группа Дж. Бакала провела измерения ke для нескольких сотен испытуемых веществ в циклогексане. Было показано, что определенные таким образом константы ke хорошо коррелируют с биологическими данными по канцерогенной активности исследованных соединений. Впоследствии эта процедура была названа ke-тестом на канцерогенность [26].
Анализ экспериментальных результатов показал: если для данного испытуемого соединения S значение ke = k(e– + S) превышает измеренную в том же циклогексане константу скорости реакции захвата электрона тетрахлоридом углерода, то есть, k(e– + S) > k(e– + CCl4), то данное соединение, как правило, является канцерогенноопасным. Этот критерий выполнялся на уровне >80% [27–29].
Рисунок 2 иллюстрирует результаты некоторых измерений группы Дж. Бакала. Из более чем 40 соединений, для которых отношение констант k(e– + S)/k(e– + CCl4) > 1, только 5 не причисляются к канцерогенам по биологическим критериям. Из 18 соединений, для которых это отношение меньше единицы, три все-таки оказались “биохимическими канцерогенами”. Неполное совпадение не удивительно. Степень электрофильности – это все-таки сродство S к электрону, а не значение константы скорости ke которая может служить только приближенной мерой сродства.
Рис. 2.
Классификация канцерогенных и неканцерогенных веществ по отношению их констант скорости захвата избыточного электрона к константе скорости захвата e– четыреххлористым углеродом в ц-гексане. Значение последней равно k(e– + CCl4) ≈ 3 × 1012 М–1 с–1.

Таким образом, более чем в 80% случаев предсказания канцерогенной опасности по ke-тесту совпали с биологическими критериями. Следует признать, что это очень высокий показатель предсказательной способности. Поэтому куратор американской программы по сравнительному сопоставлению методов оценки канцерогенного действия веществ отвел методу Бакала первое место по эффективности среди других физико-химических методов [30, 31].
Однако для реализации метода Бакала необходима сложная установка импульсного радиолиза, включающая электронный ускоритель на энергию в несколько МэВ и громоздкую гамма-защиту.
7) В качестве еще одного примера применения подхода, опирающегося на физико-химические свойства веществ и представления о молекулярных механизмах, отметим предложенный в работе [32] электрохимический метод выявления антиоксидантов. Авторы исходят из предположения, что основным оксидантом в человеческом организме является супероксид анион-радикал, ${\text{O}}_{2}^{ - }$. Суть метода сводится к исследованию конкуренции идущих в электрохимической ячейке реакций
8) Канцерогенная активность металлических макрообразцов, имплантированных в организмы животных. Хотя известно, что введение в организм ряда металлов в свободном состоянии вызывает канцерогенное действие, сейчас речь пойдет об установлении признака одноэлементных веществ (большей частью металлов), макрообразцы которых в виде кусочков, порошков, перфорированных пластинок, вводимые подкожно в ткани животных, производят канцерогенные эффекты [33].
Классифицируем одноэлементные вещества по соответствующим атомным номерам и значениям работы выхода из них электрона (Work Function, WF), рис. 3. Символы веществ, проявивших канцерогенный эффект в экспериментах [34], выделены красным цветом, а символы веществ, не проявивших такового действия или вообще не испытанных экспериментально, показаны черным. Как видно, по канцерогенной активности все вещества распадаются на две группы. Нижнюю группу составляют вещества с “малыми” значениями WF, меньшими 4 эВ. Они не производят канцерогенного эффекта. Напротив, по крайней мере, половина из веществ верхней группы, где WF превышает 4 эВ, признана канцерогенами. Реальная их доля наверняка больше половины, поскольку не все вещества данной группы были испытаны на канцерогенность в [34]. Следовательно, канцерогенная активность макрообразца характеризуется величиной его работы выхода. Последняя, помимо энергии ионизации макрообразца, имеет также смысл его сродства к электрону. Таким образом, канцерогенные свойства присущи и металлам с высокими значениями сродства к электрону (как это было постулировано Миллерами в отношении молекулярных веществ). Эти вещества прочно удерживают свои собственные электроны, а кроме того, эффективно захватывают и сторонние [35].
Рис. 3.
Разбиение элементов Таблицы Менделеева на две группы в зависимости от значений работы выхода электронов из них. Красным цветом показаны элементы, которые являются канцерогенами [34].

Из приведенного рассмотрения различных методов детектирования канцерогенов следует, что ни один из них (включая даже и эпидемиологические испытания на людях) не позволяет утверждать со стопроцентной вероятностью, является ли данное вещество канцерогеном или нет, а если да, то в какой степени. Поэтому различные методы приходится применять комплексно, создавая батареи тестов, и по сумме всех результатов делать окончательный вывод. Очевидно, что испытания на животных (будучи наиболее надежными) являются весьма дорогими и длительными. Поэтому они не позволяют охватить весь поток соединений, с которыми мы сталкиваемся на протяжении жизни (промышленные выбросы, продукты питания, лекарства, косметика и т.д.). Более того, эти соединения могут попадать в организм не в отдельно взятом виде, а в виде комбинаций, число которых оказывается непомерно большим [16]. Поэтому разработка быстрых и относительно дешевых физико-химических методов детектирования канцерогенов по-прежнему остается важной задачей.
Одной из целей настоящего обзора является показать перспективность позитронной аннигиляционной спектроскопии для детектирования канцерогенных соединений и оценки их активности.
3. НЕКОТОРЫЕ СВЕДЕНИЯ О ПОЗИТРОННОЙ АННИГИЛЯЦИОННОЙ СПЕКТРОСКОПИИ (ПАС)
Позитрон (е+), первая открытая частица антиматерии, является стабильным образованием в уединенном состоянии, но оказавшись по-соседству с электроном, позитрон аннигилирует с ним11. Энергия, включая энергию покоя e+ и e–, и импульс электрон-позитронной пары превращаются в энергию и импульс аннигиляционных фотонов.
В среде наиболее вероятен процесс двухфотонной аннигиляции: e+ + e– → 2γ по 511 кэВ. Этот процесс разрешен, когда суммарный спин позитрона и электрона равен нулю. В среде при встречах e+ и e– только с вероятностью 1/4 образуются пары с нулевым полным спином. Скорость 2γ-аннигиляции позитрона определяется величиной электронной плотности, ne, вблизи e+ [37]:
Здесь c = 3 × 1010 см/с – скорость света в вакууме, r0 = ${{e}^{2}}{\text{/}}m{{c}^{2}}$ = $\alpha \hbar {\text{/}}mc$ = 2.8 × 10–13 см – классический радиус электронов, e – заряд и m – масса электрона, α = 1/137 – постоянная тонкой структуры. В воде, например, где ne ≈ 3 × 1023 см–3, λ2γ ≈ ≈ 2 нс–1. Эта оценка применима к большинству молекулярных жидкостей.Суть ПАС состоит в инжекции e+ в образец и последующей регистрации параметров аннигиляционных γ-квантов [38]. Существует три основные разновидности e+-спектроскопии (рис. 4):
Рис. 4.
Основные экспериментальные методики позитронной аннигиляционной спектроскопии: временная, угловая и доплеровская. PALS – Positron Annihilation Lifetime Spectroscopy, ACAR – Angular Correlation of Annihilation Radiation, DBAR – Doppler Broadening of Annihilation Radiation.

1) временнáя спектроскопия, когда измеряются времена жизни позитронов в изучаемой среде;
2) угловая спектроскопия, когда измеряются углы разлета аннигиляционных фотонов (отклонение угла разлета от 180 градусов) и
3) доплеровская спектроскопия, где измеряются энергии аннигиляционных γ-квантов.
В медицине наиболее известным применением позитронной аннигиляции является позитронная эмиссионная томография (ПЭТ), когда по линиям разлета аннигиляционных фотонов восстанавливают область локализации заранее внутривенно введенного пациенту радиофармпрепарата (как правило, это дезоксифторглюказа, допированная изотопом 18F). Так как опухолевые клетки в сравнении с обычными потребляют заметно больше глюкозы, в них концентрация радиофармпрепарата накапливается до существенно бóльших значений, чем в среднем в организме.
Позитроны также широко применяются в материаловедении в качестве зонда локальной структуры конденсированных сред. С их помощью можно изучать дефекты вакансионного типа, определять нанопористость веществ (или распределение по размеру элементов свободного объема в исследуемых образцах), измеряя импульсы внутренних (коровых) e–, с которыми происходит аннигиляция, осуществлять локальный химический анализ мест аннигиляции [39, 40].
3.1. Источники позитронов
Позитроны в позитронной спектроскопии, как правило, получают в результате β+-распада подходящих протонно-избыточных радиоактивных изотопов. Для радиационно-химических и материаловедческих исследований используют долгоживущие изотопы, 22Na (T1/2 = 2.6 года) или 44Ti (T1/2 = 59 лет). Кроме большого времени жизни этих изотопов, их распад сопровождается испусканием “стартового” фотона, энергичного γ-кванта, практически одновременно рождающегося с позитроном. Последнее обстоятельство особенно важно для временнóй позитронной спектроскопии, поскольку именно оно позволяет определять времена жизни позитронов в исследуемых средах.
Напротив, в ПЭТ основное требование к источнику e+ другое. Оно состоит в минимизации воздействия излучения на пациента. Поэтому там требуются короткоживущие радиоактивные ядра с периодом полураспада около нескольких часов. Обычно это 18F (T1/2 = 110 мин) или 44Sc (T1/2 = 3.9 ч).
Позитроны, образующиеся в процессе β+-распада, имеют непрерывный энергетический спектр (от 0 до Emax). При этом их средняя начальная энергия примерно равна 0.4Emax. В зависимости от своей начальной энергии глубина проникновения позитрона в исследуемой среде (ρ – ее плотность) различна. С хорошей точностью профиль имплантированных позитронов по глубине является экспоненциальным.
Так, например, позитроны, получающиеся при распаде 22Na (Emax = 545 кэВ) проникают в воду на глубину до 1 мм, а в железо – до 100 μм. При использовании изотопа 44Ti эти величины возрастают примерно в 4 раза. Проводя эксперименты, необходимо обеспечить такую толщину исследуемой среды вокруг источника е+, чтобы все испускаемые позитроны не вылетели за ее пределы.
Обычно вещество, содержащее β+-радиоактивный источник позитронов, герметично запечатывается в тонкую (8 μм) пленку из каптона или в титановую фольгу (10 μм). В последние годы в экспериментах с жидкими средами, которые были выполнены в НИЦ КИ – ИТЭФ (см. ниже), источником е+ служил порошок тетрахлорида титана, обогащенный изотопом 44Ti (активностью 7.5 МБк) и запечатанный в титановую фольгу. Источник фиксировался в центре виалы с исследуемым раствором посредством тонкого зажима из нержавеющей стали [41].
3.2. Механизм образования атома позитрония в конденсированных молекулярных средах. Ps-пузырек
Разработанный нами механизм образования атома Ps в конденсированных молекулярных средах, наряду с выявленным Миллерами свойством электрофильности канцерогенов и установленным Бакалом диффузионным контролем скорости их реакций с внутритрековыми предсольватированными электронами, есть одна из трех опор, на которых базируется применение позитронной спектроскопии для выявления химических канцерогенов и антиканцерогенов.
Поначалу механизм образования Ps в треке быстрого позитрона обсуждался независимо от процессов образования других внутритрековых продуктов, возникающих под действием ионизирующих излучений (быстрых электронов, α-частиц, осколков деления урана). Причина этого – первоначальные представления об образовании Ps – модель Оре. Согласно этой модели горячий позитрон, е+*, с энергией в несколько эВ отрывает электрон от одной из ближайших молекул среды, образуя с ним атом Ps [42]. Этот процесс никак не увязан с другими радиолитическими процессами в треке е+*. Напротив, выдвинутый нами механизм, основанный на комбинации термализованного позитрона с одним из трековых “сухих” электронов, тесно связан с другим общим и тоже трековым процессом – образованием водорода, H2, одного из главных продуктов радиационно-химических превращений всех водородсодержащих веществ [43]. Мы исходим из того, что образование Ps имеет место в блобе22 на конечном участке трека быстрого позитрона [44]. В отличие от Ps, образование радиолитического водорода происходит не только в блобах, но также и в шпорах на всем протяжении е+ и e– треков. И атом Ps, и радиолитический водород возникают в ходе одновременно протекающих реакций (2), в которых первичные химические реагенты – потерявший энергию позитрон, е+ и многочисленные катион-радикалы (“электронные дырки”) (H2O, H2O)+ рекомбинируют с термализованными, но “сухими”, еще несольватированными электронами е–, рис. 5.
Рис. 5.
Схема трека быстрого позитрона в жидких средах (например, в воде), механизмы образования Н2 и Ps, трансформация Ps в пузырьковое состояние.

Несколько упрощая картину, оба процесса образования можно изобразить следующими реакциями:
(2)
${{{\text{e}}}^{ + }} + {{{\text{e}}}^{ - }} \to {\text{Ps}},\quad {{({{{\text{H}}}_{{\text{2}}}}{\text{O}},{{{\text{H}}}_{{\text{2}}}}{\text{O}})}^{ + }} + {{{\text{e}}}^{ - }} \to {{{\text{H}}}_{{\text{2}}}} + 2{\text{OH}}{\text{.}}$В водных средах атомы Ps образуются примерно в 40% е+-треков, тогда как доля возникающих молекул водорода по отношению к изначально образовавшимся ион-электронным парам равна ≈1/12. Достаточно эффективные акцепторы электронов способны практически полностью подавить как выходы Ps, так и водорода [43, 45].
Процесс образования Ps не оканчивается завершением реакции (2). Дело в том, электрон, входящий в состав Ps, испытывает обменное отталкивание (вследствие принципа Паули) от электронов соседних молекул. Поэтому Ps является сольвофобной частицей. Вокруг Ps возникает нанопузырек (сфероидальная полость), поверхность которого выстилают молекулы среды. Образованию Ps-пузырька способствует малая масса атома Ps, 2m. Она стоит в знаменателе выражения для энергии нулевых колебаний Ps [46, 47]. Именно по этой причине Ps-пузырек в жидких средах оказывается достаточно крупным образованием (с радиусом от нескольких ангстрем до 20 Å в жидком гелии!).
3.3. Пара- и орто-позитроний. Pick-off аннигиляция
В зависимости от значения суммарного спина (ноль или $1 \times \hbar $) позитрона и электрона в момент формирования Ps происходит образование либо пара- (p-Ps), либо орто-позитрония (o-Ps). Атом p-Ps быстро (в вакууме за 124 пс) аннигилирует на два фотона с энергиями по 511 кэВ. Атом o-Ps аннигилирует на нечетное число фотонов (обычно на три). В вакууме процесс трехфотонной аннигиляции o-Ps почти в тысячу раз более медленный, время жизни o-Ps составляет 142 нс.
В конденсированных средах время жизни o-Ps сокращается в десятки раз из-за протекания так называемого pick-off процесса. Он состоит в том, что позитрон, входящий в состав o-Ps, аннигилирует не со “своим”, а с принадлежащим одной из соседних молекул “чужим” электроном, спин которого антипараллелен спину e+ [38, 42]:
(4)
$o{\text{ - Ps}}{\kern 1pt} ( \uparrow \uparrow ) + {{{\text{e}}}^{ - }}( \downarrow ) \to 2\gamma + {{{\text{e}}}^{ - }}( \uparrow ).$Как было отмечено выше, скорость 2γ-аннигиляции позитрона и квазисвободного позитрония, находящихся в однородном континууме, определяется средней электронной плотностью, (1). В этом случае имеет место 100 процентное перекрытие волновой функции e+ с электронной плотностью молекул среды. Для Ps, находящегося в пузырьке, это перекрытие уменьшается. Если атом Ps в нанопузырьке моделировать точечной квантовой частицей с массой 2m, находящейся в сферической бесконечно-глубокой яме радиуса R∞, внутрь которой на глубину δ проникают электроны ближайших молекул (R – радиус свободного объема пузырька), то для скорости pick-off аннигиляции Ps получается следующее выражение, которое называется формулой Тао-Элдрупа [48]:
(5)
$\begin{gathered} {{\lambda }_{{po}}} = {{\lambda }_{{2\gamma }}}{{P}_{R}},\quad {\text{где}}\quad {{P}_{R}} = 1 - \frac{R}{{{{R}_{\infty }}}} + \\ + \;\frac{{\sin (2\pi R{\text{/}}{{R}_{\infty }})}}{{2\pi }} = \frac{\delta }{{{{R}_{\infty }}}} - \frac{{\sin (2\pi \delta {\text{/}}{{R}_{\infty }})}}{{2\pi }}. \\ \end{gathered} $Наличие во временнóм спектре долгоживущей компоненты (τ3 > 1.5 нс; в отсутствие химических реакций с участием позитрония τ3 = 1/λpo) – признак существования атома o-Ps в “пузырьковом” состоянии. Это время жизни достаточно велико и позволяет o-Ps принимать участие в химических реакциях. При этом его аннигиляционные параметры (в первую очередь время жизни) оказываются чувствительными к присутствию в треке позитрона радикалов, парамагнитных частиц и других примесных молекул.
3.4. Измерение времени жизни позитронов: схема эксперимента
Суть временн${\text{ы}}'$х аннигиляционных экспериментов состоит в измерении временн${\text{ы}}'$х интервалов между регистрацией “стартовых” фотонов, сопровождающих рождение позитронов, и стоп-сигналов – фотонов с энергией ≈511 кэВ, возникающих при двухфотонной аннигиляции позитронов. Принципиальная схема временнóй установки приведена на рис. 6.
Рис. 6.
Схема установки по измерению времени жизни позитронов. Сц – сцинтиллятор, (обычно это кристаллы BaF2), ФЭУ – фотоэлектронный умножитель, дискриминатор – дискриминатор постоянной доли. Приведенная энергия стартовых фотонов соответствует использованию источника e+ на основе 22Na.

Как правило, обработка временн${\text{ы}}'$х спектров сводится к разложению спектра на несколько (в средах, где возможно образование Ps, обычно на три) убывающих во времени экспоненты. В первом приближении они описывают аннигиляцию парапозитрония (τ1 = 1/λ1), “свободных” позитронов (τ2 = 1/λ2) и ортопозитрония (τ3 = 1/λ3):
(6)
$\begin{gathered} {{C}_{{{\text{theor}}}}}(t) = {{C}_{{{\text{tot}}}}}\sum\limits_i {\frac{{{{I}_{i}}}}{{{{\tau }_{i}}}}\exp \left( { - \frac{t}{{{{\tau }_{i}}}}} \right) = } \\ = {{C}_{{{\text{tot}}}}}\sum\limits_i {{{\lambda }_{i}}{{I}_{i}}\exp ( - {{\lambda }_{i}}t).} \\ \end{gathered} $Для примера на рис. 7 приведен типичный временнóй аннигиляционный спектр, измеренный в чистой воде при комнатной температуре, и его трехэкспоненциальное разложение на p-Ps, free-e+ и o-Ps компоненты.
Рис. 7.
Пример временнóго аннигиляционного спектра е+ в воде и его разложение на вклады от аннигиляций p-Ps, “свободных” е+ и o-Ps (τp-Ps = 1/λ1, ${{\tau }_{{{{{\text{e}}}^{ + }}}}}$ = 1/λ2, τo-Ps = 1/λ3; Ip-Ps = I1, ${{I}_{{{{{\text{e}}}^{ + }}}}}$ = I2, Io-Ps = I3). В спектре накоплено 23 млн старт-стоп совпадений, спектр измерялся в течение 21 часа при T = 21°C.

3.5. Применение позитронной спектроскопии в онкологии
Выше отмечалось, что время жизни o-Ps существенно зависит от наличия свободного объема в исследуемых объектах. Первые биологические приложения позитронной спектроскопии опирались именно на это свойство. Так, в работе [52] изучался рост нормальных клеточных колоний и колоний раковых клеток прямой кишки. Измерения времени жизни o-Ps в этих культурах показали, что пролиферация (разрастание) раковых клеток сопровождается образованием более “ажурной” структуры ткани, содержащей внутренние полости, нежели это имеет место в процессе роста здоровых клеток. Проявления подобного “избыточного” свободного объема способна регистрировать позитронная спектроскопия.
В работах [53, 54] исследовались здоровые и раковые клетки базального (лежащего в основании) слоя кожи одного и того же пациента. Было обнаружено, что в опухолевых клетках время жизни Ps меньше, чем в здоровых. Это является указанием на повышенную концентрацию в раковых клетках свободных радикалов, реакция с которыми сокращает длительность жизни o-Ps.
Большая группа польских авторов методом ПАС изучала образцы маточной ткани (как здоровой, так и ее доброкачественной опухоли – лейомиомы), взятые непосредственно в ходе хирургических операций [55]. Было обнаружено как удлинение времен жизни е+ и o-Ps в опухолевых тканях, так и уменьшение интенсивностей этих же компонент во временн${\text{ы}}'$х спектрах. Хотя феноменологические корреляции аннигиляционных параметров с наличием опухолей установ- лены, но причины этих взаимосвязей еще предстоит понять. Тем не менее, отмеченные исследования указывают на то, что позитронная спектроскопия может служить инструментом ранней диагностики развития рака.
Сходное исследование было проведено на хрусталиках глаза свиней [56]. Сам хрусталик устроен очень “примитивно”. Он состоит из не содержащих никаких органелл вытянутых клеток эпителия, внутри которых находятся молекулы белков. По увеличению времени жизни o-Ps в хрусталике было показано, что при T = 35–36°C в нем происходит фазовый переход, связанный с перестройкой локальной структуры хрусталика.
4. СРАВНЕНИЕ ПОЗИТРОННЫХ ЭКСПЕРИМЕНТОВ С РЕЗУЛЬТАТАМИ УСКОРИТЕЛЬНЫХ ЭКСПЕРИМЕНТОВ ДЖ. БАКАЛА
В 2006 г. ГНЦ РФ ИТЭФ им. А.И. Алиханова получил патент [57] на метод определения канцерогенности веществ, основанный на использовании позитронной аннигиляционной спектроскопии [33, 58]. Основными преимуществами использования позитронной спектроскопии для решения проблемы диагностики канцерогенов являются быстрота, простота и дешевизна. Эти качества достигаются благодаря радикальному уменьшению размеров установки, а также низкой величине потока позитронов, генерирующих “сухие” электроны, е–, реагирующие с испытуемым на канцерогенность веществом S.
Напомним, что в методе Бакала для определения константы ke необходим дорогостоящий электронный ускоритель, снабженный громоздкой гамма-защитой. В позитронном же спектрометре роль “ускорителя” быстрых электронов выполняет радиоактивный источник позитронов – запечатанное в тонкую защитную фольгу ничтожное количество радиоактивного вещества, испускающего энергичные (~1 МэВ) позитроны в количестве в несколько миллионов за секунду. По этой причине спектрометр не требует сколь-нибудь серьезной γ‑защиты. Он представляет собой сравнительно небольшую установку, умещающуюся на столе.
Может показаться, что различие между позитронной диагностикой и ke-тестом Бакала сводится лишь к разным способам генерации избыточных электронов в исследуемом растворе. Однако, это не вполне так. Методики предъявляют разные требования к концентрациям испытуемых веществ в измерительных ячейках детекторов.
В экспериментах по импульсному радиолизу растворенное вещество S присутствует обычно в концентрации cS порядка миллимолей/литр. Поэтому время жизни электрона до захвата его акцептором S в этих условиях достаточно большое: 1/k(e– + S)cS ≈ 10–8 с. Напротив, в позитронных экспериментах реакция захвата трековых электронов испытуемым веществом S должна завершаться гораздо быстрее, за времена порядка времени жизни е+ до аннигиляции, 1 нс. В противном случае эффект от присутствия электронного акцептора в растворителе не будет замечен. Необходимое ускорение реакции достигается повышением концентрации испытуемого вещества S в растворе. Следствием этого в процессах с участием позитронов реагируют, вообще говоря, предсольватированные, “сухие” электроны в квазисвободном состоянии, со значениями констант скорости реакции e– + S → S–, превышающими значения констант скорости, присущих сольватированному электрону в условиях наносекундного импульсного радиолиза. Таким образом, позитронная диагностика является новым физико-химическим методом оценки канцерогенной опасности химических соединений.
Итак, выявление канцерогенных свойств веществ базируется на свойстве сильной электрофильности большинства канцерогенов, их способности прочно связывать “cухие” электроны, возникающие в ходе облучения исследуемой среды быстрыми ионизирующими частицами. Эксперименты показывают, что в растворах веществ, не являющихся канцерогенно-опасными, вероятность образования Ps велика – 30–60%. Напротив, при добавлении канцерогенно-опасного вещества выход Ps падает почти до нуля [59] (в этом суть эффекта ингибирования образования Ps). Таким образом, измеряя коэффициенты ингибирования образования Ps, можно провести ранжирование канцерогенно-опасных веществ более просто и легко.
В позитронных исследованиях, как правило, определялся феноменологический коэффициент ингибирования, qS, образования Ps, см. ниже формулу (19):
(7)
${{P}_{{{\text{Ps}}}}}({{c}_{S}}) = \frac{{{{P}_{{{\text{Ps}}}}}({{c}_{S}} = 0)}}{{1 + {{q}_{S}}{{c}_{S}}}}.$Экспериментальные данные, рис. 8, демонстрируют наличие хорошей корреляции между коэффициентами qS ингибирования o-Ps, производимого рядом тестируемых химических соединений в растворах циклогексана и константами скорости ke их реакций с “сухими” электронами, порождаемыми в условиях наносекундного импульсного радиолиза. Прямая пропорциональность коэффициентов qS и ke друг другу означает, что они одинаково способны характеризовать степень электрофильности, а следовательно, и канцерогенность электронных акцепторов S. И позитронный, и метод импульсного радиолиза приводят к сходному порядку распределения тестируемых соединений по степени канцерогенности.
Рис. 8.
Корреляция между коэффициентами qS ингибирования образования Ps растворенными в циклогексане электронными акцепторами S и константами скорости захвата ими же трековых электронов ${\text{e}}_{{{{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{{\text{12}}}}}}}^{ - }$ в том же растворителе (циклогексане) [33, 58].

Коэффициенты Ps ингибирования некоторыми химическими соединениями, растворенными в этаноле, также пропорциональны их канцерогенной активности, определенной независимо, рис. 9.

Примечательно, что сами вероятности образования Ps, измеренные непосредственно в химических соединениях без их растворения в каком-либо растворителе, также хорошо коррелируют с их канцерогенными свойствами. В веществах, не являющихся канцерогенами, выход Ps оказывается большим, тогда как в сильных канцерогенах он не превосходит 2–3%, рис. 10. Данный факт может упростить выявление канцерогенно-опасных соединений (хотя и не во всех случаях) за счет сокращения числа измерений при разных концентрациях тестируемого вещества.

5. Ps-АНТИИНГИБИТОРЫ – ВОЗМОЖНЫЕ АНТИКАНЦЕРОГЕНЫ!?
Позитронный метод не только облегчает и ускоряет выявление канцерогенов (в сравнении с методом импульсного радиолиза), но и открывает новые возможности в понимании механизма канцерогенного действия. Проиллюстрируем это примерами.
К химическим соединениям, эффективно реагирующим с трековыми электронами, относятся антрацен, пирен и фенантрен. Согласно критерию Бакала (рис. 2) их следовало бы отнести к канцерогенам, но биологические данные не подтверждают этого [60]. Напротив, биологи относят эти соединения к антиканцерогенам. Так называют вещества, введение которых в организм подавляет вредное действие находящихся в нем канцерогенов [60]. При наличии в организме фенантрена канцерогены теряют способность к образованию злокачественных опухолей (подопытные животные, которым давали это вещество, оказывались устойчивыми к канцерогенным препаратам). Интересно, что введение фенантрена в систему н-гексан + 0.2 М C6H5Br (в которой канцероген C6H5Br более чем в 2 раза подавляет выход Ps) восстанавливает выход Ps, рис. 11 [61]. Это означает, что влияние ингибирующего действия C6H5Br ослабевает по мере роста концентрации фенантрена, то есть антиканцероген фенантрен является и антиингибитором образования Ps.
Рис. 11.
Эффект антиингибирования Ps: введение фенантрена в н-гексан, содержащий 0.2 М C6H5Br, восстанавливает выход Ps в чистом гексане [61]. Верхняя кривая демонстрирует неизменность выхода Ps при добавлении фенантрена в чистый гексан.

Причина подобного поведения состоит в следующем. Ингибирование образования Ps бром-бензолом связано с необратимым захватом электрона, сопровождающимся образованием аниона C6H5Br– и его последующей быстрой диссоциацией на радикал C6H5 (EA = 2.2 эВ) и анион Br–:
(8)
${{{\text{e}}}^{ - }} + {{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{\text{5}}}}{\text{Br}} \to {{({{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{\text{5}}}}{\text{Br}})}^{ - }}{\kern 1pt} * \to {{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{\text{5}}}} + {\text{B}}{{{\text{r}}}^{ - }}.$Напротив, захват e– молекулой фенантрена ведет к образованию сравнительно долгоживущего возбужденного состояния, (C14H10)–*. Этот анион удерживает e– весьма слабо (энергия сродства мала: EA = 0.1 эВ). Потому при встрече этого аниона с позитроном электрон переходит к e+ с образованием атома Ps. Просто добавление фенантрена к гексану заметно не сказывается на выходе Ps, рис. 11 [61]. Напротив, добавление фенантрена к раствору 0.2 М C6H5Br предотвращает необратимый захват трековых e– молекулами C6H5Br (электроны сохраняются для реакции с позитроном). Конечно, анион фенантрена ${\text{C}}_{{14}}^{{}}{\text{H}}_{{10}}^{ - }{\kern 1pt} *$ мог бы отдать захваченный e– не только позитрону, но и электрофильной молекуле C6H5Br (EA = 3.4 эВ), но коэффициенты диффузии C6H5Br и аниона фенантрена столь низки, что за время образования o-Ps они просто не успевают встретиться. В результате выход Ps в системе н-гексан + 0.2 М C6H5Br при добавлении фенанрена увеличивается. Это и есть проявление эффекта антиингибирования образования Ps.
По-видимому, проникая в клетку живого организма, подобные “слабые” акцепторы электронов не образуют сильную ковалентную связь с молекулами ДНК и не приводят к ее мутациям. Однако, адсорбция таких акцепторов на нуклеофильной поверхности молекулы ДНК может создавать “защитный” эффект. Поэтому можно ожидать, что присутствие в клетках антиингибиторов Ps типа фенантрена будет иметь антиканцерогенное действие [33, 58].
Рассмотрим биологические эффекты, производимые еще одним антиингибитором Ps – диоксидом углерода, CO2. Его антиингибирующее действие в н-гексане с предварительно добавленным туда Ps-ингибитором – йодбензолом, C6H5I, в концентрации 0.03 M – показано на рис. 12. Если отмеченная выше связь антиканцерогенных эффектов, производимых Ps антиингибиторами (напр., фенантреном) в организме человека не является случайной, то следует ожидать, что и диоксид углерода должен проявлять себя как антиканцероген.

Опыт использования углекислоты при онкологических заболеваниях оправдывает этот прогноз. Так, в работе [62] описаны эксперименты, в которых мышам прививали аденокарциному. Контрольную группу мышей поили обычной водой, а подопытных – раствором NaHCO3. Оказалось, что у животных, которые пили исключительно раствор бикарбоната (12 г/л), метастазирование было резко подавлено, рис. 13, слева). В итоге спустя 4 месяца после прививки опухоли в подопытной группе выжило 80% мышей, тогда как в контрольной осталось в живых только 30%.
Рис. 13.
Слева: антиканцерогенное действие бикарбоната натрия. Подавление метастазирования у мышей с перевитой аденокарциномой. Пунктирная линия – выживаемость мышей, потреблявших только раствор бикарбоната, сплошная линия – выживаемость мышей, которых поили обычной водой [62]. Справа: лечение почечной недостаточности посредством ежедневного перорального приема питьевой соды (2 г/день) [63].
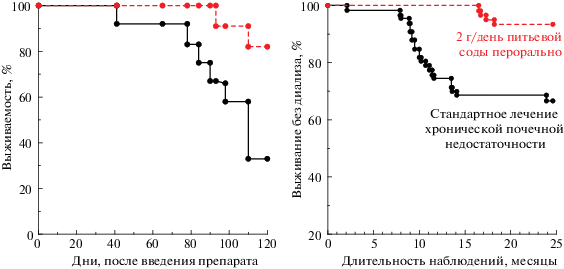
Из литературы известно и о других биологических эффектах, производимых диоксидом углерода. Оказывается, что это химическое соединение при введении в организм в составе минеральной воды или в виде содового раствора ослабляет и даже устраняет ряд протекающих в организме паталогических процессов. Так, например, замедляется развитие хронических заболеваний почек (рис. 13, справа) [63].
Таким образом, присутствие в растворах химических соединений, подобных фенантрену и углекислоте (которые эффективно реагируют с трековыми электронами, но имеют к ним низкое сродство), восстанавливает выход Ps при наличии Ps-ингибитора. Это очень похоже на поведение антиканцерогенов в живом организме, ослабляющих действие содержащихся в нем канцерогенов и иных патогенов.
Третьим примером, демонстрирующим одновременное наличие как антиингибирующих (в отношении Ps), так и антиканцерогенных свойств, являются соединения класса перфторуглеродов (перфторан, гексафторбензол). Еще в 60–70-х годах прошлого века появилась идея о создании кровезаменителей донорской крови для возмещения кровопотерь людей. Исследования велись большими группами институтов как в СССР, так и в США. Перфторан представляет собой мелкодисперсную эмульсию (коллоид), обладающую огромной поверхностью с большой сорбционной активностью, обеспечивающую эффективный газообмен. Высокая растворимость газов в перфторуглеродах обусловлена наличием в них многочисленных пустот (на молекулярном масштабе), в которые внедряются молекулы газов (O2, CO2) и где слабо связываются за счет Ван-дер-Ваальсовых сил. При этом с химической точки зрения фторуглероды – достаточно инертные и потому безопасные для организма соединения.
При травмах и стрессах ответной реакцией организма является сжатие кровеносных сосудов. В результате ухудшается кровоток и снабжение клеток кислородом. В бескислородной среде начинает преобладать гликолиз – расщепление глюкозы до молочной кислоты. При этом в клетках накапливаются ионы водорода (сильные электрофилы). Это закисление среды создает условия для развития онкологических заболеваний. В этом случае перфторановая эмульсия проявляет себя как “антиканцероген”, восстанавливая pH среды и снабжая ткани кислородом.
С другой стороны известно, что некоторые из перфторуглеродов являются антиингибиторами образования Ps. Так, например, гексафторбензол (C6F6) является эффективным акцептором квазисвободных электронов [64–66]. Энергия сродства C6F6 к электрону невелика (~0.5 эВ). Поэтому не удивительно, что наличие антиингибирующих свойств у C6F6 в отношении Ps было подтверждено в циклогексановых растворах C6F6–C2H5Br.
6. МОДЕЛЬ ОБРАЗОВАНИЯ Ps ПРИ НАЛИЧИИ ИНГИБИТОРОВ И АНТИИНГИБИТОРОВ
Итак, эффект ингибирования образования Ps проявляется в уменьшении выхода Ps при добавлении в исследуемый раствор акцептора S квазисвободных (трековых) электронов. Эффект антиингибирования состоит в восстановлении выхода Ps при добавлении в раствор ингибитора S другого вещества – антиингибитора, A (это “слабый” акцептор электронов, характеризующийся малой энергией связи). Антиингибитор перехватывает трековые электроны и образуется слабосвязанное состояние A–. При встрече A– с позитроном электрон от A– передается позитрону, что и приводит к увеличению выхода атома Ps.
Выше отмечалось, что наиболее подходящими растворителями, моделирующими внутриклеточную среду, по-видимому, являются органические среды. Дж. Бакал проводил свои эксперименты в циклогексане. Для позитронных экспериментов одним из наиболее удобных оказался 1,4-диоксан. Он относится к простым эфирам, апротонным веществам, малореакционным по отношению к электрону. Сольватированный e– в диоксане участвует в тех же реакциях, что и в других жидкостях. Скорость захвата его эффективными акцепторами контролируется диффузией или приближается к этому пределу [67].
В простейшем виде интерпретацию экспериментов по ингибированию и антиингибированию Ps можно провести в рамках следующего набора реакций, протекающих на конечном участке трека позитрона. А именно,
– захват трековых электронов молекулами ингибитора и антиингибитора:
(9)
${{{\text{e}}}^{ - }} + {\text{S}} \to {{{\text{S}}}^{ - }},\quad {{{\text{e}}}^{ - }} + {\text{A}} \to {{{\text{A}}}^{ - }},$– сольватация е– и е+:
(10)
${{{\text{e}}}^{ - }} \to {\text{e}}_{{{\text{solv}}}}^{ - },\quad {{{\text{e}}}^{ + }} \to {\text{e}}_{{{\text{solv}}}}^{ + }$– образование Ps посредством комбинации е+ с “сухим” электроном, а также в результате отрыва слабосвязанного электрона от аниона A–:
(11)
${{{\text{e}}}^{ + }} + {{{\text{e}}}^{ - }} \to {\text{Ps}},\quad {{{\text{e}}}^{ + }} + {{{\text{A}}}^{ - }} \to {\text{Ps}} + {\text{A}}.$Математически все реакции можно описать следующей системой уравнений для населенностей соответствующих состояний (ne – число электронов, порожденных в е+-блобе; ${{n}_{{{{{\text{A}}}^{ - }}}}}$ – число анионов A–). Ниже принято, что момент времени t = 0 отвечает не только окончанию процесса ионизационного замедления позитрона, но также завершению процессов термализации всех трековых частиц:
(12)
$\begin{gathered} {{{\dot {n}}}_{e}} = - ({{W}_{s}} + {{k}_{{eS}}}{{c}_{S}} + {{k}_{{eA}}}{{c}_{A}}){{n}_{e}} \to {{n}_{e}}(t) = {{n}_{0}} \cdot {{{\text{e}}}^{{ - {{W}_{e}}t}}}, \\ {{W}_{e}} = {{W}_{s}} + {{k}_{{eS}}}{{c}_{S}} + {{k}_{{eA}}}{{c}_{A}}, \\ \end{gathered} $(13)
${{\dot {n}}_{{{{A}^{ - }}}}} = {{k}_{{eA}}}{{c}_{A}}{{n}_{e}} = {{k}_{{eA}}}{{c}_{A}}{{n}_{0}} \cdot {{{\text{e}}}^{{ - {{W}_{e}}t}}},\quad {{n}_{{{{A}^{ - }}}}}(0) = 0.$(14)
${{n}_{{{{A}^{ - }}}}}(t) = \frac{{{{k}_{{eA}}}{{c}_{A}}{{n}_{0}}}}{{{{W}_{e}}}}(1 - {{{\text{e}}}^{{ - {{W}_{e}}t}}}).$Для описания судьбы позитрона необходимо учесть его сольватацию (полагаем, что ее скорость Ws примерно та же, что и для е–), аннигиляцию со скоростью λp (в (6) эта величина обозначалась как λ2) и два канала образования атома Ps – реакцию (ре)комбинации с одним из трековых квазисвободных электронов, а также реакцию отрыва электрона от аниона А–:
(15)
$\begin{gathered} {{{\dot {n}}}_{p}} = - ({{W}_{s}} + {{\lambda }_{p}}){{n}_{p}} - \frac{{{{k}_{{pe}}}{{n}_{e}}(t){{n}_{p}}}}{{V(t)}} - \frac{{{{k}_{{p{{A}^{ - }}}}}{{n}_{{{{A}^{ - }}}}}(t){{n}_{p}}}}{{V(t)}}, \\ {{n}_{p}}(0) = 1. \\ \end{gathered} $В этом уравнении V(t) имеет смысл объема блоба, который со временем увеличивается в результате амбиполярной диффузии заряженных частиц. Используя метод предписанной диффузии, упрощающий описание негомогенной кинетики внутритрековых процессов, можно показать, что V(t) ∝ t3/2. Однако, поскольку длительность реакций образования Ps малá, мы пренебрежем изменением объема блоба: V(t) ≈ V.
Помимо этого сделаем еще одно упрощение, позволяющее получить аналитическое выражение для кинетики гибели е+. В его правой части (15) будем считать, что фигурирующие там величины ne(t) и ${{n}_{{{{A}^{ - }}}}}(t)$ берутся не в произвольный момент времени, а в момент времени $\tilde {t}$, когда $\exp ( - {{W}_{e}}\tilde {t})$ = 1/2. Тогда кинетика исчезновения позитронов становится чисто экспоненциальной и характеризуется скоростью Wp:
(16)
$\begin{gathered} {{n}_{p}}(t) = {{e}^{{ - {{W}_{p}}t}}},\quad {{W}_{p}} = {{W}_{s}} + {{\lambda }_{p}} + {{W}_{{pe}}} + {{W}_{{p{{A}^{ - }}}}}, \\ {{W}_{{pe}}} \approx \frac{{{{k}_{{pe}}}{{n}_{0}}}}{{2V}},\quad {{W}_{{p{{A}^{ - }}}}} \approx \frac{{{{k}_{{p{{A}^{ - }}}}}{{n}_{0}}}}{{2V}}\frac{{{{k}_{{eA}}}{{c}_{A}}}}{{{{W}_{e}}}}. \\ \end{gathered} $В результате вероятность образования атома Ps выражается суммой двух членов:
(17)
$\begin{gathered} {{P}_{{{\text{Ps}}}}}({{c}_{S}},{{c}_{A}}) = {{k}_{{pe}}}\int\limits_0^\infty {\frac{{{{n}_{e}}{{n}_{p}}}}{V}dt} + {{k}_{{pA}}} - \int\limits_0^\infty {\frac{{{{n}_{{{{A}^{ - }}}}}{{n}_{p}}}}{V}dt} = \\ = \frac{{{{W}_{{pe}}}}}{{{{W}_{e}} + {{W}_{p}}}} + \frac{{{{W}_{{p{{A}^{ - }}}}}}}{{{{W}_{p}}}} - \frac{{{{W}_{{p{{A}^{ - }}}}}}}{{{{W}_{e}} + {{W}_{p}}}}. \\ \end{gathered} $Фитирование экспериментальных данных и определение входящих сюда подгоночных параметров проводим в два этапа. На первом этапе рассматриваем данные, когда в изучаемом растворе нет антиингибитора (cA = 0). Тогда второй и третий члены в (17) отсутствуют:
(18)
$\begin{gathered} {{P}_{{{\text{Ps}}}}}({{c}_{S}}) = \frac{{{{W}_{{pe}}}}}{{W_{e}^{0} + W_{p}^{0}}},\quad W_{e}^{0} = {{W}_{s}} + {{k}_{{eS}}}{{c}_{S}}, \\ W_{p}^{0} = {{W}_{s}} + {{\lambda }_{p}} + {{W}_{{pe}}}. \\ \end{gathered} $(19)
$\begin{gathered} {{P}_{{{\text{Ps}}}}}({{c}_{S}}) = \frac{{{{W}_{{pe}}}}}{{2{{W}_{e}} + {{W}_{{pe}}} + {{\lambda }_{p}} + {{k}_{{eS}}}{{c}_{S}}}}, \\ {{I}_{3}} = {{P}_{{{\text{Ps}}}}}({{c}_{S}}) \times 100\% . \\ \end{gathered} $Рис. 14.
Эффекты ингибирования (слева) и антиингибирования (справа) образования Ps в диоксане. В качестве ингибитора использовался CCl4 (черная кривая). В качестве антиингибиторов использовались фенольные соединения (см. табл. 1).

Фитирование кривых антиингибирования, когда в растворе помимо ингибитора CCl4 с фиксированной концентрацией cS (0.05 и 0.1 М) присутствовал также и антиингибитор с переменной концентрацией cA (вплоть до 0.4 М), проводилось на основе общего выражения (17), рис. 14. C ростом CA вероятность отрыва позитроном электрона от аниона A– возрастает, что приводит к увеличению выхода Ps:
(20)
$\begin{gathered} {{P}_{{{\text{Ps}}}}}({{c}_{S}},{{c}_{A}}) = \frac{{{{W}_{{pe}}}}}{{W_{e}^{0} + {{k}_{{eA}}}{{c}_{A}} + W_{p}^{0} + {{W}_{{p{{A}^{ - }}}}}}} + \\ + \;\frac{{{{W}_{{p{{A}^{ - }}}}}}}{{W_{p}^{0} + {{W}_{{p{{A}^{ - }}}}}}} - \frac{{{{W}_{{p{{A}^{ - }}}}}}}{{W_{e}^{0} + {{k}_{{eA}}}{{c}_{A}} + W_{p}^{0} + {{W}_{{p{{A}^{ - }}}}}}}. \\ \end{gathered} $Если в выражении (16) обозначить комбинацию ${{k}_{{p{{A}^{ - }}}}}{{n}_{0}}{\text{/}}2V$ посредством ${{\omega }_{{p{{A}^{ - }}}}}$ (смысл этой величины есть скорость реакции e+ с анионами A– молекул антиканцерогена), то
(21)
$\begin{gathered} {{P}_{{{\text{Ps}}}}}({{c}_{S}},{{c}_{A}}) = \frac{{{{W}_{{pe}}} - \frac{{{{\omega }_{{p{{A}^{ - }}}}}{{k}_{{eA}}}{{c}_{A}}}}{{W_{e}^{0} + {{k}_{{eA}}}{{c}_{A}}}}}}{{W_{e}^{0} + W_{p}^{0} + {{k}_{{eA}}}{{c}_{A}}\left( {1 - \frac{{{{\omega }_{{p{{A}^{ - }}}}}}}{{W_{e}^{0} + {{k}_{{eA}}}{{c}_{A}}}}} \right)}} + \\ + \;\frac{1}{{1 + \frac{{W_{p}^{0}}}{{{{\omega }_{{p{{A}^{ - }}}}}}}\left( {1 + \frac{{W_{e}^{0}}}{{{{k}_{{eA}}}{{c}_{A}}}}} \right)}}. \\ \end{gathered} $(22)
${{P}_{{{\text{Ps}}}}}({{k}_{{eA}}}{{c}_{A}} \gg 1) \to \frac{{{{\omega }_{{p{{A}^{ - }}}}}}}{{W_{p}^{0} + {{\omega }_{{p{{A}^{ - }}}}}}}.$Данные по эффекту антиингибирования при введении в диоксан, содержащий небольшое количество CCl4, некоторых фенольных соединений показаны на рис. 14. При фитировании данных мы использовали выражение (21). В результате для каждого вещества были определены константы скорости keA и скорости реакции е+ с А–, табл. 1.
Таблица 1.
Значения констант скоростей захвата е– молекулами антиингибиторов A и скоростей реакций е+ с анионами A–
| Aнтиингибитор | [CCl4], M | keA, M–1 c–1 | ${{\omega }_{{p{{A}^{ - }}}}}$, нс–1 |
|---|---|---|---|
| Бензойная кислота | 0.1 | 1.2 × 1013 | 2900 |
| 4-Гидроксибензойная кислота | 0.1 | 1.2 × 1013 | 2900 |
| Бензойная кислота | 0.05 | 1.3 × 1013 | 2900 |
| Салициловая кислота | 0.05 | 1.2 × 1013 | 2900 |
| Фталевая кислота | 0.05 | 7.3 × 1012 | 2900 |
| 3,4-Дигидроксибензойная к-та | 0.05 | ~0 | ~0 |
| 4-Гидроксибензойный альдегид | 0.05 | ~0 | ~0 |
Несмотря на большую погрешность полученных значений (30–50%), видно, что константы скорости захвата предсольватированных электронов этими соединениями в разы меньше соответствующей константы для CCl4. Параметры keA и ${{\omega }_{{p{{A}^{ - }}}}}$ для бензойной кислоты, измеренные при различных концентрациях CCl4, достаточно хорошо совпадают, чего и следовало ожидать.
Если принять, что n0 ≈ 30 и V ≈ 4πR3/3, где R ≈ ≈ 50 Å, то полученное значение ${{\omega }_{{p{{A}^{ - }}}}}$ ≈ 3 × 1012 с–1 приводит к константе скорости отрыва позитроном электрона от А–${{k}_{{p{{A}^{ - }}}}}$ ≈ 6 × 1013 М–1 с–1. Большая величина этой константы связана с высокой подвижностью термализованного позитрона и c кулоновским притяжением между e+ и A–. Заметим, что величина ${{k}_{{p{{A}^{ - }}}}}$ практически совпадает с найденной выше величиной константы скорости захвата квазисвободного e– молекулой CCl4 в диоксане.
Найденные значения констант скорости захвата е– фенольными кислотами примерно такие же, как и в воде. Это указывает, на то, что в этих случаях имеет место захват именно “сухого” электрона.
В литературе содержатся указания о способности бензойной кислоты тормозить возникновение и развитие злокачественных опухолей [68]. Это может служить еще одним доводом в пользу наличия связи между эффектом антиингибирования Ps с антиканцерогенными свойствами химических соединений. Разумеется, пока мы не представляем себе механизмов антиканцерогенного действия, рассмотренные варианты носят гипотетический характер. Тем не менее, существование подобных корреляций представляет интерес, и их поиск заслуживает дальнейшего продолжения.
7. ЗАКЛЮЧЕНИЕ
Цель настоящего обзора состоит в том, чтобы обосновать перспективность метода аннигиляции позитронов для оценки канцерогенных свойств химических соединений и их активности. Главное преимущество позитронной спектроскопии перед использованием метода импульсного радиолиза заключается в том, что генерация высокоэнергичных позитронов, их ионизационное торможение, протекание химических трековых реакций, а также наблюдение за их кинетикой и образующимися продуктами – все это в буквальном смысле протекает в “одном флаконе” (объемом около 10 см3). Такая компактность ускоряет и удешевляет процесс исследования.
Позитронный метод базируется на наличии тесной корреляции между степенью канцерогенности тестируемого вещества и константой скорости захвата его молекулами квазисвободных (“сухих”) электронов, порождаемых воздействием ионизирующего излучения. Трековые электроны, образующиеся в измерительной ячейке позитронного спектрометра после завершения термализации в течение какого-то времени находятся в предсольватированном состоянии. Именно они являются непосредственными предшественниками атома позитрония. Поэтому добавление в раствор, моделирующий внутриклеточную среду, канцерогенных веществ подавляет выход атома Ps. При этом значения коэффициентов ингибирования образования Ps коррелируют со степенью канцерогенности молекул. Это подтверждается независимыми медико-биологическими данными.
Вопрос о выборе “оптимального” растворителя, наилучшим образом моделирующего внутриклеточную среду, в котором и следует проводить тестирование веществ, пока не получил однозначного ответа. Помимо рекомендованного Бакалом циклогексана, мы проводили измерения также в воде и спиртах, где эффективно протекают процессы сольватации электронов и позитронов, а также в неполярных растворителях (циклогексан, н-гексан, диоксан), где эффект акцептирования электронов растворенными добавками проявляется сильнее. В этом случае параметры Ps-ингибирования оказываются бóльшими по величине и их легче измерять. Мнение о том, что именно неполярные растворители лучше моделируют внутриклеточную среду, подкрепляется также тем соображением, что из-за большого количества присутствующих в клетке органических соединений, находящаяся там вода “структурирована”, что заметно уменьшает как ее собственные сольватационные свойства, так и внутриклеточной среды в целом.
В позитронной спектроскопии хорошо известен не только эффект ингибирования образования Ps, но и эффект антиингибирования. Антиингибиторами, как правило, являются акцепторы квазисвободных электронов с малой энергией сродства, около 0.1 эВ. При добавлении антиингибитора в раствор, содержащий некоторое количество ингибитора, молекулы антиингибитора перехватывают трековые электроны, уберегая их от захвата ингибитором. При появлении поблизости позитрона анион антиингибитора легко отдает ему захваченный электрон и выход Ps увеличивается. Мы собрали несколько примеров, из которых видно (с привлечением соответствующих медико-биологических данных), что Ps антиингибиторы при их введении в живой организм ведут себя как антиканцерогены. Это явление нуждается в дальнейшем исследовании. С увеличением числа подобных примеров оно наверняка приобретет практический интерес и будет использовано для выявления потенциальных антиканцерогенов.
Таким образом, видно, что изучение явления аннигиляции позитронов применительно к медико-биологическим приложениям может принести пользу не только в части позитронной эмиссионной томографии, но и в онкологии для быстрого выявления химических канцерогенов и антиканцерогенов, изучения механизма их действия, понимания первичных процессов канцерогенеза.
Исследования методами позитронной аннигилляционной спектроскопии выполнены на оборудовании Центра коллективного пользования КАМИКС (http://kamiks.itep.ru/) НИЦ “Курчатовский институт” – ИТЭФ.
Авторы выражают благодарность А.А. Фенину (РХТУ им. Д.И. Менделеева, Москва) и А.Н. Тарновскому (BGSU, Ohio, USA) за помощь в проведении экспериментов.
Исследование выполнено при финансовой поддержке Российского фонда фундаментальных исследований в рамках научного проекта № 20-14-50150\20. Funding: The reported study was funded by RFBR, project number 20-14-50150\20.
Список литературы
Ободовский И.М. Основы радиационной и химической безопасности. Долгопрудный, Изд. дом “Интеллект”, 2013.
Scoccianti C. “Carcinogens: Identification of Carcinogens”. Encyclopedia of Food and Health, 2016. P. 658–662.
Турусов В.С., Белицкий Г.А., Пылев Л.Н., Кобляков В.А. “Механизмы действия и классификация химических канцерогенов” в книге “Канцерогенез” под ред. Д.Г. Заридзе. М.: Медицина, 2004. 576 с.
Биомаркеры и оценка риска: концепции и принципы / Всемирная организация здравоохранения, 1996 https://apps.who.int/iris/handle/10665/139694
Белицкий Г.А., Якубовская М.Г. // Биохимия. 2008. Т. 73. № 5. С. 675.
Miller E.C., Miller J.A. // Annual Rev Biochem. 1959. V. 28. P. 291.
Miller J.A., Miller E.C. // J. of the National Cancer Institute. 1971. V. 47. № 3. v-xiii.
Miller E.C. // Cancer Research. 1978. V. 38. P. 1479.
Miller E.C., Miller J.A. // Cancer. 1981. V. 47. № 5 Suppl. P. 1055.
Белицкий Г.А., Турусов В.С. “Выявление и мониторинг химических канцерогенов” в книге “Канцерогенез” под ред. Д.Г. Заридзе. М.: Медицина, 2004, 576 с.
Cohen S.M., Ellwein L.B. // Science. 1990. V. 249. P. 1007.
Gupta S., Sharma R.S., Singh R. // Int. J. Environmental Health Research. 2020.
Jargin S.V. // J. of Modern Med. Chem. 2020. V. 8. P. 85.
Editorial “When is a carcinogen not a carcinogen?” // The Lancet Oncology. 2016. V. 17. № 6. P. 681.
Rothman K.J. Epidemiology : An introduction (2-d ed.) N.Y.: Oxford University Press, 2012.
Bopp K., Barouki R., Brack W. et al. // Environment International. 2018. V. 120. P. 544.
MacGregor J.T., Huff J., Shane B., Spalding J. “Carcinogenicity”. Chapter 3 in “Screening and Testing Chemicals in Commerce”, 1995.
Madden J.C., Enoch S.J., Paini A., Cronin M.T.D. // Alternatives to Laboratory Animals. 2020. V. 48. № 4. P. 146.
Fond for the Replacement of Animals in Medical Experiments. https://frame.org.uk/
Reiter L.T., Potocki L., Chien S., Gribskov M., Bier E. // Genome Research. 2001. V. 11. № 6. P. 1114.
Konovalov F., Shaturova A., Mitrofanova O., Kudryavtsev A. // Euphytica. 2012. V. 183. P. 173.
Richard A.M., Wagner P., Purdy R., Veith G. // “SAR and Modeling” in Screening and Testing Chemicals in Commerce / OTA-BP-ENV-166. 1995. P. 101. https://archive.org/details/DTIC_ADA337665/page/n1/mode/2up
Cariello N.F., Wilson J.D., Britt B.H., Wedd D.J., Burlinson B., Gombar V. // Mutagenesis. 2002. V. 17. № 4. P. 321.
Bakale G., McCreary R.D., Gregg E.C. // Int. J. Quantum Chemistry. 1982. V. 22. № S9. P. 15.
Verberne J.B., Loman H., Warman J.M., de Haas M.P., Hummel A., Prinsen L. //Nature. 1978. V. 272. P. 343.
Bakale G., McCreary R.D. // Carcinogenesis. 1987. V. 8. P. 253.
Bakale G., McCreary R.D. // Ibid. 1990. V. 11. № 10. P. 1811.
Bakale G., McCreary R.D. // Ibid. 1992. V. 13. № 8. P. 1437.
Ennever F.K., Bakale G. // Ibid. 1992. V. 13. № 11. P. 2059.
Benigni R. // Mutation Research. 1997. V. 387. P. 35.
Bakale G. A carcinogen-screening test based on electrons. In the book Linking the Gaseous and Condensed Phases of Malter / Ed. by L.G. Christophorou Plenum Press, New York, 1994.
Ignatov S., Shishniashvili D., Ge B., Scheller F.W., Lisdat F. // Biosensors and Bioelectronics. 2002. V. 17. P. 191.
Byakov V.M., Stepanov S.V., Stepanova O.P. // Physica Status Solidi C. V. 6. № 11. P. 2503.
Williams D.R. // Chem. Rev. 1972. V. 72. P. 202.
Тимашев С.Ф. Физико-химия мембранных процессов. М.: Химия, 1988 240 с.
P.A.M. Dirac // Proceedings of the Royal Society of London. 1930. Ser. A. V. 126. P. 360.
Berestetskii V.B., Lifshitz E.M., Pitaevskii L.P. Quantum electrodynamicsics. Landau and Lifshitz. Course of theoretical physics. V. 4. Oxford, Butterworth-Heinemann, 2008.
Krause-Rehberg R., Leipner H.S. Positron Annihilation in Semiconductors. Defect Studies. Springer series in solid-state sciences. V. 127. Berlin: Springer, 1999.
Goworek T. // Annales Universitatis Mariae Curie-Sklodowska. Lublin – Polonia. 2014. V.LXIX. 1-2. SECTIO AA. P. 1.
Principles and Applications of Positron and Positronium Chemistry / Eds. Y.C. Jean, P.E. Mallone, D.M. Schrader. World Scientific Publications, Singapore, 2003.
Stepanov P.S., Selim F.A., Stepanov S.V., Bokov A.V., Ilyukhina O.V., Duplatre G., Byakov V.M. // Physical Chemistry Chemical Physics. 2020. V. 22. P. 5123.
Positron and Positronium Chemistry / Eds.: D.M. Schrader, Y.C. Jean. In “Studies in physical and theoretical chemistry”. Elsevier: Amsterdam [The Netherlands]; New York, 1988.
Byakov V.M., Stepanov S.V. // J. Radioanalyt. Nucl. Chem., Articles. 1996. V. 210. № 2. P. 371.
Stepanov S.V., Byakov V.M., Zvezhinskiy D.S., Duplatre G., Nurmukhametov R.R., Stepanov P.S. // Advances in Physical Chemistry. 2012. V.2012. Article ID 431962. 17 pages.
Byakov V.M. // Int. J. Radiat. Phys. Chem. 1976. V. 8. P. 283.
Ferrell R.A. // Physical Review. 1957. V. 108. № 2. P. 167.
Михин К.В., Степанов С.В., Бяков В.M. // Химия высоких энергий. 2005. Т. 39. № 1. С. 44.
Eldrup M., Lightbody D., Sherwood J.N. // Chem. Phys. 1981. V. 63. P. 51.
Stepanov S.V., Zvezhinskiy D.S., Byakov V.M. // Materials Science Forum. 2013. V. 733. P. 7.
http://prac.us.edu.pl/ kansy/index.php?id=lt10
http://palsfit.dk/
Axpe E., Lopez-Euba T., Castellanos-Rubio A. et al. // PLoS ONE. 2014. V. 9. № 1. e83838.
Liu G., Chen H., Chakka L., Gadzia J.E., Jean Y.C. // Phys. Stat. Sol. (c). 2007. V. 4. № 10. P. 3912.
Liu G., Chen H., Chakka L., Cheng Mei-Ling et al. // Applied Surface Science. 2008. V. 255. P. 115.
Jasinska B., Zgardzinska B., Cholubek G. et al. // Acta Phys. Polon. B. 2017. V. 48. P. 1737.
Sane P., Tuomisto F., Wiedmer S.K. et al. // Biochimica et Biophysica Acta. 2010. V. 1798. P. 958.
Бяков В.М., Степанов С.В., Степанова О.П. Способ тестирования веществ на канцерогенность. Патент на изобретение RU 2307342. http://www.findpatent.ru/patent/230/ 2307342.html
Byakov V.M., Stepanov S.V., Stepanova O.P. // Materials Science Forum. V. 607. P. 223.
Stepanov S.V., Dubov L.Yu., Akmalova Yu.A. et al. // Engineering and Technologies for Biomedicine, KnE Energy Physics. 2018. P. 476.
Глемзер Б. Человек против рака. М.: Мир. 1972. 74 с.
Ito Y., Mohamed H.F.M., Byakov V.M., Stepanov S.V. // J. of Radioanalyt. and Nucl. Chem. 1996. V. 210. № 2. P. 607.
Robey I.F., Baggett B.K., Kirkpatrick N.D. et al. // Cancer Res. 2009. V. 69. № 6. P. 2260.
de Brito-Ashurst I., Varagunam M., Raftery M.J., Yaqoob M.M. // J. American Soc. Nephrology. 2009. V. 20. № 9. P. 2075.
Ito Y., Miyake Y., Tabata Y. // Radiat. Phys. Chem. 1982. V. 19. № 4. P. 315.
Ito Y., Hasegawa M., Miyake Y., Tabata Y. // Radiat. Phys. Chem. 198. V. 21. № 1–2. P. 217.
Anisimov O.A., Vocel S.V., Molin Yu.N. // Chemical Physics. 1981. V. 56. P. 261.
Пикаев А.К. Современная радиационная химия. Радиолиз газов и жидкостей. М.: Наука, 1986.
Стражев С.В., Зубавин П.М., Мехтиев В.Н. и др. Роль питания в генезе и профилактике онкологических заболеваний. Учебно-методическое пособие. Волгоград, 2018.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии