

Журнал физической химии, 2022, T. 96, № 10, стр. 1513-1519
Проблемы теории ионного обмена I. Особенности описания ионообменных сил в классических системах
a Институт геохимии и аналитической химии им. В.И. Вернадского РАН
Москва, Россия
* E-mail: amdolgo@mail.ru
Поступила в редакцию 25.01.2022
После доработки 08.02.2022
Принята к публикации 09.02.2022
- EDN: ISVBUJ
- DOI: 10.31857/S0044453722100089
Аннотация
Рассмотрено влияние сил различной природы на ионный обмен в классических системах, представляющих собой полимерные иониты в водных растворах. Введена каноническая форма энергии взаимодействия, при которой главный вклад принадлежит электростатической энергии. Проведен теоретический анализ экспериментальных данных по электропроводности ионообменных мембран с учетом характера внутренней диффузии. Описана зависимость локальной диэлектрической проницаемости от межионного расстояния, которая учитывается при установлении конфигурации ионов и воды в области взаимодействия. Определены электростатические потенциалы некоторых сильных ионообменников.
Основное свое развитие теория ионного обмена получила в 50–60-е годы, что отражено в ряде блестящих монографий и обзоров [1, 2]. Вновь интерес к этой теме возник благодаря статье Шапошника [3], где он приводит свою интерпретацию интересных экспериментальных исследований в области ионного обмена. Ряд моментов указанной статьи, касающихся предположений автора о дробных зарядах взаимодействующих ионов, доминировании водородных связей в таких взаимодействиях, а также о природе энергии активации ионного транспорта в ионообменной мембране, породил дискуссию в журнале “Сорбционные и хроматографические процессы” [4–7].
Теоретические подходы в литературе представлены двумя концепциями: термодинамической и молекулярно-статистической. К первой относится модель Грегора, объясняющая влияние гидратации на селективность ионного обмена [8]. Его изначально физически прозрачная модель, описывающая влияние осмотического давления внутри полимера на сорбцию гидратированных ионов, постепенно обросла поправками, связанными с неидеальностью полимеров, растворов и, в целом, с несовершенством принятой концепции [9]. Модели второго типа более информативны, так как рассматривают параметры микросистемы. Например, Райс и Гаррис объясняют набухание полимера не осмосом, а электростатическими силами отталкивания между функциональными группами (ФГ), степень набухания связывается с эффективностью экранирования их зарядов противоионами [10]. В результате явление набухания ими рассматривается не как причина, а как следствие процессов, приводящих к селективности. Развивая молекулярно-статистическую концепцию, автор одного из наиболее эффективных на тот момент подходов, Эйзенман [11] смог указать основные факторы, влияющие на селективность в классических ионообменных системах, однако его полуэмпирической модели не хватает точности в количественных оценках.
Новые возможности в описании ионообменных систем появились благодаря современным вычислительным методам. Интерпретация взаимодействия ионита и противоиона, приведенная в [3], опирается на результаты молекулярного моделирования ионообменных взаимодействий с помощью программного комплекса, работающего на основе квантово-химических расчетов. Однако необходимо учитывать, что, во-первых, погрешность полуэмпирических расчетов, применяемых в области межмолекулярных взаимодействий, еще очень высока, во-вторых, в зависимости от задания исходной конфигурации микросистемы, можно получить решения в широком диапазоне, и велика вероятность выбора нетипичного результата, отсеиваемого при усреднении. Выводы, сделанные в [3] о решающем вкладе во взаимодействие водородных связей (Н-связей), не имеют подтверждения не только в классической теории, но и в эксперименте.
Проверочный опыт, результат которого сильно зависит от баланса вкладов электростатики и Н-связей, заключается в измерении коэффициентов селективности при постепенной замене воды спиртом. В этом опыте понижается способность среды к образованию Н-связей. Если в результате сорбция растет, то главный вклад вносит электростатика, усиливающаяся вследствие снижения диэлектрической проницаемости среды; если сорбция уменьшается, то, вероятно, она обусловлена Н-связями. В упомянутых в книге [12] экспериментальных исследованиях влияния спирта на ионный обмен отмечается рост селективности ионита при добавлении в водный раствор спирта, что свидетельствует о малозначимом участии Н-связей во взаимодействии.
Цель предлагаемого теоретического исследования – изучить равновесные и кинетические особенности взаимодействия фиксированных ионов ионообменника с противоионами, количественно охарактеризовать участие электростатических сил и найти характеристики ионитов из известных экспериментов по электропроводности ионообменных мембран.
ТЕОРЕТИЧЕСКИЙ АНАЛИЗ
Форма энергии взаимодействия фиксированного иона и противоиона
Электростатический характер взаимодействия однозарядных ФГ и противоионов с зарядами ${{z}_{1}}e$ и ${{z}_{2}}e$ ($e$ – элементарный заряд) отражается в уравнении реакции ионного обмена с целочисленными стехиометрическими коэффициентами ${{z}_{1}}$ и ${{z}_{2}}$. Поделив обе части на произведение ${{z}_{1}}{{z}_{2}}$, реакцию можно записать в форме ионного обмена на одной ФГ:
Реакция ионного обмена с точностью до постоянного слагаемого, поддерживающего условие электронейтральности системы, может быть разбита на моноионные полуреакции сорбции и десорбции. В основе обсуждаемой модели лежит полуреакция сорбции противоиона:
Энергия содержит три основных вклада – за счет сил Кулона, Ван-дер-Ваальса и водородных связей: Полная энергия отрицательна, что соответствует притяжению. Первый член – кулоновское взаимодействие противоиона с однозарядной ФГ – отрицательный. За ван-дер-ваальсово взаимодействие ответственен второй член, который положителен, так как представляет собой энергию дегидратации иона в процессе сорбции при пересечении межфазной границы. Эта величина приходится на z функциональных групп, поэтому вклад, приходящийся на 1 ФГ, равен: где ${{a}_{w}}$ – стандартная энергия гидратации, которая равна энергии взаимодействия иона с молекулой воды первого гидратного слоя; n – число молекул первого гидратного слоя вокруг иона, эквивалентное массе воды, теряемой гидратной оболочкой при сорбции (так называемый “гидратный дефект”). Относительный вклад дипольных взаимодействий в энергию сорбции иона, приходящийся на 1 молекулу гидратной воды, оценим как(3)
$\frac{{\Delta W}}{{n\left| {{{U}_{C}}} \right|}} \approx \frac{{{{\mu }_{w}}}}{{ze{{d}_{w}}}} = \frac{{0.142}}{z} \ll 1,$Третьим членом, отражающим участие Н-связей, обычно можно пренебречь вследствие насыщенности системы по Н-связям, как в водной среде, так и в полимерной. Этот фактор проявляется лишь в случае гидрофобного сорбата, выталкиваемого силой поверхностного натяжения в менее плотную по Н-связям среду внутри полимерной фазы. Такое взаимодействие, называемое гидрофобным, вызвано разрушением Н-связей при растворении гидрофобной молекулы и вносит отрицательный вклад в полную энергию сорбции. Гидрофобным механизмом объясняется так называемая необменная сорбция, или твердофазная экстракция, не подчиняющаяся стехиометрии ионного обмена. Необменная сорбция – обычно очень слабый побочный процесс, который в нашей модели не рассматривается.
Итак, основной член энергии взаимодействия – кулоновский. Рассмотрим его описание:
(4)
${{U}_{C}} = - \frac{{{{e}^{2}}}}{{4\pi {{\varepsilon }_{0}}\varepsilon }}\frac{z}{{{{r}_{R}} + {{r}_{I}}}},$(5)
${{U}_{0}} = - \frac{{{{e}^{2}}}}{{4\pi {{\varepsilon }_{0}}\varepsilon }}\frac{1}{{{{r}_{R}} + a}}$Энергия активации ионного транспорта внутри ионита
Согласно [3], в эксперименте измерялась электропроводность ионообменной мембраны $\kappa $ на фоне воды в зависимости от ионной формы и температуры. Изучался процесс транспорта ионов в этой системе как источник информации о взаимодействии. При этом найденная по Аррениусу энергия активации приравнивалась к энергии взаимодействия между сорбентом и сорбатом. Покажем, что такая интерпретация неточна.
Перенос ионов в мембране определяется коэффициентом внутренней диффузии. Ниже приводится модель, описанная в книге [15] и ряде других работ автора.
Согласно теореме Эйнштейна–Смолуховского, удвоенный коэффициент диффузии является коэффициентом пропорциональности между квадратом расстояния ${{\rho }^{2}}$ и временем $\tau $ стохастического движения, совершаемого частицей для такого перемещения. Период движения $\tau $ состоит из двух интервалов: времени неподвижного контакта с ФГ ${{\tau }_{s}}$ и времени “перескока” – перехода иона к другой ФГ ${{\tau }_{l}}$ (рис. 1а). Отношение этих интервалов равно коэффициенту распределения иона $\Gamma = \frac{{{{\tau }_{s}}}}{{{{\tau }_{l}}}}$, а квадрат расстояния определяется через время и коэффициент диффузии в поровой жидкости: ${{\rho }^{2}} = 2{{D}_{l}}{{\tau }_{l}}$. В результате для коэффициента внутренней диффузии получим выражение [15, 16]:
Рис. 1.
Схема, поясняющая модель внутренней диффузии противоиона (“–”): а – движение иона в поре ионита между фиксированными ионами (“+”) в режиме “перескоков”; б – потенциальная функция поля в поре сорбента (обозначения см. в тексте).
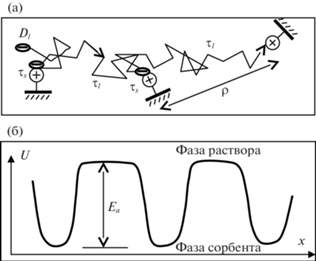
Потенциальная функция внутри ионита имеет минимумы возле функциональных групп и максимумы между ними (рис. 1б). Разность этих уровней равна величине энергии активации перескока ${{E}_{{\text{a}}}}$. Применяя законы Больцмана для доли свободных (не сорбированных) ионов: ${{\left( {\Gamma + 1} \right)}^{{ - 1}}} = \exp \left( { - \frac{{{{E}_{{\text{a}}}}}}{{RT}}} \right)$, и Нернста–Эйнштейна [17]: $\kappa \propto \frac{D}{T}$, можно выразить отношение электропроводностей мембраны при разных температурах через отношения коэффициентов диффузии и, в конечном счете, согласно модели перескоков, – через энергию активации [4, 7]:
(9)
${{E}_{{\text{a}}}} = \frac{{R\left( {\ln \frac{{{{\kappa }_{2}}}}{{{{\kappa }_{1}}}} - \ln \frac{{{{\kappa }_{{{\text{liq2}}}}}}}{{{{\kappa }_{{{\text{liq1}}}}}}}} \right)}}{{\frac{1}{{{{T}_{1}}}} - \frac{1}{{{{T}_{2}}}}}} = E_{{\text{a}}}^{*} - \Delta E.$(10)
$\Delta E = {{R\ln \frac{{{{\kappa }_{{{\text{liq2}}}}}}}{{{{\kappa }_{{{\text{liq1}}}}}}}} \mathord{\left/ {\vphantom {{R\ln \frac{{{{\kappa }_{{{\text{liq2}}}}}}}{{{{\kappa }_{{{\text{liq1}}}}}}}} {\left( {\frac{1}{{{{T}_{1}}}} - \frac{1}{{{{T}_{2}}}}} \right)}}} \right. \kern-0em} {\left( {\frac{1}{{{{T}_{1}}}} - \frac{1}{{{{T}_{2}}}}} \right)}}.$В табл. 1 приведены данные работы [3], преобразованные в соответствии с формулами (9) и (7).
Таблица 1.
Расчет стандартных потенциалов ($ - {{U}_{0}}$) ФГ ионитов из экспериментальных данных
| Противоион | Заряд z | $E_{{\text{a}}}^{*}$, кДж/моль | ${{E}_{{\text{a}}}} = E_{{\text{a}}}^{*} - 14.0$, кДж/моль | ${{{{E}_{{\text{a}}}}} \mathord{\left/ {\vphantom {{{{E}_{{\text{a}}}}} z}} \right. \kern-0em} z}$, кДж/моль | $ - {{U}_{0}}$, кДж/моль |
|---|---|---|---|---|---|
| Na+ | 1 | 21.7 | 7.7 | 7.7 | сульфогруппа 7.5 ± 0.7 |
| Ca2+ | 2 | 27.2 | 13.2 | 6.6 | |
| Al3+ | 3 | 38.4 | 24.4 | 8.1 | |
| Cl- | 1 | 20.4 | 6.4 | 6.4 | четвертичное аммониевое основание 5.5 ± 0.7 |
| ${\text{SO}}_{4}^{{2 - }}$ | 2 | 24.6 | 10.6 | 5.3 | |
| ${\text{PO}}_{4}^{{3 - }}$ | 3 | 28.8 | 14.8 | 4.9 |
Первые три столбца табл. 1 соответствуют статье [3]. С учетом поправки и с использованием канонической формы для энергии получили потенциалы ионитов с сульфогруппами (СГ, SG) и четвертичными аммониевыми основаниями (ЧАО, QAB) как средние величины, отклонение от которых для разных ионов составляет ~10%, что соответствует допустимой величине малого параметра. Из формулы (5) для потенциала ${{U}_{0}} = - \frac{{{\text{Fe}}}}{{4\pi {{\varepsilon }_{0}}\varepsilon }}\frac{1}{{{{r}_{R}} + a}}$ можно найти характерные размеры фиксированных зарядов, увеличенные на пробную константу: ${{r}_{R}} + a = \left\{ \begin{gathered} 2.3\;{\AA}\;{\text{(SG)}} \hfill \\ 3.1\;{\AA}\;{\text{(QAB)}} \hfill \\ \end{gathered} \right.$. Полученные расстояния не допускают нахождения молекул воды диаметром ${{d}_{w}} = 2.7$ Å между ионом и ФГ, что абсурдно, так как при расчете использовалась диэлектрическая проницаемость (ДЭП) воды $\varepsilon = 81$. Противоречие разрешается, если в таких микросистемах учитывать локальное отклонение ДЭП от предельной величины.
Локальная диэлектрическая проницаемость
К сожалению, в литературе [18–23] нет удовлетворительного теоретического разбора случая локальной ДЭП в молекулярных системах без применения эмпирических соотношений. Наиболее близок к представленному ниже подходу вывод, описанный в [22], однако полученное там соотношение является эмпирическим, что лишает подход универсальности и требуемой точности.
Рассмотрим разрабатываемую автором неэмпирическую модель, опирающуюся на явление диэлектрического насыщения. Легко поляризуемая среда при внесении точечного заряда изменяет картину случайно расположенных векторов диполей, выстраивая их вдоль силовых линий. При этом исходный заряд полностью или частично компенсируется в точке нахождения, а его плотность переносится на границу окружающего монослоя из диполей среды. Подобный перенос плотности заряда может происходить и в следующих слоях. Ввиду дискретности эта картина в случае единичных зарядов носит вероятностный характер фазового переноса целого заряда на расстояние нескольких слоев диполей. Такой эстафетный процесс объясняет аномальную подвижность ионов воды (гидроксония и гидроксила) с длиной эстафеты в 2–3 молекулы воды.
Граница области переноса заряда (ОПЗ) – это поверхность с минимальным потенциалом ${{\varphi }_{0}}$, которая доступна для перемещения плотности заряда из центра. Увеличение центрального заряда на величину $dq$ равносильно переносу такого малого заряда из удаленной точки с нулевым потенциалом на границу ОПЗ. Прирост энергии составит ${{\varphi }_{0}}dq$, что приведет к увеличению размера ОПЗ на $dr$, согласно уравнению
где $\mu $ – дипольный момент элемента среды, q – заряд внутри сферы радиусом r, $g\sim 1$ – коэффициент, зависящий от угловых характеристик диполей на границе ОПЗ. Процесс уширения ОПЗ можно представить как замену на пограничном участке шириной $dr$ внешнего элемента среды со случайной ориентацией диполя на элемент ОПЗ с радиальной ориентацией диполя. Модуль силы взаимодействия диполя с зарядом в первом случае вдвое ниже, чем во втором. Разность этих сил совершает отрицательную работу по увеличению размера ОПЗ на $dr$, что записано в правой части уравнения.Представленная модель базируется на уравнении Пуассона для диэлектрической среды [21, 22], которое в [23] приводится в более простой эквивалентной форме:
(12)
$\rho = - \frac{{{{\varepsilon }_{0}}}}{\varepsilon }\left( {{\mathbf{E}}\operatorname{grad} \varepsilon } \right).$Учитывая дискретные свойства водной среды, выразим расстояние как $r = {{d}_{w}}\left( {\tfrac{1}{2} + i} \right)$, ${\text{ }}i = {\text{0, 1, 2,}}...$ и получим итоговое выражение для локальной ДЭП, которое удовлетворяет граничным условиям и температуре 293 К (рис. 2):
Точки на графике рассчитаны по результатам экспериментов, приведенным в работе [17], где изучалась зависимость статической диэлектрической проницаемости от концентрации электролита.Рис. 2.
Зависимость локальной ДЭП от числа гидратных оболочек вокруг иона с размером молекулы воды. Экспериментальные точки нанесены в соответствии с табл. 2.
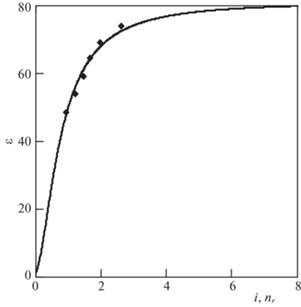
Число молекул воды, окружающих ионы в растворе соли NaCl, рассчитывалось из модельных соображений по распределению ионов в узлах кубической решетки. Соседними к выбранному иону являются ионы, связанные с ним не только ребрами, но и диагоналями граней и объемов граничащих ячеек, поэтому среднее расстояние между ближайшими ионами определялось как сторона кубической ячейки умноженная на коэффициент $\chi = \frac{{6 \times 1 + 12\sqrt 2 + 8\sqrt 3 }}{{26}} = 1.416$. Половина этого расстояния – средний радиус объема, приходящегося на один ион, связан с молярной концентрацией раствора формулой: ${{r}_{1}} = {{c}^{{ - {1 \mathord{\left/ {\vphantom {1 3}} \right. \kern-0em} 3}}}}\chi \times $ $ \times \;9.4{\text{/}}2 = 6.66{{c}^{{ - {1 \mathord{\left/ {\vphantom {1 3}} \right. \kern-0em} 3}}}}$ Å. По длине этого отрезка за вычетом радиуса иона, оцениваемого здесь как радиус молекулы воды, укладывается число ${{n}_{r}}$ молекул воды:
(15)
${{n}_{r}} = \frac{{{{r}_{1}}}}{{{{d}_{w}}}} - \frac{1}{2} = 2.47{{c}^{{ - {1 \mathord{\left/ {\vphantom {1 3}} \right. \kern-0em} 3}}}} - 0.5.$(16)
$\varepsilon = 80\exp ( - 0.18{{c}^{{{2 \mathord{\left/ {\vphantom {2 3}} \right. \kern-0em} 3}}}}).$Таблица 2.
Диэлектрическая проницаемость растворов NaCl в зависимости от концентрации (с) при 21°C [17]; аппроксимация зависимости формулами (14) и (16) и радиальное число гидратных слоев (${{n}_{r}}$) по формуле (15)
| с, моль/л | ${{n}_{r}}$ (15) | ε (14) для $i = {{n}_{r}}$ | ε (16) | ε ± 2 (экспе-римент) |
|---|---|---|---|---|
| 0.5 | 2.61 | 72.3 | 71.4 | 73.9 |
| 1.0 | 1.97 | 67.6 | 66.8 | 69.1 |
| 1.5 | 1.66 | 64.0 | 63.2 | 64.3 |
| 2.0 | 1.46 | 60.9 | 60.1 | 59.0 |
| 3.0 | 1.21 | 55.7 | 55.0 | 54.0 |
| 5.0 | 0.944 | 47.8 | 47.3 | 48.4 |
Стандартный потенциал ионита
Рассмотрим выражение для локальной ДЭП вблизи фиксированных зарядов ионита. Гидратные слои между зарядами функциональной группы и противоиона складываются, и в этом случае в формуле (14) у числа слоев исчезает двойка:
(17)
$\begin{gathered} {{\varepsilon }_{i}} = {{81}^{{1 - {{{\left( {1 + i} \right)}}^{{ - 2}}}}}} = 1,\;27,\;50,\;62,\;... \\ {\text{для}}\;\;i = 0,\,1,\,2,\,3,\,... \\ \end{gathered} $Модельные потенциалы быстро падают при росте числа слоев воды между зарядами, имеющими одинаковый размер, равный размеру молекулы воды,
(18)
$\begin{gathered} {{E}_{i}} \approx \frac{{Fe}}{{4\pi {{\varepsilon }_{0}}{{d}_{w}}}}\frac{1}{{{{\varepsilon }_{i}}\left( {1 + i} \right)}} = \\ = \mathop {514}\limits_{i = 0} ,\;\mathop {9.53}\limits_{i = 1} ,\;\mathop {3.43}\limits_{i = 2} ,\;...\;\left( {{\text{кДж/моль}}} \right), \\ \end{gathered} $(19)
$\frac{{{{r}_{R}}}}{{{{d}_{w}}}} \approx \frac{{2{{E}_{1}}}}{{\left| {{{U}_{0}}} \right|}} - \frac{1}{2} - 1 = \left\{ {\begin{array}{*{20}{c}} {1.0{\text{ SG}}} \\ {2.0{\text{ QAB}}} \end{array} } \right..$Подстановка полученных размеров в расчет потенциалов ионитов с разной степенью гидратации дает три варианта (табл. 3): без воды – соответствует хемосорбции; с одной молекулой воды – основной вариант, отражающий присутствие растворителя как макрокомпонента в системе “ионит–противоион”; с двумя молекулами воды – для редких случаев сильно гидратированных систем, в которых возможна термическая десорбция ионов. Большее число промежуточных молекул воды не реализуется при ионообменном равновесии в условиях существования жидкой воды.
Таблица 3.
Стандартные потенциалы ($ - {{U}_{{0i}}}$, кДж/моль) функциональных групп сильных ионитов с различной степенью гидратации при 293 К (i – число молекул воды между ФГ и противоионом)
| i | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| СГ | 372 | 7.6 | 3.0 | 1.9 |
| ЧАО | 273 | 5.4 | 2.3 | 1.5 |
Итак, стандартный потенциал ФГ сильных ионитов при 293 К равен 7.6 кДж/моль для СГ и 5.4 кДж/моль для ЧАО.
ЗАКЛЮЧЕНИЕ
Проведенный теоретический анализ показал, что
• основная сила, благодаря которой осуществляется ионный обмен, – кулоновское взаимодействие ионов, что позволяет представить энергию в канонической форме (7);
• электропроводность ионообменных мембран, определяемая внутренней диффузией ионов, содержит информацию об энергии взаимодействия фиксированных ионов с противоионами в величинах отношения коэффициентов диффузии ионов вне и внутри сорбента;
• при описании электростатического взаимодействия на молекулярном масштабе следует учитывать различие величин локальной и макроскопической диэлектрической проницаемости, которые связаны уравнениями типа (14) и (16);
• понятие области переноса заряда является новой интерпретацией известного эффекта насыщения полярных диэлектриков, удобной для рассмотрения энергетического баланса при внесении в полярную среду электрического заряда.
Основные выводы работы:
• найденная закономерность для локальной диэлектрической проницаемости от концентрации электролита имеет важное значение не только для теории ионного обмена, но и в целом для теории межмолекулярных взаимодействий;
• из анализа данных эксперимента по электропроводности ионитовых мембран, опирающегося на представления о внутренней диффузии и локальной диэлектрической проницаемости, следует, что энергия взаимодействия функциональной группы с противоионом определяется кулоновским притяжением при конфигурации с одной молекулой воды между ионами;
• развитая электростатическая модель позволила найти и объяснить величины стандартных потенциалов сильных ионитов; ее применение для неэмпирического описания равновесия ионного обмена будет рассмотрено во второй части заявленной серии.
Работа выполнена в рамках Программы РАН № 0137-2019-0023.
Список литературы
Helfferich F. Ion Exchange. N.Y.: McGraw-Hill, 1962. 624 p.
Ion Exchange: A Series of Advances / Ed. by J.A. Malinsky. N.Y.: Buffalo, 1966. V. 1. 423 p.
Шапошник В.А. // Сорбц. хроматогр. процессы. 2020. Т. 20. № 1. С. 48. https://doi.org/10.17308/sorpchrom.2020.20/2379
Хамизов Р.Х, Долгоносов А.М. // Там же. 2020. Т. 20. № 5. С. 641.
Шапошник В.А. // Там же. 2021. Т. 21. № 2. С. 285.
Хамизов Р.Х. // Там же. 2021. Т. 21. № 4. С. 600. https://doi.org/10.17308/sorpchrom.2021.21/3645
Долгоносов А.М. // Там же. 2021. Т. 21. № 4. С. 606. https://doi.org/10.17308/sorpchrom.2021.21/3646
Gregor H.P., Bregman J.I. // J. Colloid Sci. 1951. V. 6. P. 323.
Marinsky J.A. Interpretation of Ion-Exchange Phenomena. In: Ion Exchange: A Series of Advances / Ed. by J.A. Marinsky. N.Y.: Buffalo, 1966. V. 1. 423 p. Ch.1.
Harris F.E., Rice S.A. // J. Chem. Phys., 1956. V. 24. P. 1258.
Eisenman G. // Biophys. J., Suppl., 1962. V. 2. № 2. P. 259.
Rieman W., Walton H.F. Ion Exchange in Analytical Chemistry. Oxford: Pergamon Press, 1970. 310 p.
Никольский Б.П., Парамонова В.И. // Успехи химии. 1939. Т. 8. С. 1535.
Сенявин М.М., Рубинштейн Р.Н., Веницианов Е.В. и др. Основы расчета и оптимизации ионообменных процессов. М.: Наука, 1972. 175 с.
Долгоносов А.М., Сенявин М.М., Волощик И.Н. Ионный обмен и ионная хроматография. М.: Наука, 1993. 222 с. С. 28.
Долгоносов А.М., Гарбар А.М. // Журн. физ. химии. 1986. Т. 60. № 1. С. 199.
France-Lanord A., Grossman J.C. // Phys. Rev. Lett. 2019. V. 122. P. 136001-1-6. https://doi.org/10.1103/PhysRevLett.122.136001
Hasted J.B., Ritson D.M., Collie C.H. // J. Chem. Phys. 1948. V. 16. P. 1
Shellman J. A. // J. Chem. Phys. 1957. V. 26. № 5. P. 1225.
Robinson R.A., Stokes R.H. Electrolyte Solutions. L.: Butterworths Sci. Publ., 1959. 559 p.
Vasilyev V. // J. Comput. Chem. 2002. V. 23. P. 1254. https://doi.org/10.1002/jcc.10131
Mallik B., Masunov A., Lazaridis Th. // J. Comput. Chem. 2002. V. 23. P. 1090. https://doi.org/10.1002/jcc.10104
Ландау Л.Д., Лифшиц Е.М. Электродинамика сплошных сред: Т. 8. Курс теоретической физики. М.: Наука, 1982. 632 с. Ч. 2.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии