

Журнал физической химии, 2022, T. 96, № 10, стр. 1458-1465
Квантово-химическое моделирование радикальных реакций изопренового каучука с серой и N-циклогексил-2 бензтиазолилсульфенамидом
М. Е. Соловьев a, *, В. В. Власов a
a Ярославский государственный технический университет
150023 Ярославль, Россия
* E-mail: soloviev56@gmail.com
Поступила в редакцию 18.02.2022
После доработки 28.03.2022
Принята к публикации 06.04.2022
- EDN: YBTQYQ
- DOI: 10.31857/S0044453722100302
Аннотация
Квантово-химическим методом функционала плотности DFT B3LYP/6-311G** проведены расчеты изменения полной электронной энергии при реакциях инициирования и сшивания при серной вулканизации изопренового каучука с использованием ускорителя N-циклогексил-2 бензтиазолилсульфенамида в присутствии кислорода воздуха. Проанализировано влияние числа атомов серы на активность радикалов сульфидирующего комплекса, возникающих при акцептировании серой радикалов, образующихся при распаде ускорителя.
В процессе вулканизации эластомеров линейный полимер – каучук превращается в сетчатый – резину в результате реакций вулканизации под действием определенных химических агентов. Среди различных методов вулканизации наиболее старым и распространенным в промышленности является вулканизация серой [1], предложенная Чарльзом Гудьиром (Charles Goodyear) в США и независимо от него Томасом Хенкоком (Thomas Hancock) в Англии в 1841–1842 г. Скорость вулканизации элементарной серой достаточно мала, и до открытия органических ускорителей вулканизации процесс вулканизации автомобильных шин длился при 140°С более 4 ч. Ситуация кардинально изменилась с открытием в 1906 г. органических ускорителей. С тех пор режимы вулканизации резин постоянно сокращались и сейчас время вулканизации шин массового ассортимента составляет несколько десятков минут, а время вулканизации тонкостенных резиновых изделий не превышает нескольких минут. Среди десятков типов ускорителей, применяемых для вулканизации карбоцепных каучуков в настоящее время, сульфенамидные ускорители, к числу которых относится рассматриваемый в настоящей работе N-циклогексил-2-бензтиазолилсульфенамид, являются одними из наиболее широко распространенных. Помимо ускорителей в состав серной вулканизующей группы могут входить также активаторы: оксиды металлов и их соли и жирные кислоты.
Несмотря на более чем 180-летнюю историю серной вулканизации, до сих пор многие вопросы ее механизма остаются не до конца изученными, что неоднократно отмечалось различными авторами [2–5]. В силу сложности и большого количества реакций, протекающих при серной вулканизации каучуков в присутствии органических ускорителей и активаторов, вплоть до настоящего времени, для моделирования ее кинетики все еще используются эмпирические уравнения, отражающие лишь вид зависимости сдвигового момента вулканизуемой смеси от времени, измеряемой на виброреометре [6–9], и не учитывающие баланса по отдельным компонентам вулканизующей группы. Вместе с тем, по мере накопления экспериментальных данных, полученных с применением современных физико-химических методов анализа, появилась возможность применения методов формальной кинетики химических реакций для описания кинетики серной вулканизации. Первоначально в таких моделях выделялись лишь некоторые обобщенные стадии реакции без выделения отдельных реагирующих веществ [10]. Однако в 2003 г. в работе Гоша (Ghosh P.) с соавторами была предложена кинетическая модель, в который строго учитывался материальный баланс по всем химическим соединениям, участвующим в реакции. В настоящее время подходы, основанные на методах формальной кинетики химических реакций, вызывают наибольший интерес [11–15]. В этой связи приобретают актуальность исследования детального механизма отдельных стадий реакций, в том числе с применением вычислительных методов физической химии. Примеры таких расчетов, выполненных с применением полуэмпирического квантово-химического метода PM3, приведены в работах [11, 16].
Сера, имея сходную с кислородом электронную структуру внешней электронной оболочки, проявляет, аналогично кислороду, способность к акцептированию свободных радикалов. В связи с этим следует ожидать определенной конкурентности кислорода и серы в радикальных реакциях, протекающих при серной вулканизации, поскольку на практике вулканизация производится в присутствии кислорода воздуха. Вместе с тем при рассмотрении вулканизации эти вопросы обычно не затрагиваются.
В настоящей статье мы рассматриваем только радикальные реакции, которые протекают при вулканизации без использования в качестве активаторов оксидов или солей металлов и жирных кислот, поскольку в этом случае реакции имеют гетерогенный характер, и использованный в работе подход является недостаточным для моделирования данной ситуации.
МЕТОДЫ РАСЧЕТА
В настоящей статье реакции, протекающие при взаимодействии серы с ускорителем и каучуком, моделировались методом функционала плотности [17, 18] c гибридным обменно-корреляционным функционалом Беке, Ли, Янга и Парра (Becke, Lee, Yang & Parr) [19, 20] на уровне теории DFT B3LYP/6-311G** с открытыми оболочками (метод ODFT). Вычисления проводились с использованием программных комплексов NWChem [21] и ORCA [22].
Ранее использованный в настоящей работе тип функционала применялся для моделирования реакций окисления углеводородов в работах [23–25]. При этом наблюдалась хорошая корреляция между кинетическими параметрами окисления и изменениями термодинамических функций в результате реакций, рассчитанных данным квантово-химическим методом. В этой связи предполагалось, что и при моделировании радикальных реакций серной вулканизации можно ожидать адекватных результатов. Хотя, разумеется, данный функционал, являясь одним из наиболее популярных, уступает по точности дважды гибридным функционалам и ряду других [26]. Выбор его в существенной степени был ограничен доступными компьютерными ресурсами, поскольку помимо расчетов по оптимизации геометрии проводилось также молекулярно-динамическое моделирование в квантово-классическом приближении для того, чтобы оценить устойчивость исследуемых молекулярных структур.
При проведении расчетов проводилась оптимизация геометрии молекулярных моделей. Поиск конформаций с минимальной энергией осуществлялся квазиньютоновским методом BFGS (Broyden–Fletcher–Goldfarb–Shanno) [27–30]. Оптимизация производилась в системе внутренних координат молекулы в формате Z-матрицы. Критерием окончания поиска являлось достижение среднеквадратичной нормы градиента (RMS Gradient) полной электронной энергии молекулярной системы значения 0.00001 атомных единиц (Eh/bohr). Поскольку градиентный поиск не является достаточным для нахождения экстремума, оптимизация производилась из нескольких разных стартовых конформаций молекулы. Кроме того, при вычислении термодинамических функций производилась проверка наличия экстремума на основании вибрационного анализа [31] – по отсутствию отрицательных собственных значений матрицы Гессе в точке предполагаемого экстремума.
В конформации с минимальной энергией рассчитывались термодинамические функции (энтальпия и свободная энергия Гиббса) как сумма полной электронной энергии DFT в описанном выше приближении и термодинамической составляющей в приближении “жесткий ротатор–гармонический осциллятор” при температуре реакции, вычисленной по стандартной методике на основе вибрационного анализа в состоянии идеального газа [32]. Свободные энергии реакций вычисляли как разности соответствующих величин для продуктов реакций и реагентов при температуре реакции.
В процессе молекулярно-динамического моделирования энергия и градиент на каждом шаге интегрирования классического уравнения движения вычислялась тем же квантово-химическим методом DFT B3LYP/6-311G**. Начальное распределение скоростей отвечало Максвелл-Больцмановскому распределению, для поддержания температуры использовался термостат [33], параметр релаксации составлял 100 шагов интегрирования по времени.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В отличие от кислорода, который распространен в природе в виде двухатомной молекулы, основное состояние которой представляет собой триплет, то есть бирадикал, сера распространена в природе в форме восьмичленных колец, которые образуют в твердом состоянии при комнатной температуре орторомбическую кристаллическую решетку. Для двухатомной молекулы серы S2 основным состоянием, так же как и для молекулы кислорода, является триплет. Согласно расчету, разница в энергии между основным триплетным состоянием и возбужденным синглетным для молекулы S2 составила –94.6 кДж/моль. Однако природная сера диамагнитна, то есть восьмичленные кольца находятся в синглетном состоянии. Представляло интерес оценить влияние размеров кольца на энергию перехода из синглетного в триплетное состояние, которое сопровождается разрывом кольца с образованием цепочки атомов. На рис. 1 представлены рассчитанные зависимости энергии разрыва связей молекул серы из состояния с мультиплетностью m = 1 в состояние с мультиплетностью m = 3 от числа атомов n в молекуле. За исключением n = 4 для всех молекул основным состоянием является синглет и форма молекул в виде деформированного кольца, так что атомы, чередуясь, располагаются по разные стороны от средней плоскости. Из всех изученных колец выпадает структура с числом атомов, равным четырем, для которой энергия разрыва отрицательна, то есть ее основным состоянием является триплет. Анализ показал, что в триплетном состоянии после оптимизации геометрии четырехатомная цепочка распадается на две, практически параллельно расположенные, двухатомные молекулы. Таким образом, единственной структурой с триплетным основным состоянием является молекула S2.
Рис. 1.
Зависимость энергии разрыва связей молекул серы из состояния с мультиплетностью m = 1 в состояние с мультиплетностью m = 3 от числа атомов в молекуле.
Как видно из рисунка, восьмичленное кольцо является наиболее устойчивым, что согласуется с тем, что в природе сера имеет именно эту структуру. Энергия разрыва связи восьмичленного кольца, полученная в данном расчете, существенно меньше энергии серной связи в органических сульфидах [34]. Помимо длины серной цепочки, это объясняется тем, что в данном случае появляется дополнительный выигрыш за счет разности энергий синглетного и триплетного состояний. Сера обладает интересным свойством спонтанной полимеризации при температуре выше 432 К, которое имеет все черты фазового перехода. При полимеризации происходит разрыв восьмичленных колец и образование цепных молекул с высокой степенью полимеризации. Вычисленная из термодинамических данных энтальпия разрыва связи при переходе кольца–линейная цепь составляет по экспериментальным данным [35] менее 146 кДж/моль, что согласуется с настоящим расчетом.
Обсуждая вопрос о механизме разрыва кольца можно отметить, что этот процесс, возможно, происходит через промежуточную стадию синглетного бирадикала. Однако используемый в работе уровень теории не позволяет корректно описать данный механизм. Для этого необходимы более ресурсоемкие методы, например, многодетерминантные методы. Хотя по спиновому запрету реакция разрыва колец протекать не должна, но на практике из-за того, что разница в энергиях основного синглетного состояния (кольцо) и триплетного (бирадикал, цепочка) не слишком большая, эта реакция происходит, о чем свидетельствует собственно образование поперечных связей при вулканизации чистой серой. Кроме того, описанное выше явление термополимеризации серы было бы невозможно без синглет-триплетной конверсии. Процессы триплет-синглетной конверсии протекают при свето-инициированной полимеризации [36, 37], с ними связаны магнитные эффекты при окислении непредельных соединений [38]. Так что, в данном случае мы просто рассматриваем начальную и конечную стадии процесса, которые, вполне возможно, протекают через стадию образования синглетного радикала, но детальное рассмотрение этого процесса выходит за рамки статьи.
Разница в энергии открытых и закрытых циклов с разным числом атомов, хотя и имеет косвенное отношение к механизму вулканизации, но дает полезную дополнительную информацию. Дело в том, что при вулканизации, помимо образования поперечных связей, имеет место циклическое присоединение серы, причем для некоторых вариантов технологии (получение эбонитов) эти реакции являются основными. Поэтому на основании данных рис. 1 можно сделать вывод о том, что преимущественное число атомов серы в цикле должно варьироваться от 6 до 10.
Возвращаясь к вулканизации, на основании полученных результатов возникает вопрос, что может быть инициатором вулканизации в отсутствии ускорителя? Молекулярный кислород может инициировать радикально-цепное окисление углеводородов по би- или тримолекулярной реакции [39], поскольку сам в основном состоянии является бирадикалом и, поэтому, не нарушая спинового запрета, может, например, отрывать водород от углеводорода. В случае серы, восьмичленное кольцо которой в основном состоянии является синглетом, такая реакция приводила бы к нарушению спинового запрета. Молекулярно-динамическое моделирование показало, что восьмичленное кольцо при температурах вулканизации является устойчивым и без изменения спинового состояния не распадается в цепочку или на более мелкие фрагменты. Даже, если в качестве стартовой конфигурации выбрать разорванное кольцо, то после оптимизации геометрии оно восстанавливается. В то же время, если в процессе молекулярно-динамического моделирования изменить мультиплетность системы, то за достаточно короткое время происходит самопроизвольный разрыв цепочки. В качестве примера на рис. 2 приведена зависимость потенциальной энергии молекулы S8 в процессе молекулярно-динамического моделирования при T = = 433 К. В качестве стартовой конфигурации было выбрано оптимизированное по геометрии кольцо с мультиплетностью m = 1. Видно, что, несмотря на флуктуации, конфигурация кольца сохраняется достаточно длительное время, превышающее время колебательной релаксации системы. В момент времени интегрирования, равный 1750 фс, параметры системы были изменены путем установки мультиплетности m = 3. При этом, как видно, за достаточно короткое время произошел самопроизвольный разрыв кольца, и далее динамика модели отвечала флуктуациям разорванной цепи. Таким образом, причиной термополимеризации серы, а также вулканизации в отсутствии инициаторов являются, по-видимому, синглет-триплетные переходы, приводящие к разрыву серных колец.
Рис. 2.
Зависимость потенциальной энергии молекулы S8 от времени молекулярно-динамического моделирования при T = 433 K.

При вулканизации на воздухе в отсутствии органических ускорителей инициатором могут являться органические пероксиды, образующиеся при окислении каучука. Чистые пероксиды используются в технологической практике в качестве ускорителей серной вулканизации и при этом являются одним из наиболее активных типов ускорителей. При окислении каучука кислородом происходит накопление гидропероксидов, которые, распадаясь, инициируют вулканизацию серой. Согласно расчету, изменение энергии при реакции образования гидропероксида полиизопрена составило –55.6 кДж/моль. Однако скорость данной реакции, протекающей по радикально-цепному механизму, в отсутствии инициаторов достаточно мала, поскольку автоинициирование имеет достаточно высокую энергию активации [39], поэтому концентрация пероксидов и гидропероксидов при автоокислении каучука возрастает достаточно медленно, и скорость вулканизации серой без ускорителей невысока. Если же пероксиды или гидропероксиды включить в состав резиновой смеси в нужном количестве, то скорость вулканизации резко возрастает. Наиболее активными при распаде гидропероксидов являются гидропероксильные радикалы OH*. Изменение энергии при реакции
по данным расчета составило ΔЕ = –110 кДж/моль. То есть сера является акцептором радикалов и может участвовать в реакциях передачи цепи.Перейдем к рассмотрению реакций, протекающих в присутствии сульфенамидного ускорителя. Ускоритель N-циклогексил-2 бензтиазолилсульфенамид :
в дальнейшем для удобства будем обозначать как CG–NH–SBA. С точки зрения радикально-цепного механизма реакции вулканизации данный ускоритель представляет собой радикально-цепной инициатор. Вместе с тем, поскольку в технологической практике все радикально-цепные инициаторы принято называть ускорителями вулканизации, мы также будем использовать этот термин. В качестве модели каучука при вычислениях использовали цис-3-метилгекс-3-ен ((Z)-3-Methyl-3-hexene). Для него использовалось обозначение RH, подразумевая под символом “Н” водород у альфа-углеродного атома к двойной связи.
Первым этапом инициирования является распад ускорителя на радикалы, который может протекать по двум направлениям. Первое направление связано с образованием бензтиазолильного и циклогексиламинного радикалов, второе направление связано с отрывом водорода от атома азота сульфенамидного ускорителя
(2)
$\begin{gathered} {\text{CG}}{\kern 1pt} --{\kern 1pt} {\text{NH}}{\kern 1pt} --{\kern 1pt} {\text{SBA}} \to {\text{CG}}{\kern 1pt} --{\kern 1pt} {\text{NH}}{\kern 1pt} * + *{\kern 1pt} {\text{SBA}}, \\ \Delta Е = 251\;{\text{кДж/моль}}, \\ \end{gathered} $(3)
$\begin{gathered} {\text{CG}}{\kern 1pt} --{\kern 1pt} {\text{NH}}{\kern 1pt} --{\kern 1pt} {\text{SBA}} \to {\text{CGN}}{\kern 1pt} *{\kern 1pt} --{\kern 1pt} {\text{SBA}} + {\text{Н}}, \\ \Delta Е = 38{\text{7}}\;{\text{кДж/моль}}{\text{.}} \\ \end{gathered} $Сравнение энергий этих реакций показывает, что наиболее вероятной является реакция распада ускорителя на два радикала, что подтверждается направлением реакции, наблюдающимся на практике.
Образовавшиеся после распада ускорителя радикалы могут разрывать кольцо S8(ring) по реакции, аналогичной (1)
(4)
$\begin{gathered} {{{\text{S}}}_{{{\text{8(ring)}}}}} + *{\kern 1pt} {\text{SBA}} \to *{\kern 1pt} {{{\text{S}}}_{{\text{8}}}}{\text{SBA}}, \\ \Delta Е = - 87\;{\text{кДж/моль}}, \\ \end{gathered} $(5)
$\begin{gathered} {{{\text{S}}}_{{{\text{8(ring)}}}}} + {\text{CG}}{\kern 1pt} --{\kern 1pt} {\text{NH}}{\kern 1pt} * \to {\text{CG}}{\kern 1pt} --{\kern 1pt} {\text{NHS}}_{8}^{*}, \\ \Delta Е = --55\;{\text{кДж/моль}}. \\ \end{gathered} $Из сравнения изменений энергий этих реакций с изменением энергии при реакции (1) можно сделать вывод, что фрагменты сульфенамидного ускорителя являются не такими активными инициаторами по сравнению с гидропероксидным радикалом, причем наименее активным является циклогексиламинный радикал. Данный результат согласуется с известным из технологической практики фактом, что гидропероксиды являются более активными ускорителями серной вулканизации по сравнению с сульфенамидами.
Помимо взаимодействия с серой радикалы ускорителя дегидрируют каучук по реакциям
(6)
$\begin{gathered} {\text{RH}} + *{\kern 1pt} {\text{SBA}} \to {\text{HSBA}} + {\text{R}}*, \\ \Delta Е = --9\;{\text{кДж/моль}}, \\ \end{gathered} $(7)
$\begin{gathered} {\text{RH}} + {\text{CG}}{\kern 1pt} --{\kern 1pt} {\text{NH}}{\kern 1pt} * \to {\text{CG}}{\kern 1pt} --{\kern 1pt} {\text{N}}{{{\text{H}}}_{2}} + {\text{R}}*, \\ \Delta Е = --6{\text{3}}\;{\text{кДж/моль}}. \\ \end{gathered} $Как видим, в данной реакции циклогексиламинный радикал является более активным. Можно было бы ожидать взаимодействия образовавшихся в результате этих реакций углеродцентрированных радикалов с серой аналогично реакциям (1), (4), (5)
(8)
${{{\text{S}}}_{{{\text{8(ring)}}}}} + {\text{R}}* \to *{\kern 1pt} {{{\text{S}}}_{8}}{\text{R}}.$(9)
${\text{R}}{\kern 1pt} * + *{\kern 1pt} {{{\text{S}}}_{{\text{8}}}}{\text{X}} \to {\text{R}}{{{\text{S}}}_{{\text{8}}}}{\text{X}},$Полисульфидные связи относительно непрочные, так что в результате обменных реакций число атомов серы в персульфидных подвесках и, соответственно, в серных поперечных связях не равно строго восьми, а варьируется. В табл. 1 приведены значения ΔЕ при реакциях вида
(10)
$*{{{\text{S}}}_{{\text{8}}}}{\text{X}} \to *{{{\text{S}}}_{y}}{\text{X}} + *{\text{S}}_{{8 - y}}^{*}$Таблица 1.
Влияние числа атомов серы y и типа радикала на изменение энергии (ΔЕ, кДж/моль) при реакции (10) с выделением бирадикала серы $*{\text{S}}_{{8 - y}}^{*}$
| Тип радикала | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| *SyH | 234 | 128 | 127 | 188 | 245 | 240 | 430 |
| *SySBA | 126 | 123 | 126 | 96 | 166 | 179 | 309 |
| CGNHS$_{y}^{*}$ | 123 | 124 | 116 | 98 | 163 | 76 | 228 |
| RS$_{y}^{*}$ | 42 | –19 | –10 | 13 | 78 | 62 | 297 |
Прочность поперечных связей зависит от их сульфидности. При изменении числа атомов серы в поперечной связи изменяется потенциальная энергия разрыва связи, но при этом также может изменяться их динамическая подвижность. В табл. 2 приведены данные, иллюстрирующие зависимость изменения энергии при реакции разрыва сшивки по разным связям с различным числом атомов
(11)
${\text{R}}{\kern 1pt} --{\kern 1pt} {{{\text{S}}}_{y}}{\text{R}} \to {\text{R}}{\kern 1pt} * + *{\kern 1pt} {{{\text{S}}}_{y}}{\text{R}},$(12)
${\text{R}}{{{\text{S}}}_{z}}{\kern 1pt} --{\kern 1pt} {{{\text{S}}}_{{y - z}}}{\text{R}} \to {\text{RS}}_{z}^{*} + *{{{\text{S}}}_{{y - z}}}{\text{R}}$Таблица 2.
Изменения энергии (ΔЕ, кДж/моль) при реакциях (11), (12) для различного числа атомов серы, образующих сшивку и различных типов разрываемых связей
| Число атомов серы | Разрываемая связь | ||||
|---|---|---|---|---|---|
| ${\text{R}}{\kern 1pt} --{\kern 1pt} {{{\text{S}}}_{y}}{\text{R}}$ | ${\text{RS}}{\kern 1pt} --{\kern 1pt} {{{\text{S}}}_{{y--1}}}{\text{R}}$ | ${\text{R}}{{{\text{S}}}_{2}}{\kern 1pt} --{\kern 1pt} {{{\text{S}}}_{{y--2}}}{\text{R}}$ | ${\text{R}}{{{\text{S}}}_{3}}{\kern 1pt} --{\kern 1pt} {{{\text{S}}}_{{y--3}}}{\text{R}}$ | ${\text{R}}{{{\text{S}}}_{4}}{\kern 1pt} --{\kern 1pt} {{{\text{S}}}_{{y--4}}}{\text{R}}$ | |
| 1 | 180 | ||||
| 2 | 150 | 213 | |||
| 3 | 155 | 151 | |||
| 4 | 205 | 164 | 98 | ||
| 5 | 198 | 201 | 98 | ||
| 6 | 293 | 212 | 152 | 115 | |
| 7 | 361 | 295 | 151 | 157 | |
| 8 | 299 | 374 | 245 | 167 | 210 |
Как видно, наименьшей энергией разрыва связи обладают полисульфидные связи RS2–S2R и RS2–S3R. Этот результат согласуется также с тем, что в реакциях распада бирадикалов
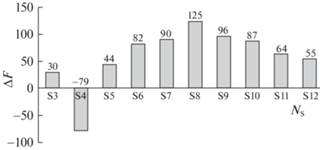
отрыв радикалов с двумя атомами серы требует наименьшей энергии, что связано с тем, как отмечено выше, что для двухатомной молекулы серы основным состоянием является триплет.Степень динамической подвижности поперечной связи можно оценить по величине коэффициента вариации ее длины при молекулярно-динамическом моделировании. Такие данные приведены на рис. 3, где показана зависимость коэффициента вариации расстояния между атомами углерода двух молекул изопрена, между которыми образована серная поперечная связь от числа атомов серы в связи. Несмотря на статистические флуктуации, видно, что с увеличением числа атомов серы в поперечной связи ее подвижность изменяется не монотонно. Наибольшей подвижностью обладают связи с числом атомов серы, равным четырем. Далее, с увеличением числа атомов серы в поперечной связи имеется тенденция к уменьшению подвижности. Это объясняется тем, что при большой длине связи атомы серы начинают формировать собственную, более жесткую структуру путем складывания серной цепочки, подобно тому, как показано на рис. 2 после разрыва кольца.
Рис. 3.
Зависимость коэффициента вариации (γ) длины серной поперечной связи от числа атомов серы (N) в ней при молекулярно-динамическом моделировании при T = 373 K.

Заканчивая рассмотрение реакций серной вулканизации, протекающих при инициировании N-циклогексил-2 бензтиазолилсульфенамидом, отметим, что обсуждение различных вариантов реакций основывалось, главным образом, на изменении электронной энергии реагентов, а вклады в изменение свободной энергии, связанные с изменением колебательно-вращательных составляющих внутренней энергии и энтропией не учитывались. Это было обусловлено тем, что используемая в настоящее время теория для вычисления таких вкладов основана на приближении молекулы “жесткий ротатор – гармонический осциллятор” в состоянии идеального газа. Для реакции вулканизации, протекающей в конденсированном состоянии с реагентами, имеющими большое число внутренних вращательных степеней свободы и, кроме того, имеющих склонность к образованию ассоциатов, такая теория является заведомо неприменимой. В то же время квантово-химический расчет электронной составляющей не связан с такими приближениями и является вполне адекватным. Реакция распада инициатора (2), лимитирующая протекание всего процесса, характеризуется сильным положительным значением изменения электронной энергии. Расчет изменения свободной энергии в приближении “жесткий ротатор – гармонический осциллятор” дает для этой реакции поправку ‒75.2 кДж/моль при температуре 298.15 К и ‒108.4 кДж/моль при температуре 448 К, которая выше обычно применяемой температуры вулканизации 416–433 К. То есть согласно этой оценки изменение свободной энергии реакции будет даже при высокой температуре иметь достаточно большое положительное значение. Это означает, что формально реакция (2) в данных условиях термодинамически невозможна. Однако на самом деле она протекает, поскольку в действительности является лишь первой стадией радикально-цепного процесса, последующие стадии которого (4)–(7), (9) характеризуются отрицательными значениями ΔЕ, так что весь процесс вулканизации, как известно, достаточно сильно экзотермический. Для описания этого процесса необходимо кинетическое рассмотрение, что выходит за рамки настоящей работы. Вместе с тем, сравнительный анализ изменений электронной энергии отдельных реакций, проведенный в настоящей работе, позволяет из большого числа возможных реакций выбрать наиболее вероятные и построить приемлемую в вычислительном отношении формальную кинетическую схему.
Таким образом, в настоящей работе с использованием квантово-химического метода функционала плотности DFT B3LYP/6-311G** исследованы реакции, протекающие при серной вулканизации изопренового каучука с ускорителем N-циклогексил-2 бензтиазолилсульфенамидом.
Показано, что в отсутствии ускорителя вулканизация может инициироваться кислородом воздуха. В результате расчета зависимости энергии разрыва серного кольца от числа атомов в нем установлено, что наиболее устойчивыми являются восьмичленные кольца серы, что согласуется с тем фактом, что именно такая форма является наиболее распространенной в природе.
Установлено, что при инициировании вулканизации ускорителем, наиболее активными в реакции дегидрирования каучука являются циклогексиламинные радикалы, образующиеся при распаде ускорителя. В то же время в реакции с серным циклом эти радикалы уступают в активности бензтиазолильным.
Показано, что основной реакцией образования персульфгидрильных подвесок, участвующих в образовании поперечных связей, является реакция рекомбинации углеводородных и пертиильных радикалов, образующихся при атаке серного кольца радикалами, возникающими при распаде ускорителя.
В результате исследования зависимости прочности поперечных полисульфидных связей от числа атомов серы, образующих связь, выявлено, что наиболее вероятными продуктами распада будут радикалы, содержащие два атома серы. Следствием этого будет преобладание поперечных связей, содержащих четыре атома серы при их рекомбинации.
Выполненные в настоящей работе расчеты позволяют при составлении кинетической схемы вулканизации и расчете кинетики выбрать именно те реакции, которые согласно данному расчету являются более вероятными.
Список литературы
Coran A.Y. Vulcanization // The science and technology of rubber / Ed. by Mark J.E., Erman B., Roland C.M., 4th ed. Academic Press: Elsevier, 2013. P. 337.
Scheele W., Lorenz O., Dummer W. // Rubber Chemistry and Technology. 1956. V. 29 (1) P. 1. https://doi.org/10.5254/1.3542510
Saville B., Watson A.A. // Ibid. 1967. V. 40 (1). P. 100. https://doi.org/10.5254/1.3539039
Nieuwenhuizen P.J., Reedijk J., van Duin M., McGill W.J. // Ibid. 1997. V. 70 (3). P. 368. https://doi.org/10.5254/1.3538436
Ghosh P., Katare S., Patkar P. et al. //Rubber Chemistry and Technology. 2003. V. 76 (3). P. 592. https://doi.org/10.5254/1.3547762
Wang P., Qian H., Yu H. // J. of Applied Polymer Science 2003. V. 88. P. 680. https://doi.org/10.1002/app.11632
Khang T.H., Ariff Z.M. // J. Therm. Anal. Calorim. 2012. V. 109. P. 1545. https://doi.org/10.1007/s10973-011-1937-3
Fathurrohman M.I., Maspanger D.R., Sutrisno S. // Bulletin of Chemical Reaction Engineering & Catalysis. 2015. V. 10 (2). P. 104. http://bcrec.undip.ac.id.
Hosseini S.M., Razzaghi-Kashani M. // Soft Matter. 2018. V. 14. P. 9194–9208. https://doi.org/10.1039/c8sm01953c
Ding R., Leonov A.I. // J. of Applied Polymer Science. 1996. V. 61. P. 455. https://doi.org/10.1002/(SICI)1097-4628(19960718)61: 3<455::AID-APP8>3.0.CO;2-H.
Likozar B., Krajnc M. // Ibid. 2007. V. 103. P. 293. https://doi.org/10.1002/app.25284
Маркелов В.Г., Соловьев М.Е. // Изв. вузов. Сер. Химия и хим. технология. 2007. Т. 50. № 4. С. 95.
Zhao F., Wu C., Shi X. // J. of Macromolecular Science, Part B: Physics. 2011. V. 50: 2. P. 398. https://doi.org/10.1080/00222341003772282
Leroy E., Souid A., Deterre R. // Polymer Testing. 2013. V. 32. P. 575. https://doi.org/10.1016/j.polymertesting.2013.01.003
Milani G., Milani F. // J. Math. Chem. 2017. V. 55. P. 552. https://doi.org/10.1007/s10910-016-0695-7
Маркелов В.Г., Раухваргер А.Б., Соловьев М.Е. // Изв. вузов. Сер. Химия и хим. технология. 2009. Т. 52. № 5. С. 119.
Hohenberg P., Kohn W. Inhomogeneous Electron Gas // Phys. Rev. 1964. V. 136. 3B. P. B864. https://doi.org/10.1103/PhysRev.136.B864
Kohn W., Sham L.J. //Phys. Rev. 1965. V. 140. 4A. P. A1133. https://doi.org/10.1103/PhysRev.140.A1133
Becke A.D. // J. Chem. Phys. 1993. V. 98. № 7. P. 5648. https://doi.org/10.1063/1.462066
Miehlich B., Savin A., Preuss H. // Chem. Phys. Lett. 1989. V. 157. № 3. P. 200. https://doi..https://doi.org/10.1016/0009-2614(89)87234-3
Valiev M. et al. // Comput. Phys. Commun. 2010. V. 181. № 9. P. 1477.https://doi.org/10.1016/j.cpc.2010.04.018
Neese F. // Wiley Interdiscip. Rev.: Comput. Mol. 2017 Sci.8. e1327. https://doi.org/10.1002/wcms.1327
Pliss E.M., Soloviev M.E., Tikhonov I.V. et al. // Rus. J. of Phys.Chem.B. 2016 V. 10. № 3. P. 417.https://doi.org/10.1134/S1990793116030052.
Solov’ev M.E., Kurganova E.A., Frolov A.S., Koshel’ G.N. // Rus. J. of Phys. Chem. A. 2019. V. 93. № 3. C. 470. https://doi.org/10.1134/S0036024419030191
Soloviev M., Moskalenko I., Pliss E. // Reaction Kinetics, Mechanisms and Catalysis. 2019. V. 127. № 2. P. 561. https://doi.org/10.1007/s11144-019-01613-w.
Goerigk L., Mehta N. //Australian J. of Chem. 2019. V. 72 (8). P. 563. https://doi.org/10.1071/CH19023
Broyden C.G. // J. of the Institute of Mathematics and Its Applications, 1970. V. 6. P. 76. https://doi.org/10.1093/imamat/6.1.76
Fletcher R. // Computer J. 1970. V. 13 (3). P. 317. https://doi.org/10.1093/comjnl/13.3.317
Goldfarb D. // Mathematics of Computation. 1970. V. 24 (109). P. 23. https://doi.org/10.1090/S0025-5718-1970-0258249-6
Shanno D.F. // Ibid. 1970. V. 24 (111). P. 647. https://doi.org/10.1090/S0025-5718-1970-0274029-X
Mueller M. Fundamentals of Quantum Chemistry. Molecular Spectroscopy and Modern Electronic Structure Computation. New York (NY): Kluwer Academic. 2002. https://doi.org/10.1063/1.1535013
Levine I.A. Quantum Chemistry. 5-th ed / I.A. Levine. New Jersey, Upper Sadle River: Prentice-Hall, 2000. 740 p.
Bussi G., Donadio D., Parrinello M. // J. Chem Phys. 2007. V. 126. P. 014101. https://doi.org/10.1063/1.2408420
Degirmenci I., Coote M.L. // J. Phys. Chem. A 2016. 120. 37. P. 7398–7403. https://doi.org/10.1021/acs.jpca.6b08223
Fairbrother F., Gee G., MerrallL G.T. // J. of Polymer Science. 1955. V. 26. P. 459. https://doi.org/10.1002/pol.1955.120168231
Khudyakov I.V., Arsu N., Jockusch S., Turro J. Magnetic and Spin Effects in the Photoinitiation of Polymerization. Designed Monomers and Polymers .2003. V. 6. P. 91. https://doi.org/10.1163/156855503321127565
Rintoul I. Kinetic Control of Aqueous Polymerization Using Radicals Generated in Different Spin States. Processes. Open Access Chem. Biolog. Proc. Eng. J. 2017. V. 5. P. 1. https://doi.org/10.3390/pr5020015
Pliss E.M., Grobov A.M., Kuzaev A.K., Buchachenko A.L. // Mendeleev Commun. 2020. V. 30. P. 433. https://doi.org/10.1016/j.mencom.2020.07.009
Denisov E.T., Afanas’ev I.V. Oxidation and antioxidants in organic chemistry and biology. 2005. Boca Raton: CRC Press. 992 p.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии