

Журнал физической химии, 2022, T. 96, № 11, стр. 1535-1546
Энтальпийно-энтропийная компенсация в реакциях раскрытия оксиранового цикла
И. В. Шпанько a, *, И. В. Садовая b
a Донецкий национальный университет имени В. Стуса
Винница, Украина
b Донецкий национальный университет
Донецк, Украина
* E-mail: shpanko16@ukr.net
Поступила в редакцию 18.11.2021
После доработки 26.01.2022
Принята к публикации 28.01.2022
- EDN: NLYSVW
- DOI: 10.31857/S0044453722110309
Аннотация
Обобщены результаты систематического исследования энтальпийно-энтропийного компенсационного эффекта в некаталитических и катализируемых пиридинами реакциях арилоксиранов с органическими кислотами разных классов. Этот эффект проявляется в изопараметрических (изокинетических, изоэнергетических) реакционных сериях вследствие взаимодействия (неаддитивности) совместных эффектов температуры и структуры. Приведены экспериментальные доказательства его физической реальности в ряде перекрестных реакционных серий. В рамках компенсационного эффекта осуществлены переходы от одного состояния реакционных систем, при котором энтальпийный терм свободной энергии активации приобретает нулевое значение (∆H≠ = = 0, ∆G≠ = –TΔS≠), к другому состоянию, при котором исчезает вклад в свободную энергию активации энтропийного терма (ΔS≠ = 0, ∆G≠ = ΔH≠). Обсужден характер активационных процессов в отсутствие энтальпийно-энтропийной компенсации.
ВВЕДЕНИЕ
Для эффективного управления химическими процессами необходимо знать количественные закономерности совместного влияния внешних и внутренних факторов (структура, катализатор, растворитель, давление, pH среды и т.д.) на их кинетические, активационные, термодинамические и другие характеристики. Для решения этой фундаментальной проблемы химии широко привлекаются методы корреляционного анализа. Известны десятки эмпирических корреляционных соотношений [1–8], успешно используемых для количественной оценки влияния различных факторов на химические процессы. Они демонстрируют поразительную универсальность принципа линейности в изменении свободных энергий, который, как показал Пальм, является частным случаем более общей закономерности – принципа полилинейности [4, 9]. Примером простейших соотношений полилинейности являются двухпараметровые уравнения типа
(1)
${{F}_{{ij}}} = {{F}_{{00}}} + q_{i}^{0}{{x}_{i}} + q_{j}^{0}{{x}_{j}} + {{q}_{{ij}}}{{x}_{i}}{{x}_{j}}.$Уравнение (1) обладает таким замечательным свойством, как изопараметричность [4, 9]. Она выражается в том, что при критических значениях параметра фактора i $x_{i}^{{{\text{ИП}}}}$ = –$q_{j}^{0}$/qij или фактора j $x_{j}^{{{\text{ИП}}}}$ = –$q_{i}^{0}$/qij, названных изопараметрическими точками (ИПТ) [4], коррелируемая величина Fij имеет одно и то же значение $F_{{ij}}^{{{\text{ИП}}}}$ = F00 – ‒ $q_{i}^{0}q_{j}^{0}$/qij, которое не изменяется при варьировании соответственно xj ($q_{j}^{i}$ = 0) или xi ($q_{i}^{j}$ = 0).
Первоначально понятие изопараметричности возникло в рамках формальной теории взаимодействия [4]. Впоследствии выяснилось, что свойство изопараметричности присуще реальным реакционным сериям (РС). На практике изопараметричность проявляется в равенстве нулю угловых коэффициентов чувствительности к эффектам одного из факторов в эмпирических однопараметровых корреляциях, например, α (β) в уравнении Бренстеда, ρ в уравнении Гаммета, пропорциональный энергии активации коэффициент в уравнении Аррениуса и т.д., в ИПТ по параметру другого фактора. После перехода через ИПТ происходит инверсия знаков соответствующих коэффициентов чувствительности (парадокс изопараметричности).
Соотношения полилинейности показали свою эффективность при изучении в многофакторных условиях процессов нуклеофильного замещения у бензоильных, бензильных и бензгидрильных электрофильных центров, а также при интерпретации их механизмов [10–12]. Благодаря интенсивному взаимодействию эффектов структуры в этих процессах были получены первые в истории химии экспериментальные доказательства феномена изопараметричности.
Прогресс в изучении изопараметричности связан с поиском таких РС, в которых в перекрестные взаимодействия были бы вовлечены, помимо структурных, и другие факторы. Из всего многообразия факторов, оказывающих влияние на химические, физические, биологические и другие процессы, следует выделить такой универсальный фактор, как температура. Изучению эффектов температуры в химических процессах посвящено огромное количество публикаций со времен Аррениуса. Интерес к исследованию температурных зависимостей значительно возрос после того, как была обоснована концепция изокинетических (изоравновесных) соотношений, базирующихся на энтальпийно-энтропийном компенсационном эффекте (КЭ) [13]. Важнейшей количественной характеристикой химических процессов, описываемых этими соотношениями, является изокинетическая (изоравновесная) температура Tизо, при которой имеет место полная компенсация в изменении энтальпийной и энтропийной составляющих свободной энергии активации (реакции) при варьировании параметра какого-либо отличного от температуры фактора j, поэтому при Tизо$\Delta G_{{jT}}^{ \ne }$ (∆GjT) = const, вследствие чего наблюдается изокинетический (изоравновесный) феномен: $\lg {{k}_{{jT}}}$ = const ($\lg {{K}_{{jT}}}$ = = const). Если в уравнении (1) один из переменных факторов, например, i является температурой (xi = Т), то тогда ИПТ по температуре TИП равна Tизо в изопараметрических (в частном случае в изокинетических) РС с энтальпийно-энтропийной компенсацией.
Несмотря на широкий фронт исследования энтальпийно-энтропийного КЭ в различных областях естественных наук случаи экспериментального наблюдения TИП встречаются крайне редко. Как правило значения TИП попадают в область далекой экстраполяции, т.е. TИП имеет скорее виртуальный, чем экспериментальный характер. В связи с экспериментальной недоступностью TИП, статистически ненадежными расчетами компенсационных корреляций, отсутствием приемлемых теоретических обоснований КЭ, концепция энтропийно-энтальпийной компенсации является предметом перманентных дебатов в течение многих десятилетий (см., например, [14–21]).
Целью настоящей статьи является обобщение результатов проведенного нами систематического исследования энтальпийно-энтропийного КЭ в реакциях арилаксиранов с органическими кислотами разной природы в некаталитических и каталитических условиях.
Энтальпийно-энтропийный КЭ как аспект изопараметричности (формальный анализ)
Неаддитивное влияние температуры T и какого-либо фактора j на свободную энергию активации описывается полилинейным соотношением
(2)
$\Delta G_{{jT}}^{ \ne } = \Delta G_{{00}}^{ \ne } + a_{j}^{0}{{x}_{j}} + a_{T}^{0}T + {{a}_{{jT}}}{{x}_{j}}T.$Иное интригующее свойство соотношения (2) проявляется, если представить его в форме уравнения
(3)
$\Delta G_{{jT}}^{ \ne } = \Delta G_{{00}}^{ \ne } + a_{T}^{0}T + (a_{j}^{0} + {{a}_{{jT}}}T){{x}_{j}}.$Итак, в ИПТ по температуре TИП(G) величина $\Delta G_{{jT}}^{{ \ne {\text{ИП}}}}$ сохраняет постоянство при варьировании параметра xj фактора j. Это происходит вследствие энтальпийно-энтропийного КЭ, а именно, из-за компенсации в изменении энтальпийной и энтропийной части свободной энергии активации δjΔH≠ = TИП(G)δjΔS≠, в результате чего δj$\Delta G_{{jT}}^{{ \ne {\text{ИП}}}}$ = = δjΔH≠ – TИП(G)δjΔS≠ = 0, $\Delta G_{{jT}}^{{ \ne {\text{ИП}}}}$ = const и, следовательно, $a_{j}^{T}$ = 0 в уравнении (4) при Тconst = TИП(G). Поскольку при TИП(G) δjΔH≠ – TИП(G)δjΔS≠ = 0, то при переходе через TИП(G) будет изменяться соотношение вкладов δjΔH≠ и TδjΔS≠ в изменение δj$\Delta G_{{jT}}^{ \ne }$ (δjΔH≠ – TδjΔS≠ > 0, δjΔH≠ – TδjΔS≠ < 0), вследствие чего произойдет обращение порядка влияния фактора j на $\Delta G_{{jT}}^{ \ne }$. Это выразится в инверсии знака коэффициента чувствительности $a_{j}^{T}$.
С другой стороны, в ИПТ $х_{j}^{{{\text{ИП}}(G)}}$ свободная энергия активации $\Delta G_{{jT}}^{{ \ne {\text{ИП}}}}$ не зависит от температуры T: $\Delta G_{{jT}}^{{ \ne {\text{ИП}}}}$ = $\Delta G_{{00}}^{ \ne }$ – $a_{j}^{0}a_{T}^{0}$/ajT. Это возможно, если в выражении $\Delta G_{{jT}}^{{ \ne {\text{ИП}}}}$ = $\Delta H_{j}^{ \ne }$ – T$\Delta S_{j}^{ \ne }$ энтропия активации равна нулю ($\Delta S_{j}^{ \ne }$ = 0) и величина $\Delta G_{{jT}}^{{ \ne {\text{ИП}}}}$ определяется энтальпийным термом ($\Delta G_{{jT}}^{{ \ne {\text{ИП}}}}$ = $\Delta H_{j}^{ \ne }$). Переход через $х_{j}^{{{\text{ИП}}(G)}}$, где $\Delta S_{j}^{ \ne }$ = 0, будет вызывать изменение знака $\Delta S_{j}^{ \ne }$ ($\Delta S_{j}^{ \ne }$ > 0, $\Delta S_{j}^{ \ne }$ < 0) и, следовательно, обращение порядка влияния T на $\Delta G_{{jT}}^{ \ne }$, что отразится в инверсии знака коэффициента чувствительности $a_{T}^{j}$.
После деления на T левой и правой части уравнения (2), получим изопараметрическое соотношение
(5)
$\Delta G_{{jT}}^{ \ne }{\text{/}}T = \Delta G_{{00}}^{ \ne }{\text{/}}T + a_{j}^{0}{{x}_{j}}{\text{/}}T + a_{T}^{0} + {{a}_{{jT}}}{{x}_{j}}.$В связи с вышеприведенными интригующими предсказаниями состояния активационного процесса в ИПТ TИП(G) (TИП(G/Т)), $х_{j}^{{{\text{ИП}}(G)}}$, $x_{j}^{{{\text{ИП}}(G/Т)}}$ и после перехода через эти точки, основанными на анализе абстрактных полилинейных соотношений (2) и (5), возникает вопрос о том, являются ли они лишь следствием формальных математических свойств этих соотношений, или же они предстают перед нами как физическая реальность в химических процессах. Ответ на этот вопрос мы получили при систематическом исследовании совместных эффектов структуры и температуры в реакциях раскрытия оксиранового цикла, отчет о котором представлен в настоящей статье.
Следует отметить, что в случае аддитивного характера совместных эффектов температуры и фактора j в соотношениях типа (2) исчезает перекрестный член (ajT = 0) и поэтому проявление изопараметричности, а, следовательно, и энтальпийно-энтропийного КЭ становится в принципе невозможным. В рамках принципа линейности в изменении свободных энергий в этом случае РС могут быть либо изоэнтальпийными ($\Delta H_{j}^{ \ne }$ = const, δj∆H≠ = 0, δj∆G≠ = –Tδj∆S≠), либо изоэнтропийными ($\Delta S_{j}^{ \ne }$ = const, δj∆S≠ = 0, δj∆G≠ = δj∆H≠). Примеры таких РС приведены в этой статье и в обзоре [22].
Перекрестные эффекты структуры и температуры. Физическая реальность энтальпийно-энтропийного КЭ
Количественные аспекты перекрестных эффектов структуры и температуры изучены в представленных схемами 1, 2 реакциях X-замещенных 2-арилоксиранов 1а–е [X = H (1а), 3-Br (1б), 4-Br (1в), 4-Cl (1г), 3-NO2 (1д), 4-NO2 (1е), 4-Br-3-NO2 (1ж), 3,5-(NO2)2 (1з)] и симметрично Х-замещенных транс-2,3-диарилоксиранов 4а–г [Х = H (4а), 3-Br (4б), 4-NO2 (4в), 3-Br-5-NO2 (4г)] с Y-замещенными аренсульфоновыми кислотами 2а–е [Y = 4-OCH3 (2а), 4-CH3(2б), H (2в), 4-Cl (2г), 4-Br (2д), 3-NO2 (2е)] [23–27] и аренкарбоновыми (бензойными) кислотами 3а–д [Y = 4-OCH3 (3а), H (3б), 3-Br (3в), 3-NO2 (3г), 3,5-(NO2)2 (3д)] [28, 29], а также в реакциях оксирана 1а, с такими представителями NH-кислот, как аренсульфонимиды 5а–в [Y = 4-OCH3 (5а), 4-CH3(5б), H (5в)] (схема 3 ) [30] и N-ароилбензолсульфонамиды 6а–д [Y = 4-CH3(6а), H (6б), 4-Cl (6в), 3-F (6г), 4-NO2 (6д)] (схема 4 ) [31]. Кроме того, в реакциях оксирана 1а с кислотами 3б-г и 6а, б, д, катализируемых Z-замещенными пиридинами Z-Py 7а–д [Z = 4-OMe (7a), 4-Et (7б), H (7в), 3-COOEt (7г), 3-CN (7д), рассмотрены совместные эффекты температуры и структуры катализатора (схема 5 ), а также температуры и структуры кислотного реагента (схема 6 ) [32–34].

Схема 1

Схема 2

Схема 3

Схема 4

Схема 5

Схема 6
В реакциях с участием оксиранов 1а–е происходит α-раскрытие цикла с образованием первичных спиртов (схема 1 ), а в реакциях оксиранов 1ж, з образуются вторичные спирты, продукты β-раскрытия цикла [24, 35].
Влияние температуры на скорость реакций оценивалось с помощью уравнения Эйринга:
(6)
$\lg ({{k}_{{jT}}}{\text{/}}T) = A_{{T = \infty }}^{j} + B_{T}^{j}{\kern 1pt} {{10}^{3}}{\text{/}}T.$Для учета электронных эффектов заместителей Х и Y при фиксированных температурах T использовалось уравнение Гаммета в виде соотношений
(7)
$\lg {{k}_{{\text{X}}}}_{Т} = \lg {{k}_{{{\text{H}}T}}} + \rho _{{\text{X}}}^{Т}{{\sigma }_{{\text{X}}}},$(8)
$\lg {{k}_{{\text{Y}}}}_{Т} = \lg {{k}_{{{\text{H}}T}}} + \rho _{{\text{Y}}}^{T}{{\sigma }_{{\text{Y}}}}.$Оценка совместного влияния структуры и температуры Т на скорость реакций (схемы 1–4 ) осуществлялась с использованием полилинейных уравнений
(9)
$\begin{gathered} \lg {{k}_{{{\text{Y}}Т}}} = \lg {{k}_{{{\text{H}}T = \infty }}} + \rho _{{\text{Y}}}^{{Т = \infty }}{{\sigma }_{{\text{Y}}}} + \\ + \;q_{T}^{{{\text{Y}} = {\text{H}}}} \times {{10}^{3}}{\text{/}}T + {{q}_{{{\text{Y}}T}}}{{\sigma }_{{\text{Y}}}} \times {{10}^{3}}{\text{/}}T, \\ \end{gathered} $(10)
$\begin{gathered} \lg {{k}_{{{\text{X}}Т}}} = \lg {{k}_{{{\text{H}}T = \infty }}} + \rho _{{\text{X}}}^{{Т = \infty }}{{\sigma }_{{\text{X}}}} + \\ + \;q_{T}^{{{\text{X}} = {\text{H}}}} \times {{10}^{3}}{\text{/}}T + {{q}_{{{\text{X}}T}}}{{\sigma }_{{\text{X}}}} \times {{10}^{3}}{\text{/}}T. \\ \end{gathered} $Уравнение (9) описывает эффекты температуры и заместителей Y в кислотном реагенте при фиксированных заместителях Х в оксиране, а уравнение (10) – эффекты температуры и заместителей Х при фиксированных заместителях Y.
Коэффициенты уравнения (9) и рассчитанные на их основе значения ИПТ по параметрам варьируемых факторов $\sigma _{{\text{Y}}}^{{{\text{ИП}}}}$ = –$q_{T}^{{{\text{Y}} = {\text{H}}}}$/qYT и ТИП = = ‒qYT × 103/$\rho _{{\text{Y}}}^{{Т = \infty }}$ приведены в табл. 1 для РС 1–3. В этой таблице также представлены коэффициенты уравнений (9) и (10), в которых отсутствуют перекрестные члены (qYT = 0, qXT = 0), для РС 4–7.
Таблица 1.
Коэффициенты уравнений (9)а, (10)а и значения ИПТ по параметрам варьируемых факторов для реакций оксиранов 1, 4 с кислотами 2, 3, 5, 6 (ДО – диоксан, ДГ – диглим, АН – ацетонитрил, 1,2-ДХЭ – 1,2-дихлорэтан)
| РС | Оксиран | Кислота | Среда | $\lg {{k}_{{{\text{H}}T = \infty }}}$ | $\rho _{{\text{Y}}}^{{Т = \infty }}$ | $q_{T}^{{{\text{Y}} = {\text{H}}}}$ | qYT | $\sigma _{{\text{Y}}}^{{{\text{ИП}}}}$ | ТИП, K | Ссылки |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1з | 2а–г, е | ДО : ДГ, 1 : 1 | 8.4 ± 0.2 | 9.5 ± 0.6 | –3.21 ± 0.07 | –2.5 ± 0.2 | –1.28 | 263б | [24] |
| 2 | 1е | 2а–в, д, е | ДО | 10.4 ± 0.3 | 21 ± 1 | –3.4 ± 0.1 | –5.8 ± 0.4 | –0.59 | 276 | [23] |
| 3 | 4г | 2а–г | ДО : 1,2-ДХЭ, 7 : 3 | 11.7 ± 0.1 | 8.4 ± 0.7 | –4.61 ± 0.04 | –2.2 ± 0.2 | –2.09 | 262б | [27] |
| 4в | 1а | 3а–д | АН | –4.87 ± 0.03 | 1.79 ± 0.04 | –4.3 ± 0.3 | – | – | – | [28] |
| 5 | 1а | 5а–в | ДО | 6.1 ± 0.2 | 1.69 ± 0.03 | –2.91 ± 0.07 | – | – | – | [30] |
| 6 | 1а | 6а–д | АН | 8.4 ± 0.5 | 1.45 ± 0.04 | –4.6 ± 0.2 | – | – | – | [31] |
| $\lg {{k}_{{{\text{H}}T = \infty }}}$ | $\rho _{{\text{X}}}^{{Т = \infty }}$ | $q_{T}^{{{\text{X}} = {\text{H}}}}$ | qХT | |||||||
| 7 в,г | 1а–е | 3д | АН | –2.42 ± 0.04 | –2.99 ± 0.08 | –4.3 ± 0.3 | – | – | – | [29] |
а Коэффициенты перекрестной корреляции R ≥ 0.995. б Экспериментально наблюдаемая ИПТ. в В уравнениях (9), (10) использована внутренняя шкала температуры τТ = (1/Т – 1/333) × 103. г В уравнении (10) использованы константы $\sigma _{{\text{X}}}^{ + }$ заместителей Х.
Значения ТИП в РС 1-3 соответствуют наклонам компенсационных зависимостей (11)–(13) в изменении энтальпии $\Delta H_{{\text{Y}}}^{ \ne }$ и энтропии $\Delta S_{{\text{Y}}}^{ \ne }$ активации под влиянием заместителей Y в кислоте 2.
(11)
$\begin{gathered} \Delta H_{{\text{Y}}}^{ \ne } = (83.3 \pm 0.3) \times {{10}^{3}} + (268 \pm 3)\Delta S_{{\text{Y}}}^{ \ne }, \\ S = 382,\quad r = 0.999,\quad n = 5, \\ \end{gathered} $(12)
$\begin{gathered} \Delta H_{{\text{Y}}}^{ \ne } = (75 \pm 2) \times {{10}^{3}} + (260 \pm 14)\Delta S_{{\text{Y}}}^{ \ne }, \\ S = 1092,\quad r = 0.998,\quad n = 3, \\ \end{gathered} $(13)
$\begin{gathered} \Delta H_{{\text{Y}}}^{ \ne } = (93.2 \pm 0.3) \times {{10}^{3}} + (266 \pm 6)\Delta S_{{\text{Y}}}^{ \ne }, \\ S = 290,\quad r = 0.999,\quad n = 4. \\ \end{gathered} $Можно использовать и другие альтернативные методы расчета ТИП, основанные на принципе полилинейности. Так, например, линейная зависимость коэффициента чувствительности $\rho _{{\text{Y}}}^{T}$ в уравнении (8) от обратной температуры для РС 3 имеет следующий вид [27]: $\rho _{{\text{Y}}}^{T}$ = (8.4 ± 0.7) + (–2.2 ± ± 0.2) × 103/T (r = 996). Ее угловой наклон совпадает со значением коэффициента перекрестной корреляции qYT этой серии в табл. 1. Из данной зависимости можно определить значение изокинетической температуры ТИП = 262 K, при которой исчезает чувствительность к эффектам заместителей Y ($\rho _{{\text{Y}}}^{T}$ = 0). Эта температура согласуется со значениями ТИП в табл. 1 и в уравнении (13).
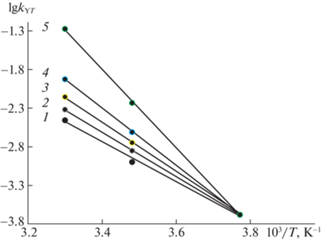
В РС 1, 3 осуществлена экспериментальная реализация ТИП. Их значения близки к температуре 265 K в эксперименте. При этой температуре в соответствии с закономерностями изопараметрических зависимостей должно наблюдаться отсутствие чувствительности процесса к эффектам заместителей Y. Действительно, в этих РС значения $\rho _{{\text{Y}}}^{T}$ стремятся к нулю с уменьшением температуры до 265 K [24, 26]: 1.18 ± 0.05 (303 K), 0.81 ± 0.01 (287 K), 0 (265 K); 1.01 ± 0.09 (298 K), 0.50 ± 0.04 (281 K), 0.10 ± 0.05 (265 K). Рисунок 1 иллюстрирует реализацию ТИП = 263 K в РС 1.
Рис. 1.
Пересечение прямых в координатах уравнения Аррениуса при 103/Т = 3.77 K–1 (Т = 265 K) вблизи ИПТ ТИП = 263 K (103/ТИП = 3.80 K–1) в РС 1 (табл. 1), включающей реакции оксирана 1з с аренсульфоновыми кислотами 2а (1), 2б (2), 2в (3), 2г (4), 2е (5).
В РС 2 реализация формально доступной TИП = = 276 K оказалась невозможной вследствие твердого состояния используемого растворителя (диоксан, Тпл = 284.7 K).
В ИПТ $\sigma _{{\text{Y}}}^{{{\text{ИП}}}}$ скорость процесса не должна зависеть от температуры. Однако эти точки не были реализованы в РС 1–3 вследствие дефицита электронодонорных заместителей Y неаминного характера с константой σY, равной или меньшей –0.59.
В РС 4–7 (табл. 1) отсутствует взаимодействие эффектов температуры и структуры (qYT = 0, qXT = 0), вследствие чего в них не проявляется энтальпийно-энтропийный КЭ. Они являются изоэнтальпийными относительно вариации структуры оксиранового субстрата (заместители Х) и кислотного реагента (заместители Y). Так, например, в РС 4 энтальпия активации незначительно изменяется при переходе от одного заместителя Y к другому в ряду кислот 3б–д [28]: $\Delta H_{{\text{Y}}}^{ \ne }$, кДж/моль (Y): 75.5 (H), 77.5 (3-Br), 76.5 (3-NO2), 84.0 (3,5‑(NO2)2). Вместе с тем в этом же ряду кислот происходит значительное изменение энтропии активации $\Delta S_{{\text{Y}}}^{ \ne }$, Дж/(моль K): –114, –92.6, –84.7, ‒39.6. В РС 7 наблюдается аналогичное поведение активационных параметров: в ряду оксиранов 1а, г, е $\Delta H_{{\text{X}}}^{ \ne }$, кДж/моль (Х) = 84 (H), 71 (4-Cl), 68 (4-NO2); $\Delta S_{{\text{X}}}^{ \ne }$, Дж/(моль K) = –39, –87, –134 [29]. Таким образом, влияние структурных факторов на свободную энергию активации, а, следовательно, и на скорость процесса, осуществляется путем изменения в основном энтропии активации. В соответствии с принципом полилинейности в РС 4–7 выполняются линейные зависимости $\Delta S_{{\text{Y}}}^{ \ne }$ ($\Delta S_{{\text{X}}}^{ \ne }$) от σY (σX) и $\Delta G_{{{\text{Y}}T}}^{ \ne }$ ($\Delta G_{{{\text{Х}}Т}}^{ \ne }$) от $\Delta S_{{\text{Y}}}^{ \ne }$ ($\Delta S_{{\text{X}}}^{ \ne }$). Так, в РС 4 эти зависимости имеют следующий вид [28]: $\Delta S_{{\text{Y}}}^{ \ne }$ = (–115 ± 4) + (52 ± 2)σY (r = 0.991), $\Delta G_{{{\text{Y}}Т = 333}}^{ \ne }$ = (88 ± 2) × 103 + (210 ± 20)$\Delta S_{{\text{Y}}}^{ \ne }$ (r = 0.993).
В терминах активационных параметров взаимодействие эффектов структуры и температуры в реакциях оксирановых субстратов с кислотными реагентами (схемы 1–4 ) описывается полилинейными уравнениями
(14)
$\Delta G_{{{\text{Y}}T}}^{ \ne } = \Delta G_{{{\text{Н}}Т = 0}}^{ \ne } + Q_{{\text{Y}}}^{{Т = 0}}{{\sigma }_{{\text{Y}}}} + Q_{T}^{{{\text{Y}} = {\text{H}}}}T + {{Q}_{{{\text{Y}}Т}}}{{\sigma }_{{\text{Y}}}}T,$(15)
$\Delta G_{{{\text{Х}}T}}^{ \ne } = \Delta G_{{{\text{Н}}Т = 0}}^{ \ne } + Q_{{\text{Х}}}^{{Т = 0}}{{\sigma }_{{\text{Х}}}} + Q_{T}^{{{\text{Х}} = {\text{H}}}}T + {{Q}_{{{\text{Х}}Т}}}{{\sigma }_{{\text{Х}}}}T.$В табл. 2 приведены коэффициенты уравнения (14) для РС 1–3 с экспериментально реализованными ИПТ по температуре ТИП(G) = –$Q_{{\text{Y}}}^{{Т = 0}}$/QYТ и по константе заместителя Y $\sigma _{{\text{Y}}}^{{{\text{ИП(}}G{\text{)}}}}$ = –$Q_{T}^{{{\text{Y}} = {\text{H}}}}$/QYТ. Здесь также представлены уравнения (14), (15) без перекрестных членов (QYТ = 0, QХТ = 0) для РС 4–7. Значения ТИП(G) в РС 1, 3 совпадают с приведенными для этих серий в табл. 1. При ТИП(G) исчезает влияние заместителей Y на свободную энергию активации $\Delta G_{{{\text{Y}}T}}^{ \ne }$, а, следовательно, и на скорость процесса вследствие ранее обсужденного энтальпийно-энтропийного КЭ. Так, в РС 3 величина $\Delta G_{{{\text{Y}}T}}^{ \ne }$ становится практически неизменной при варьировании заместителя Y в кислотах 2а–г за счет полной компенсации в изменении энтальпийного и энтропийного терма при температуре 265 K, близкой к ТИП(G) 261 K [27]: $\Delta G_{{{\text{Y}}T = 265}}^{ \ne }$(Y) = 93.3 (4-OCH3), 92.9 (4-CH3), 93.0 (H), 93.1 (4‑Cl) кДж/моль. Графическая иллюстрация реализации ТИП(G) = 263 К в РС 1 представлена на рис. 2, где прямая 3 с нулевым наклоном показывает отсутствие влияния заместителей Y на свободную энергию активации $\Delta G_{{{\text{Y}}T}}^{ \ne }$ при температуре 265 K в эксперименте (ρY = 0).
Таблица 2.
Коэффициенты уравнений (14)а, (15)а и значения ИПТ по параметрам варьируемых факторов для реакций оксиранов 1, 4 с кислотами 2, 3, 5, 6 (ДО – диоксан, ДГ – диглим, АН – ацетонитрил, 1,2-ДХЭ – 1,2-дихлорэтан)
| РС | Оксиран | Кислота | Среда | $\Delta G_{{{\text{Н}}Т = 0}}^{ \ne }$ | $Q_{{\text{Y}}}^{{Т = {\text{ }}0}}$ | $Q_{T}^{{{\text{Y}} = {\text{H}}}}$ | QYТ | $\sigma _{{\text{Y}}}^{{{\text{ИП(}}G{\text{)}}}}$ | ТИП(G), К | Ссылки |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1з | 2а–г, е | ДО : ДГ, 1 : 1 | 60.7 ± 1 | 41 ± 2 | 0.084 ± 0.003 | –0.156 ± 0.008 | 0.54б | 263б | [24] |
| 2 | 4в | 2а–г, е | ДО : 1,2-ДХЭ, 7 : 3 | 67 ± 1 | 39 ± 3 | 0.083 ± 0.004 | –0.16 ± 0.01 | 0.52б | 244 | [25] |
| 3 | 4г | 2а–г | ДО : 1,2-ДХЭ, 7 : 3 | 83 ± 1 | 47 ± 5 | 0.036 ± 0.003 | –0.18 ± 0.02 | 0.20б | 261б | [27] |
| 4 | 1а | 3а–д | АН | 86 ± 5 | –11.4 ± 0.2 | 0.08 ± 0.01 | – | – | – | [28]в |
| 5 | 1а | 5а–в | ДО | 52 ± 0.3 | 12.1 ± 0.5 | 0.16 ± 0.01 | – | – | – | [30]в |
| 6 | 1а | 6а–д | АН | 89 + 5 | –7.8 + 0.3 | 0.08 + 0.01 | – | – | – | [31]в |
| $\Delta G_{{{\text{Н}}Т = 0}}^{ \ne }$ | $Q_{{\text{X}}}^{{Т = {\text{ }}0}}$ | $Q_{T}^{{{\text{Y}} = {\text{H}}}}$ | QXТ | |||||||
| 7г | 1а, г, е | 3д | АН | 69 ± 8 | 19.4 ± 0.5 | 0.09 ± 0.02 | – | – | – | [29]в |
Рис. 2.
Влияние температуры 303 K (1), 287 K (2), 265 K (3) на чувствительность свободной энергии активации $\Delta G_{{{\text{Y}}T}}^{ \ne }$ к эффектам заместителей Y в реакциях оксирана 1з с кислотами 2а–г, е (РС 1, табл. 2).

ИПТ $\sigma _{{\text{Y}}}^{{{\text{ИП(}}G{\text{)}}}}$ в РС 1–3 попадают в экспериментальный интервал варьирования σY-констант заместителей Y в кислотах 2а–г, е (σY = –0.27–0.71). В этих точках $\Delta G_{{{\text{Y}}T}}^{{ \ne {\text{ИП}}}}$ не зависит от температуры. Рисунок 2 демонстрирует реализацию в РС 1 $\sigma _{{\text{Y}}}^{{{\text{ИП(}}G{\text{)}}}}$ = 0.54, в которой исчезает влияние Т на величину $\Delta G_{{{\text{Y}}T}}^{{ \ne {\text{ИП}}}}$. Это возможно, если в выражении $\Delta G_{{{\text{Y}}T}}^{{ \ne {\text{ИП}}}}$ = $\Delta Н_{{\text{Y}}}^{ \ne }$ – Т$\Delta S_{{\text{Y}}}^{ \ne }$ энтропия активации $\Delta S_{{\text{Y}}}^{ \ne }$ = = 0 и свободная энергия активации равна энтальпии активации ($\Delta G_{{{\text{Y}}T}}^{{ \ne {\text{ИП}}}}$ = $\Delta Н_{{\text{Y}}}^{ \ne }$). Такую уникальную ситуацию подтверждает, например, уравнение $\Delta S_{{\text{Y}}}^{ \ne }$ = (–87 ± 1) + (187 ± 3)σY (r = 0.999) [25] для РС 2, из которого следует, что $\Delta S_{{\text{Y}}}^{ \ne }$ = 0 при значении σY = 0.46, которое соответствует величине $\sigma _{{\text{Y}}}^{{{\text{ИП(}}G{\text{)}}}}$ = 0.52.
В РС 1–3 не только продемонстрирован редкий в химических процессах случай реализации ИПТ по структурному параметру $\sigma _{{\text{Y}}}^{{{\text{ИП(}}G{\text{)}}}}$, но и осуществлены переходы через эти точки при варьировании заместителей Y в кислоте 2. Пример перехода через $\sigma _{{\text{Y}}}^{{{\text{ИП(}}G{\text{)}}}}$ = 0.54 в РС 1 показан на рис. 2. В соответствии с закономерностями изопараметрических зависимостей при таких переходах происходит инверсия знака энтропии активации $\Delta S_{{\text{Y}}}^{ \ne }$. В РС 2 этот феномен наблюдается при переходе через $\sigma _{{\text{Y}}}^{{{\text{ИП(}}G{\text{)}}}}$ = 0.52 в ряду кислот 2а–г, е: $\Delta S_{{\text{Y}}}^{ \ne }$, Дж/(моль K) (Y, σY) = –140 (4-OMe, –0.27), –119 (4-Me, –0.17), –85 (H, 0), –42 (4-Cl, 0.23), 44 (3‑NO2, 0.71). Инверсия знака энтропии активации вызывает обращение влияния температуры на величину $\Delta G_{{{\text{Y}}T}}^{ \ne }$. Это явление демонстрирует рис. 3, где показано обращение наклонов температурных зависимостей свободной энергии активации после перехода через $\sigma _{{\text{Y}}}^{{{\text{ИП(}}G{\text{)}}}}$ = 0.54 в РС 1. Точка пересечения корреляционных прямых на этом рисунке, где отсутствует влияние заместителей Y на величину $\Delta G_{{{\text{Y}}T}}^{ \ne }$, соответствует ранее упомянутой ИПТ по температуре ТИП(G) = 263 K.
Рис. 3.
Обращение влияния температуры на свободную энергию активации $\Delta G_{{{\text{Y}}T}}^{ \ne }$ после перехода через ИПТ $\sigma _{{\text{Y}}}^{{{\text{ИП(}}G{\text{)}}}}$ = 0.54 в РС 1 (табл. 2), включающей реакции оксирана 1з с кислотами 2a, Y = 4-OCH3, σY = = –0,27 (1), 2в, Y = H, σY = 0 (2), 2г, Y = 4-Cl, σY = = 0.23 (3), 2е, Y = 3-NO2, σY = 0.71 (4).

В РС 4–7 отсутствует взаимодействие эффектов структуры и температуры (QYТ = 0, QХТ = 0). Как уже отмечалось при обсуждении этих серий в табл. 1, они являются изоэнтальпийными относительно эффектов структурных факторов.
Яркое проявление феномена изопараметричности наблюдается в катализируемых пиридинами 7а–д реакциях оксирана 1а с аренкарбоновыми кислотами 3б–г (схема 5 ) [32, 34]. Для описания совместного влияния на скорость этих реакций температуры и заместителей Z в катализаторе Z-Py использовано уравнение
(16)
$\begin{gathered} \lg {{k}_{{{\text{Z}}Т}}} = \lg {{k}_{{{\text{H}}T = \infty }}} + \rho _{{\text{Z}}}^{{Т = \infty }}{{\sigma }_{{\text{Z}}}} + \\ + \;q_{T}^{{{\text{Z}} = {\text{H}}}} \times {{10}^{3}}{\text{/}}T + {{q}_{{{\text{Z}}T}}}{{\sigma }_{{\text{Z}}}} \times {{10}^{3}}{\text{/}}T. \\ \end{gathered} $Коэффициенты уравнения (16) и рассчитанные на их основе значения ИПТ $\sigma _{{\text{Z}}}^{{{\text{ИП}}}}$ = –$q_{T}^{{{\text{Z}} = {\text{H}}}}$/qZT и ТИП = –qZT × 103/$\rho _{{\text{Z}}}^{{Т = \infty }}$ приведены в табл. 3 для РС 1–3. Здесь также представлены коэффициенты уравнений (9) и (16), в которых отсутствуют перекрестные члены (qYT = 0, qZТ = 0), для РС 4, 5.
Таблица 3.
Коэффициенты уравнений (9)а, (16)а и значения ИПТ по параметрам варьируемых факторов для катализируемых пиридинами 7а–д реакций оксирана 1а с кислотами 3б–г и 6а, б, д в ацетонитриле
| РС | Кислота | Py | $\lg {{k}_{{{\text{H}}T = \infty }}}$ | $\rho _{{\text{Z}}}^{{Т = \infty }}$ | $q_{T}^{{{\text{Z = H}}}}$ | qZT | $\sigma _{{\text{Z}}}^{{{\text{ИП}}}}$ | ТИП, K | Ссылки |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3б | 7а–г | 5.0 ± 0.1 | –15.6 ± 0.8 | –2.81 ± 0.04 | 4.6 ± 0.2 | 0.61 | 295б | [32]в |
| 2 | 3в | 7а–д | 5.6 ± 0.1 | –15.5 ± 0.5 | –2.86 ± 0.04 | 4.5 ± 0.2 | 0.62б | 295б | [32]в |
| 3 | 3г | 7а–д | 5.6 ± 0.2 | –16.9 ± 0.8 | –2.80 ± 0.06 | 5.0 ± 0.2 | 0.56б | 296б | [34] |
| 4 | 6б | 7б–д | 7.8 ± 0.1 | –0.81 ± 0.02 | –3.83 ± 0.04 | – | – | – | [33]в |
| $\lg {{k}_{{{\text{H}}T = \infty }}}$ | $\rho _{{\text{Y}}}^{{Т = \infty }}$ | $q_{T}^{{{\text{Y}} = {\text{H}}}}$ | qYT | ||||||
| 5 | 6а, б, д | 7в | 7.7 ± 0.3 | 1.21 ± 0.03 | –3.79 ± 0.09 | – | – | – | [33]в |
ИПТ по температуре ТИП в РС 1–3 соответствуют наклонам компенсационных зависимостей (17)–(19) в изменении энтальпии $\Delta H_{{\text{Z}}}^{ \ne }$ и энтропии $\Delta S_{{\text{Z}}}^{ \ne }$ активации под влиянием заместителей Z в катализаторе 7 в ряду кислот 3б, в, г (r = = 0.999) [32].
(17)
$\Delta H_{{\text{Z}}}^{ \ne } = (97.8 \pm 0.6) \times {{10}^{3}} + (296 \pm 3)\Delta S_{{\text{Z}}}^{ \ne },$(18)
$\Delta H_{{\text{Z}}}^{ \ne } = (95.6 \pm 0.3) \times {{10}^{3}} + (294 \pm 2)\Delta S_{{\text{Z}}}^{ \ne },$(19)
$\Delta H_{{\text{Z}}}^{ \ne } = (93.4 \pm 0.3) \times {{10}^{3}} + (293 \pm 1)\Delta S_{{\text{Z}}}^{ \ne }.$В этих РС TИП не выходят за пределы температурного интервала 279–343 K в эксперименте, что свидетельствует об их физической реальности. Данные табл. 4 показывают, что согласно концепции энтальпийно-энтропийной компенсации при температуре эксперимента 295 K, соответствующей TИП, параметр чувствительности $\rho _{{\text{Z}}}^{T}$ к эффектам заместителей Z в пиридинах 7а–д в реакциях оксирана 1а с кислотами 3б–г приближается к нулевому значению, т.е. реакционная система становится изокинетической. Противоположные знаки $\rho _{{\text{Z}}}^{T}$ после перехода через ТИП указывают на обращение порядка влияния заместителей Z на каталитическую активность пиридинов, что являются убедительным свидетельством парадокса изопараметричности, экспериментально наблюдаемого при изменении температуры от 279 до 343 K. Графическая иллюстрация этого парадокса в РС 3 представлена на рис. 4, где в координатах уравнения Аррениуса показано, что по разные стороны от ТИП = 296 K (103/ТИП = 3.38 K–1) порядок влияния заместителей Z на каталитическую активность пиридинов является противоположным.
Таблица 4.
Значения $\rho _{{\text{Z}}}^{T}$ (r ≥ 0.995) в уравнении Гаммета для катализируемых пиридинами 7а–д реакций оксирана 1а с кислотами 3б–г (схема 5 ) в ацетонитриле при разных температурах [32]
| Кислота (Y) | $\rho _{{\text{Z}}}^{T}$ | ||||
|---|---|---|---|---|---|
| 279 K | 295 K | 308 K | 323 K | 343 K | |
| 3б(H) | 0.751 ± 0.005 | –0.078 ± 0.002 | –0.77 ± 0.06 | –1.62 ± 0.06 | –2.4 ± 0.2 |
| 3в (3-Br) | 0.81 ± 0.08 | –0.077 ± 0.009 | –0.77 ± 0.04 | –1.74 ± 0.04 | –2.4 ± 0.1 |
| 3г (3-NO2) | 0.761 ± 0.009 | –0.083 ± 0.003 | –0.75 ± 0.04 | –1.30 ± 0.07 | –2.4 ± 0.1 |
Рис. 4.
Обращение влияния температуры на каталитическую активность пиридинов 7а (1), 7б (2), 7в (3), 7д (4) после перехода через ИПТ 103/ТИП = 3.38 K–1 (ТИП = 296 K, $\rho _{{\text{Z}}}^{T}$ = 0) в РС 3 (табл. 3).

Что касается ИПТ $\sigma _{{\text{Z}}}^{{{\text{ИП}}}}$, то они фактически реализованы в РС 2 и 3 в случае пиридина 7д (Z = = 3‑CN, σZ = 0.56). В этих точках на скорость каталитического процесса не влияет температурный фактор ($q_{T}^{{\text{Z}}}$ = 0). Экспериментальное достижение $\sigma _{{\text{Z}}}^{{{\text{ИП}}}}$ = 0.56 в РС 3 показано на рис. 4, где корреляционная прямая 4 с нулевым наклоном демонстрирует отсутствие влияния температуры на каталитическую активность пиридина 7д. Р-исунок 5 демонстрирует реализацию $\sigma _{{\text{Z}}}^{{{\text{ИП}}}}$ = 0.56 в координатах уравнения Гаммета. Отсутствие влияния температуры в этой точке на скорость каталитического процесса возможно, если в уравнении (6) угловой коэффициент $B_{T}^{j}$ = = ‒$\Delta H_{j}^{ \ne }$/2.3R = 0 ($B_{T}^{{\text{Z}}}$ = –$\Delta H_{{\text{Z}}}^{ \ne }$/2.3R = 0) вследствие равенства нулю энтальпии активации $\Delta H_{j}^{ \ne }$ = 0 ($\Delta H_{{\text{Z}}}^{ \ne }$ = 0). Действительно, в РС 2, 3 величина энтальпии активации близка к нулевому значению ($\Delta H_{{\text{Z}}}^{ \ne }$ = 3.3 кДж/моль, Z = 3-CN) вблизи $\sigma _{{\text{Z}}}^{{{\text{ИП}}}}$ 0.62 и 0.56. При этом энтропия активации достигает больших отрицательных значений ($\Delta S_{{\text{Z}}}^{ \ne }$ = = –314 и –308 Дж/(моль K)) [34], а вклад энтропийного терма Т$\Delta S_{{\text{Z}}}^{ \ne }$ в величину свободной энергии активации при 298 K превышает 96%. Эти данные указывают на то, что при достижении рассматриваемых ИПТ каталитический процесс осуществляется фактически без активационного барьера.
Рис. 5.
Пересечение корреляционных прямых в координатах уравнения Гаммета в ИПТ $\sigma _{{\text{Z}}}^{{{\text{ИП}}}}$ = 0.56 ($q_{T}^{{\text{Z}}}$ = = 0) в реакции оксирана 1а с кислотой 3в, катализируемой пиридинами Z-Py 7а–д при температурах 343 (1), 323 (2), 308 (3), 295 (4), 279 (5) К (РС 3, табл. 3).

В РС 4, 5 (табл. 3) отсутствует взаимодействие эффектов структуры и температуры (qZT = 0, qYT = = 0). Они является изоэнтальпийными относительно заместителей Z в катализаторе 7 и Y в кислотном реагенте 6. Так, например, в этих сериях при закрепленном заместителе Y = H (Z = H) энтальпия активации $\Delta H_{{\text{Z}}}^{ \ne }$ ($\Delta H_{{\text{Y}}}^{ \ne }$) практически не изменяется при переходе от одного заместителя Z (Y) к другому: $\Delta H_{{\text{Z}}}^{ \ne }$, кДж/моль = 72, 71, 73, 73 в ряду пиридинов 7б, в, г, д ($\Delta H_{{\text{Y}}}^{ \ne }$, кДж/моль = 71, 71, 71 в ряду кислот 6а, б, д) [33]. Влияние структурных факторов на свободную энергию активации каталитического процесса происходит в основном за счет изменения энтропийного терма, например, $\Delta G_{{{\text{Z}}T = 308}}^{ \ne }$ (Y = H) = (44 ± 6) + (–0.58 ± ± 0.06)ΔSZ≠ (r = 0.998); $\Delta G_{{{\text{Y}}T = 323}}^{ \ne }$ (Z = H) = (66 ± 2) + + (–0.37 ± 0.02)$\Delta S_{{\text{Y}}}^{ \ne }$ (r = 0.998) (рассчитано по данным работы [33]).
Для оценки совместного влияния температуры и структуры кислотного реагента 3, 5 (заместители Y), а также структуры катализатора 7 (заместители Z) на свободную энергию активации каталитических реакций (схемы 5, 6 ) использовались уравнения (14) и (20).
(20)
$\Delta G_{{{\text{Z}}T}}^{ \ne } = \Delta G_{{{\text{Н}}Т = 0}}^{ \ne } + Q_{{\text{Z}}}^{{Т = 0}}{{\sigma }_{{\text{Z}}}} + Q_{T}^{{Z = H}}T + {{Q}_{{{\text{Z}}Т}}}{{\sigma }_{{\text{Z}}}}T.$В табл. 5 приведены коэффициенты уравнения (20) для РС 1–3 с экспериментально реализованными ИПТ по параметрам варьируемых факторов. Здесь также представлены уравнения (14), (20) без перекрестных членов (QZТ = 0, QYТ = 0), а также значения ИПТ по температуре TИП(G) = = ‒$Q_{{\text{Z}}}^{{Т = 0}}$/QZТ и по константе заместителя Z $\sigma _{{\text{Z}}}^{{{\text{ИП(}}G{\text{)}}}}$ = = –$Q_{T}^{{{\text{Z}} = {\text{H}}}}$/QZТ. Значения ТИП(G) в указанных сериях соответствуют приведенным в табл. 3. В этих точках исчезает влияние заместителей Z на свободную энергию активации $\Delta G_{{{\text{Z}}T}}^{ \ne }$ вследствие энтальпийно-энтропийной компенсации. Так, в РС 3 при температуре 295 K, близкой к изокинетической 296 K, величина $\Delta G_{{{\text{Z}}T}}^{ \ne }$ становится практически неизменной при варьировании заместителя Z в пиридинах 7а, в, г, д: $\Delta G_{{{\text{Z}}T = 295}}^{ \ne }$(Z) = 93.7 (4-OCH3), 94.1 (H), 93.8 (3-COOEt), 94.2 (3-CN) кДж/моль (рассчитано по данным работы [34]). На рис. 6 показан один из примеров перехода через ТИП(G) 296 K в РС 3, сопровождающийся обращением каталитической активности указанных пиридинов.
Таблица 5.
Коэффициенты уравнений (14)а, (20)а и значения ИПТ по параметрам варьируемых факторов для катализируемых пиридинами 7а–д реакций оксирана 1а с кислотами 3б–г и 6а, б, д в ацетонитриле
| РС | Кислота | Py | $\Delta G_{{{\text{Н}}Т = 0}}^{ \ne }$ | $Q_{{\text{Z}}}^{{Т = 0}}$ | $Q_{T}^{{{\text{Z}} = {\text{H}}}}$ | QZТ | $\sigma _{{\text{Z}}}^{{{\text{ИП(}}G{\text{)}}}}$ | ТИП(G), K | Ссылки |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3б | 7а–г | 53 ± 1 | –81 ± 6 | 0.150 ± 0.005 | 0.27 ± 0.02 | –0.56 | 300б | [32]в |
| 2 | 3в | 7а–д | 52 ± 2 | –80 ± 4 | 0.148 ± 0.005 | 0.27 ± 0.01 | –0.55 | 296б | [32]в |
| 3 | 3г | 7а–д | 53 ± 1 | –86 ± 3 | 0.138 ± 0.003 | 0.29 ± 0.01 | –0.48 | 296б | [34] |
| 4 | 6б | 7б–д | 79 ± 3 | 6.6 ± 0.6 | 0.08 ± 0.01 | – | – | – | [33]в |
| $\Delta G_{{{\text{Н}}Т = 0}}^{ \ne }$ | $Q_{{\text{Y}}}^{{Т = 0}}$ | $Q_{T}^{{{\text{Y}} = {\text{H}}}}$ | QYТ | ||||||
| 5 | 6а, б, д | 7в | 78 ± 2 | –7.8 ± 0.2 | 0.078 ± 0.007 | – | – | – | [33]в |
Рис. 6.
Переход через ИПТ ТИП(G) = 296 K ($\rho _{{\text{Z}}}^{T}$ = 0) в реакции оксирана 1а с кислотой 3в, катализируемой пиридинами 7а (1), 7в (2), 7г (3), 7д (4) (РС 3, табл. 5).

В ИПТ $\sigma _{{\text{Z}}}^{{{\text{ИП(}}G{\text{)}}}}$ величина $\Delta G_{{{\text{Z}}T}}^{{ \ne {\text{ИП}}}}$ не должна зависеть от температуры вследствие равенства нулю энтропии активации (при $\Delta S_{{\text{Z}}}^{ \ne }$ = 0 $\Delta G_{{{\text{Z}}T}}^{{ \ne {\text{ИП}}}}$ = $\Delta H_{{\text{Z}}}^{ \ne }$). Однако, эти ИПТ не были реализованы в эксперименте.
Что касается РС 4, 5 в табл. 5, то в них отсутствует взаимодействие эффектов структуры и температуры. Они являются изоэнтальпийными относительно вариации заместителей Z в катализаторе 7 (δZΔH≠ = 0) и в кислотном реагенте 6 (δYΔH≠ = 0). В РС 4 в ряду пиридинов 7б–д значения $\Delta H_{{\text{Z}}}^{ \ne }$ и $\Delta S_{{\text{Z}}}^{ \ne }$ равны соответственно 72, 71, 73, 73 кДж/моль и –97, –100, –104, –106 Дж/(моль K), а в РС 5 в ряду кислот 6а, б, д $\Delta H_{{\text{Y}}}^{ \ne }$ = 71, 71, 71 кДж/моль; $\Delta S_{{\text{Y}}}^{ \ne }$ = –104, –100, –83 Дж/(моль K).
ЗАКЛЮЧЕНИЕ
В рассмотренных реакциях раскрытия оксиранового цикла доказана физическая реальность абстрактных свойств формальных полилинейных соотношений типа (2), из которых наиболее важным является энтальпийно-энтропийная компенсация, проявляющаяся в изопараметрических РС вследствие взаимодействия эффектов температуры и структуры в активационном процессе. Количественным атрибутом этих серий являются ИПТ по параметрам варьируемых факторов (структура, температура), в которых реальная реагирующая система приобретает особые “магические” свойства. В этих точках исчезает влияние соответствующих факторов на кинетические и активационные характеристики химического процесса, а при переходе через ИПТ наблюдается парадокс изопараметричности – обращение знаков соответствующих коэффициентов чувствительности, а также знака такого активационного параметра, как энтропия активации. Последнее происходит в РС 1–3 (табл. 2) при переходе через ИПТ $\sigma _{{\text{Y}}}^{{{\text{ИП(}}G{\text{)}}}}$: $\Delta S_{{\text{Y}}}^{ \ne }$ < 0, $\Delta S_{{\text{Y}}}^{ \ne }$ = 0, $\Delta S_{{\text{Y}}}^{ \ne }$ > 0. Что касается энтальпии активации, то ее величина приближается к нулю ($\Delta H_{{\text{Z}}}^{ \ne }$ = 3.3 кДж/моль) в ИПТ $\sigma _{{\text{Z}}}^{{{\text{ИП}}}}$ в РС 2, 3 (табл. 3). В этом случае химический процесс осуществляется фактически при близком к нулю значении энергии активации. Дальнейшие исследования следует направить на поиск реакций с трудно воспринимаемой отрицательной энтальпией (энергией) активации.
Знание изопараметрических свойств РС с энтальпийно-энтропийным КЭ расширяет наши представления о малоизученных количественных аспектах органических реакций. В этом контексте следует ожидать новых интересных открытий при изучении совместного влияния структуры, температуры, среды, катализатора и других факторов на кинетические, активационные, термодинамические и другие характеристики химических процессов.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Wells P.R. // Chem. Rev. 1963. V. 63. № 3. P. 171.https://doi.org/10.1021/cr60222a005
Chapman N., Shorter J. Eds. Advances in Linear Free Energy Relationships. New York: Plenum Press, 1972. 481 p. https://doi.org/10.1007/978-1-4615-8660-9
Джонсон К. Уравнение Гаммета. М.: Мир, 1977. 240 с.
Пальм В.А Основы количественной теории органических реакций. Л.: Химия, 1977. 360 с.
Chapman N.B., Shorter J. Eds. Correlation Analysis: Recent Advances. New York: Plenum Press, 1978. 541 p. https://doi.org/10.1007/978-1-4615-8831-3
Shorter J. Correlation Analysis of Organic Reactivity, with Particular Reference to Multiple Regression. Somerset, NJ: John Wiley and Sons Inc., 1982. 235 p.
Рейнхарт К. Растворители и эффекты среды в органической химии. М.: Мир, 1991. 763 с.
Williams A. Free Energy Relationships in Organic and Bioorganic Chemistry. Cambridge: RSC, 2003. 298 p.https://doi.org/10.1039/9781847550927
Пальм В.А., Истомин Б.И. // Реакц. спос. орг. соед. 1969. Т. 6. № 2. С. 427.
Shpanko I.V., Kim S.I., Koh H.J., Lee I. // Bull. Korean Chem. Soc. 1995. V. 16. № 6. P. 533.
Шпанько И.В. // ТЭХ. 1999. Т. 35. № 2. С. 67.
Шпанько И.В. // Там же. 2001. Т. 37. № 5. С. 265.
Leffler J.E., Grunwald E. Rates and Equilibrium of Organic Reactions. New York: John Wiley and Sons Inc., 1963. 458 p.
Exner O. // Prog. Phys. Org. Chem. 1973. V. 10. P. 411.
Liu L., Guo Q.-X. // Chem. Rev. 2001. V. 101. № 3. P. 673.https://doi.org/10.1021/cr990416z
Sharp K. // Protein Sci. 2001. V. 10. № 3. P. 661.https://doi.org/10.1110/ps.37801
Norwisz J., Musielak T.J. // Therm. Anal. Calorim. 2007. V. 88. P. 751. https://doi.org/10.1007/s10973-006-8139-4
Barrie P.J. // Phys. Chem. Chem. Phys. 2012. V. 14. № 1. P. 327. https://doi.org/10.1039/ c1cp22667c
Cornish-Bowden A. // J. Biosci. 2017. V. 42. № 4. P. 665. https://doi.org/10.1007/s12038-017-9719-0
Mianowski A., Radko T., Siudyga T. // Reac. Kinet. Mech. Cat. 2021. V. 132. P. 37. https://doi.org/10.1007/s11144-020-01898-2
Sapunov V.N., Saveljev E.A., Voronov M.S., Valtiner M., Linert W. // Thermo. 2021. V. 1. № 1. P. 45. https://doi.org/10.3390/thermo1010004
Дворко Г.Ф., Пономарев Н.Е., Пономарева Э.А. // ЖОХ. 2010. Т. 80. № 1. С. 5.https://doi.org/10.1134/S1070363210010019
Шпанько И.В., Садовая И.В., Китайгородский А.М. // ТЭХ. 2000. Т. 36. № 6. С. 367. https://doi.org/10.1023/A:1005272628953
Шпанько И.В., Садовая И.В. // Там же. 2010. Т. 46. № 3. С. 171. https://doi.org/10.1007/s11237-010-9136-z
Шпанько И.В., Садовая И.В. // Журн. физ. химии. 2016. Т. 90. № 12. С. 1771.https://doi.org/10.1134/S0036024416120268
Шпанько И.В., Садовая И.В. // ЖОХ. 2017. Т. 87. № 11. С. 1810. https://doi.org/10.1134/S107036321711007X
Shpan’ko, I.V., Sadovaya, I.V. // Reac. Kinet. Mech. Cat. 2018. V. 123. P. 473.https://doi.org/10.1007/s11144-017-1340-6
Шпанько И.В., Садовая И.В., Китайгородский А.М. // Укр. хим. журн. 2003. Т. 69. № 6. С. 111.
Шпанько И.В., Садовая И.В. // ЖОрХ. 2005. Т. 41. № 7. С. 1011. https://doi.org/10.1007/s11178-005-0282-z
Шпанько И.В., Садовая И.В. // Укр. хим. журн. 2004. Т. 70. № 4. С. 104.
Шпанько И.В., Садовая И.В. // Там же. 2015. Т. 81. № 10. С. 124.
Шпанько И.В., Садавая И.В. // Кинетика и катализ. 2014. Т. 55. № 1. С. 59.https://doi.org/10.1134/S002315841401011X
Шпанько И.В., Садовая И.В. // ЖОХ. 2019. Т. 89. № 12. С. 1835. https://doi.org/10.1134/ S0044460X19120059
Шпанько И.В., Садовая И.В. // Журн. физ. химии. 2013. Т. 87. № 12. С. 1994. https://doi.org/10.1134/S0036024413120224
Шпанько И.В., Садовая И.В., Куликова Н.В. // ЖОрХ. 2011. Т. 47. № 5. С. 685. https://doi.org/10.1134/S107042801105006X
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии