

Журнал физической химии, 2022, T. 96, № 3, стр. 313-323
Механизм фотоизомеризации цис-азобензола. роль ридберговской 3s-орбитали азогруппы
a Институт биохимической физики им. Н.М. Эмануэля РАН
Москва, Россия
* E-mail: mik@sky.chph.ras.ru
Поступила в редакцию 17.09.2021
После доработки 17.09.2021
Принята к публикации 20.09.2021
- EDN: JJNNCC
- DOI: 10.31857/S0044453722030177
Аннотация
Проведен анализ данных по фемтосекундной фотоизомеризации цис-азобензола (c-AB) при действии лазерного Vis-излучения с длиной волны λex = 435 нм. Учтено, что ридберговская 3s-орбиталь (R3s) азогруппы c-AB индуцирует обратимую электронно- (e-) волновую таутомеризацию между e-нейтральной формой c-AB (E,$R_{{{\text{3s}}}}^{0}$) и e-поляризованной формой E+,$R_{{{\text{3s}}}}^{{1--}}$, подобно e-таутомеризации между нейтральной формой Z,$R_{{{\text{3s}}}}^{0}$ и поляризованной формой Z+,$R_{{{\text{3s}}}}^{{1--}}$ транс (t-) AB. Оранжевый цвет c-AB объяснен тем, что его e-таутомеры E+,$R_{{{\text{3s}}}}^{{1--}}$ обладают хромогенными катионами фениламинильного типа (PhAT), как Z+,$R_{{{\text{3s}}}}^{{1--}}$ в t-AB; более сильная окраска c-AB относительно t-AB отражает более высокую равновесную долю e-таутомеров E+,$R_{{{\text{3s}}}}^{{1--}}$ в с-AB относительно доли Z+,$R_{{{\text{3s}}}}^{{1--}}$ в t-AB. Представлен механизм фотоизомеризации c-AB, согласно которому, возбужденные импульсами с λex = 435 нм e-таутомеры E+*,$R_{{{\text{3s}}}}^{{1--}}$ превращаются в состояние E++*,$R_{{{\text{3s}}}}^{{2--}}$ с двумя катионами PhAT. Показано, что действующее в E++*,$R_{{{\text{3s}}}}^{{2--}}$ между катионами PhAT кулоновское отталкивание раздвигает их с константой τ = 47 фс в промежуточное планарное цис-состояние ($E_{{{\text{ims}}}}^{{ + + *}}$, $R_{{{\text{3s}}}}^{{2--}}$), и при этом запасенная в c-AB энергия суммируется с энергией фотовозбуждения; затем состояние $E_{{{\text{ims}}}}^{{ + + *}}$,$R_{{{\text{3s}}}}^{{2--}}$ изомеризуется с константой τ = 170 фс в планарный транс-интермедиат (Z++*,$R_{{{\text{3s}}}}^{{2--}}$), релаксирующий преимущественно в t-AB. Даны схемы каналов релаксации и объяснены причины изменения квантовых выходов c ↔ t-изомеризации AB при замене растворителя – н-гептана – на водно-спиртовую смесь.
Цель настоящей работы – корректное описание механизма с → t-фотоизомеризации цис-азобензола (c-AB) на основе накопленных в литературе экспериментальных данных по структурным и спектроскопическим свойствам c-AB. Требование реновации механизма фотоизомеризации c-AB обусловлено необходимостью учета реальных свойств участников изомеризации c-AB ↔ t-AB, а также необходимостью отказа от трактовки природы цветности изомеров азобензола по Википедии [1].
Согласно [1], в транс-изомере AB, в отличие от цис-изомера, существует единая система из семи сопряженных π-связей. “цис-Изомер AB бесцветен и менее устойчив, а транс-изомер окрашен (имеет полосу поглощения в синей части спектра). Облучение ультрафиолетом приводит к образованию цис-изомера и обесцвечиванию вещества; облучение синим светом или нагревание приводят к получению транс-изомера и появлению окраски”. Данная цитата из [1] искажает реальные свойства изомеров AB. Во-первых, она фиксирует неверное представление о семи сопряженных π-связях в t-AB (об этом см. ниже, это представление использовали в ранних работах по описанию спектроскопии и t ↔ c-фотоизомеризации в рамках метода молекулярных орбиталей Хюккеля [2, 3], оно сохранилось и в работах с более продвинутыми квантово-механическими моделями [4–10]). Во-вторых, утверждение о бесцветности c-AB [1] несостоятельно. Об этом свидетельствует давно установленный факт более сильной окраски c-AB [2, 3]. В работах [2, 3] Vis-полосы изомеров AB, ответственные за их оранжевый цвет, были отнесены к S0 → S1-переходам электронов с sp2-несвязывающих (n) орбиталей атомов азота на нижние незанятые и делокализованные по молекулам π*-орбитали изомеров AB (n → π*-переход). Это отнесение Vis-полос t-AB и c-AВ к n → π*-переходам [2, 3] стало традиционным в последующих исследованиях [4–10]. В [2, 3] было четко установлено, что в спиртовой среде у Vis-полосы c-AB (ее максимум при λm = 433 нм) коэффициент экстинкции εm = 1518 л/(моль см) в ∼3 раза больше, чем у Vis-полосы t-AB (εm = = 510 л/(моль см), λm ≈ 443.0 нм), и что первая слабо смещена в коротковолновую сторону (на ≈540 см –1) относительно второй. Согласно авторам [2], усиление интенсивности Vis-полосы c-AB может быть связано с характерным для c-AB отталкиванием электронных оболочек фенильных колец и, как следствие, со смещением их электронной плотности на связь N=N, ведущим к росту интенсивности n → π*-полосы. Ниже будет показано, что азогруппы изомеров AB, наоборот, имеют электронный дефицит.
Недавно установлено, что Vis-полосы t-AB принадлежат хромогенным центрам со строением катионов фениламинильного типа (PhAT) [11–19], а местоположение реальных полос n → → π*-перехода находится в UV-области 300–320 нм [11, 12]. Было также показано, что катионы PhAT являются ключевыми хромогенами аминоазобензольных красителей [13]. Обобщение результатов исследований структурно-спектроскопических свойств аминоазобензольных красителей в работе [13] стало началом этапа реновации научных представлений о природе цветности и фотохимии соединений азобензола [14–18]. Необходимость этой реновации обусловлена тем, что азогруппа указанных соединений служит локальным хромофором, а ее ридберговская 3s-орбиталь (R3s) обеспечивает генерацию и характер функционирования катионов PhAT, особенности спектроскопических и физико-химических свойств t-AB (и его замещенных производных), в том числе возможность образования таких супермолекул, как ридимеры [12–16, 18]. Учет роли R3s-орбитали в качестве генератора и диспетчера e-конфигураций катионов PhAT позволил раскрыть адекватный механизм t → c-фотоизомеризации t-AB в недавно опубликованной работе [20]. Теперь роль R3s-орбитали демонстрируется с использованием литературных экспериментальных данных, необходимых и достаточных для качественно нового понимания механизма фотоизомеризации c-AB → t-AB.
e-ТАУТОМЕРЫ И UV-, Vis-СПЕКТРЫ АЗОБЕНЗОЛЬНЫХ ИЗОМЕРОВ
Согласно работам [11, 13, 17, 19, 20], перекрывание sp2- и R3s-орбиталей в азогруппах в t-AB и c-AB обеспечивает обратимое промотирование одного из sp2-электронов на орбиталь R3s по схеме 1.

Здесь и далее используются символы Z и E, приближенно отражающие форму молекул t-AB и с-AB. Сочетания Z,$R_{{{\text{3s}}}}^{0}$ и E,$R_{{{\text{3s}}}}^{0}$ обозначают преобладающие по содержанию неполяризованные молекулы t-AB и c-AB с вакантными орбиталями $R_{{{\text{3s}}}}^{0}$, а Z+,$R_{{{\text{3s}}}}^{{1--}}$ и E+,$R_{{{\text{3s}}}}^{{1--}}$ – e-таутомеры, возникающие вследствие обратимого переноса электрона с несвязывающей sp2-орбитали одного из азотов азогруппы на орбиталь R3S азогруппы N=N (в кратком виде это соответствует уравнению (:sp2) + + $R_{{{\text{3s}}}}^{0}$ ↔ ($ \bullet {\kern 1pt} s{{p}^{2}}$)+ + $R_{{{\text{3s}}}}^{{1--}}$ ). По схеме 1, :sp2-орбиталь в структурах Z и E, отдавая электрон на $R_{{{\text{3s}}}}^{0}$ и получая положительный заряд ($ \bullet {\kern 1pt} s{{p}^{2}}$)+, поляризует π-связь азогруппы, локализуя оба π-электрона на pz-орбитали своего атома N. При этом другие половинки молекул t-AB, c-AB становятся катионами PhAT (ph+${\text{N}}_{{ \bullet \bullet }}^{ + }$), в которых заряды делокализуются в орто- и пара-положениях колец [11, 17, 19, 20]. Катионы PhAT имеют не только связывающие и разрыхляющие молекулярные π-орбитали (МО), но и вакантные несвязывающие МО (НСМО) с нулевой энергией. В них переход электронов с высших занятых π-орбиталей (ВЗМО) происходит на НСМО под действием света в VIS-полосе (π → π*-переход) при ≈440 нм. В результате этого изомеры AB получают оранжевый цвет, причем именно фотовозбуждение катиона ph+N+ в Z+ (схема 1) объясняет тот установленный для t-AB факт [21], что переходный момент Vis-полосы расположен в плоскости молекулы примерно вдоль ее длинной оси, а не перпендикулярно, как должно быть при n → π*-переходе.
Орбиталь R3s, акцептируя sp2-электрон того или иного атома азота, генерирует в катионах Z+ и E+ обратимый e-обмен с чередованием катионов PhAT на противоположных половинках молекул (например, в t-AB:
Характерные свойства t-AB
До сих пор оранжевую окраску молекул t-AB приписывают либо n → π*-переходу в структуре Z (схема 1), либо π-сопряжению фенильных колец через азогруппу [1]. Такое сопряжение семи π-связей отмечали авторы [24], установившие факт плоского строения молекулы t-AB методом газофазной электронной дифракции. Но в [21] показано, что в структуре Z группа N=N является локальным хромофором, не имеющим π-сопряжения с фенильными кольцами. (Подобные несопряженные (“фиксированные“) локальные π-связи достаточно распространены в полициклических ароматических углеводородах [25]).
В [21] ph-группы t-AB рассматриваются как квантово-волновые резонаторы с вырожденными π*-уровнями. Возбуждение такого единичного резонатора происходит в UV-полосе при 40 000 см–1 (250 нм). Однако их бинарная связь в t-AB снимает вырождение, расщепляя их π*-уровни (±8500 см–1) и обеспечивая энергетический резонанс с делокализацией энергии фотовозбуждения [21]. Иначе говоря, в t-AB есть возможность для π → π*-перехода электрона с ВЗМО какого-либо из бинарно связанных ph-резонаторов на нижний из расщепленных π*-уровней. Данной ситуации соответствует появление отсутствующей у алкилбензолов, но характеристической для Z,$R_{{{\text{3s}}}}^{0}$ (схема 1) UV-полосы S0 → S1 с νm = 31500 см–1, λm ≈ 320 нм. Сходные бинарные ph-резонаторы, ответственные за UV-полосы поглощения при λm ≈ 320 нм, существуют и в аминоазобензольных красителях, а принадлежащие этим красителям бинарные катионы PhAT определяют их цветность [13–16, 18].
К характерным свойствам t-AB следует отнести отсутствие дипольного e-момента (μe), установленное в [26] на его растворах в неполярном бензоле. Использованный в [26] прибор работал на переменном e-поле с частотой 1200 кГц. Судя по результату, это поле не искажало существенно формы e-облака, генерируемого чрезвычайно высокочастотным обменом электронов между sp2-орбиталями то одного, то другого азота и R3s-орбиталью азогруппы t-AB (схема 1, таутомеры Z+,$R_{{{\text{3s}}}}^{{1--}}$ ). Иначе говоря, в опытах с t-AB [26] имела место компенсация осциллирующих дипольных моментов для внешнего переменного поля [26]. Между тем, в среде полярных жидкостей форма облака R3s-орбитали азогруппы t-AB может изменяться вследствие образования комплексов с молекулами растворителя (ридиплексами) [11, 20]. Образование ридиплексов вызывает рост интенсивности Vis-полосы (λm ≈ 440 нм) t-AB при переходе от н-гептана к этанолу [11], причем не за счет водородных связей (по данным ИК-спектроскопии, принадлежащие t-AB группы N=N не образуют водородных связей с этанолом [27]). Vis-полоса t-AB в безводном этаноле (λm ≈ 443.0 нм) имеет повышенное значение εm = 510 л/(моль см) [2] вместо εm ≈ 430 л/(моль см) (при λm = 440 нм) у t-AB в безводном гептане [11]. Интенсивность Vis-полосы еще выше у t-AB в смеси “этанол (15%)/(вода 85%)”: εm = 760 л/(моль см) (λm = = 420 нм) [28], а в 12%-ном водном этаноле εm = = 900 л/(моль см) [11]. Насыщение гептана водой тоже приводит к более высокому значению εm = = 570 ± 30 л/(моль см) относительно εm ≈ ≈ 430 л/(моль см) у раствора t-AB в “сухом“ гептане [11]. Молекулы t-AB не протонируются в 15%-ном водном этаноле вплоть до концентрации серной кислоты 2.7 моль/л [11]. При этом Vis-полоса сохраняет значение λm = 416 нм (как в чистой воде), но получает значительно более высокий коэффициент εm = 1700 л/(моль см). Отсюда следует, что ридиплексы в водном спирте не только увеличивают интенсивность Vis-полосы t-AB, но и затрудняют реакцию протонирования при невысокой концентрации H2SO4. В этой связи предположено [20], что ридиплексы образуются за счет не водородной связи, а связи электронов $R_{{{\text{3s}}}}^{{1--}}$-орбитали с кислородом спирта или воды.
Характерные свойства c-AB
В изомере c-AB, с его квантово-волновой e-таутомеризацией E,$R_{{{\text{3s}}}}^{0}$ ↔ E1+,$R_{{{\text{3s}}}}^{{1--}}$ (схема 1), ph-кольца не образуют бинарно связанных (по Симпсону) резонаторов. Об этом свидетельствует значительное отличие характеристик ближних UV-полос c-AB и t-AB (растворы в этаноле). Так, форма E,$R_{{{\text{3s}}}}^{0}$ имеет показатели λm = 280, εm = = 5260 л/(моль см) [2] (λm = 281 нм, εm = = 4990 л/(моль см) [29]), а форма Z,$R_{{{\text{3s}}}}^{0}$: λm = = 319 нм, εm = 21300 л/(моль см) [2] (λm = 316, εm = = 22 300 л/(моль см) [29]). Зато в E,$R_{{{\text{3s}}}}^{0}$ sp2-орбитали образуют локальные молекулярные орбитали (mo): связывающую mos и антисвязывающую moa, как в пиридазине [30]:

Наличие по e-паре на (mos)$^{{ \bullet \bullet }}$ и (moa)$^{{ \bullet \bullet }}$ не дает энергетической выгоды для E в силу равенства энергий связывания и разрыхления. Но энергия орбитали (moa)$^{{ \bullet \bullet }}$ в E превышает энергию sp2 орбиталей в Z, увеличивая вероятность e-перехода на R3s-орбиталь и долю катиона PhAT. Это ведет к усилению интенсивности Vis-полосы c-AB и ее заметное гипсохромное смещение относительно Vis-полосы t-AB. Последнее можно связать с усилением кулоновского взаимодействия между e-облаком R3s-орбитали и катионом PhAT в c-AB и повышением энергии фотопереноса электрона в плоскости катиона PhAT. Важно также, что перемещение электрона по схеме (moa)$^{{ \bullet \bullet }}$ + R3s → → (moa)$^{{ \bullet + }}$,$R_{{{\text{3s}}}}^{{1--}}$ удаляет один разрыхляющий электрон и создает в азогруппе трехэлектронную связь между атомами N, увеличивая энергию связи между атомами азота [31, с. 173] и соответственно стабилизацию c-AB.
Ближняя UV-полоса изомера E (схема 2) с λm ≈ ≈ 280 нм практически повторяет положение ближней UV-полосы анилина: в водной среде у этой полосы λm = 280 нм (εmax = 1430 л/(моль см)) [32, с. 210], а в изооктане λm = 287 нм [32, с. 403]. Считается, что это связано с частичным смещением несвязанной e-пары азота аминогруппы анилина в фенильное кольцо и появлением на NH2-группе положительного заряда, делающего анилин более слабым основанием, чем аммиак [31, с. 281; 33, с. 85, с. 100]. Не исключено, что в E-изомере полоса с λm = 280 нм тоже появляется из-за частичного смещения в кольцо e-пары с орбитали (moa)$^{{ \bullet \bullet }}$. Между тем, акты такого смещения должны чередоваться с актами обратимого промотирования электронов по схеме: (moa)$^{{ \bullet \bullet }}$ + R3s ↔ ↔ (moa)$^{{ \bullet + }}$,$R_{{{\text{3s}}}}^{{1--}}$ . Последние приводят к появлению состояний E+,$R_{{{\text{3s}}}}^{{1--}}$ , возникающих поочередно на фенильных кольцах и создающих некоторую степень смещения e-облака орбитали $R_{{{\text{3s}}}}^{{1--}}$ от азогруппы в направлении катионов PhAT. Такой e-поляризацией можно объяснить наличие у c-AB дипольного e-момента μ = 3.0 D [26], в противоположность μ = = 0.0 у t-AB.
Акты е-промотирования по схеме^ (moa)$^{{ \bullet \bullet }}$ + + R3s ↔ (moa)$^{{ \bullet + }}$,$R_{{{\text{3s}}}}^{{1--}}$ создают в c-AB более высокий положительный заряд на азогруппе (схема 2), чем в t-AB, и это способствует понижению основности c-AB в реакции протонирования относительно основности t-AB. Но это понижение сочетается с более низкой кислотностью сопряженной кислоты с-AB+H относительно кислоты t-AB+H. Дело в том, что с-AB+H отдает свой протон растворителю труднее, чем t-AB+H вследствие образования системы локальных орбиталей (mos)$^{{ \bullet \bullet }}$ и (moa)$^{{ \bullet \bullet }}$ без энергетического выигрыша. В то же время кислота t-AB+H отдает протон легче благодаря относительно высокому энергетическому выигрышу от восстановления π-связи N=N с транс-ориентацией sp2 орбиталей. Это обеспечивает более низкую кислотность у с-AB+H (pKa = –2.25), чем у t-AB+H ((pKa = –2.95) [29].
Еще важное отличие с-AB от t-AB, связанное с более высокой e-плотностью на $R_{{{\text{3s}}}}^{{1--}}$ орбитали с-AB, – повышение интенсивности Vis-полосы ридиплексов с-AB с молекулами этанола. Так, Vis-полоса c-AB в неполярном циклогексане (λm = = 439 нм) имеет коэффициент экстинкции εm = = 1260 л/(моль см), в безводном этаноле (λm = = 433 нм) εm = 1480 л/(моль см) [29], а в 95%-ном этаноле ее интенсивность еще выше: εm = = 2000 л/(моль см) (λm = 440 нм) [28]. Последнее может быть связано с наличием 5% воды, образующей более прочные ридиплексы с с-AB.
Относительно высокая прочность ридиплексов с-AB с кислородом HO-групп позволяет использовать оксид алюминия (γ-Al2O3 с 3% воды, активность II по Брокману [34]) для разделения смесей с-AB и t-AB, полученных путем фотоизомеризации. Растворы смесей с-AB и t-AB в петролейном эфире (пентане) разделяли в колонках, заполненных порошком указанного γ-Al2O3 [2, 29]. При этом с-AB прочно сорбировался в верхней части колонки в виде оранжево-красного кольца, а t-AB элюировался вместе с углеводородом. Выделение более прочно сорбированного с-AB в [2] вели этиловым эфиром, а в [29] – смесью пентан/метанол с объемным соотношением 19/1. Для частиц γ-Al2O3 указанной формы характерна значительная дефектность кристаллической решетки [35, с. 94 ], и их поверхность покрыта преимущественно гидроксильными группами HO, которые образуют с с-AB более прочные ридиплексы, чем с t-AB, как в ситуации с этанолом.
МЕХАНИЗМ с → t ФОТОИЗОМЕРИЗАЦИИ
Теоретические работы до сих пор ограничиваются анализом модельной изомеризации Z ↔ E с ошибочным отнесением их Vis-полос с λm ≈ 440 нм к n → π* (S0 → S1)-переходам [7–10, 36, 37] и без учета индуцируемых R3s-орбиталью e-таутомеров Z+,$R_{{{\text{3s}}}}^{{ - 1}}$ и E+,$R_{{{\text{3s}}}}^{{ - 1}}$ (схема 1). С этим связана начатая в [38–40] длительная дискуссия о способности Vis-излучения (λm ≈ 440 нм) индуцировать в Z инверсное смещение фенильного кольца в плоскости угла N=N–C молекулы, а в E – ротацию группы phN – вокруг связи N–N. Делаются попытки выявить компьютерными расчетами способы перемещения фотовозбужденных состояний Z* и E* по их поверхностям потенциальной энергии S1 (n,π*) к зонам пересечения с поверхностями S0 и способы реализации актов изомеризации. Одна из наиболее продвинутых компьютерных методик моделирования (неадиабатическое квантово-химическое – молекулярно-динамическое) позволила [8] осуществить статистический анализ большого числа расчетных траекторий движения возбужденных состояний в обоих направлениях изомеризации Z и E. Для E была показана реализация только ротационного пути с неодинаковой вероятностью поворотов фенильных колец в направлении и против направления движения часовой стрелки [8]. Возможно, эти детали представляют интерес в плане длящейся дискуссии, однако они относятся к мнимой модели изомеризации E → Z, без учета роли e-таутомеров E+,$R_{{{\text{3s}}}}^{{1--}}$ (схема 1). Теперь эту роль можно осветить на основании экспериментальных данных [6].
Экспериментальные данные [6] и их авторская трактовка
В опытах [6] использовали импульсную лазерную установку с длиной волны возбуждающего света λex = 435 нм (длительность импульсов 100 фс), позволяющую регистрировать разностные транзитные сигналы. Облучали циркулирующие в замкнутой системе этанольные растворы фотостационарной смеси в балансе фотодинамического равновесия 75% c-AB/25% t-AB. Состояние баланса поддерживали воздействием на проточный раствор t-AB непрерывного UV-излучения (λm = = 366 нм) на этапе, предшествующем введению раствора в импульсное устройство. По оценке [6] действие на эти смеси импульсов с λex = 435 нм увеличивает концентрацию c-AB* до ≈90%. Ответные сигналы от возбужденных состояний определяли в виде разностной оптической плотности с использованием зондирующих импульсов с заданной длиной волны (λpr).
На рис. 1 кривые 1, 2 характеризуют транзитный (tr-) спектр фотостационарной смеси. Он получен при том, что после актов возбуждения с λex = 435 нм все зондирующие импульсы имели время задержки td = 100 фс и покрывали интервал 350–750 нм. Этот tr-спектр представлен в виде зависимости разностной оптической плотности в единицах милли-ΔD (mΔD) от λpr. Он содержит интенсивную Vis-полосу (λm = 550 нм), на спаде которой наблюдается слабая полоса с λm = 650 нм (кривая 1), и интенсивную UV-полосу с λm = 360 нм (кривая 2). (На рис. 1 приведены также tr-спектры 3–6, полученные при фотовозбуждении аминоазобензола [41], объясненные в [42] и используемые ниже как референтные для полос с λm = = 550 и 650 нм (рис. 1, кривая 1). Кроме UV-Vis полос 1, 2 (рис. 1), авторы [6] зарегистрировали кинетические кривые транзитных сигналов (зависимость mΔD от td) для 49 длин волн λpr в интервале 360–750 нм. Из них привели графики с λpr = = 360, 415, 467 и 563 нм, не обсуждая конкретную природу сигналов (рис. 2, соответственно, кривые 1–4 с линейной шкалой td в интервале “ –1, 1 фс ” и логарифмической шкалой для td > 1 фс). На этих графиках сигналы быстро достигают максимумов mΔDm (наиболее высоких при λpr = 360 и 563 нм), вслед за которыми идут быстрые спады, отнесенные в [6] непосредственно к c → t-изомеризации c-AB* в n,π*-состоянии. Кинетику этих спадов аппроксимировали моноэкспоненциальной зависимостью mΔD от времени td с характеристической константой τ = 180 фс. На кривой 4 (λpr = 563 нм) эта зависимость с константой τ = = 180 фс описывает более 80% от начального значения mΔD0, затем идет долгое медленное затухание сигнала. На кривой 1 (рис. 2, 360 нм) за быстрым спадом идет стадия, которую связали [6] с выделением имеющейся в с-AB избыточной над t-AB энергии, ведущей к длительно релаксирующему “горячему“ колебательно (v) возбужденному S0-состоянию t-ABV с измененным спектром поглощения. Полагая, что полученное для фотостационарного раствора значение τ = 180 фс не соответствует реальному значению, авторы [6] осуществили дополнительные опыты по транзитной спектроскопии t-AB с λex = 435 нм и с их учетом уточнили значение константы до величины τ = 170 фс.
Рис. 1.
Транзитные разностные спектры этанольных растворов смеси t-AB/c-AB = 75%/25% (1, 2) и t-AAB2 (3–6), полученные при возбуждении импульсами света с λех = 435 (1, 2) и 400 нм (3–6) с временами задержки зондирующих импульсов: 1, 2 – 0.1, 3 – 0.1, 4 – 0.5, 5 – 1.0, 6 – 5.0 пс. Пояснения в тексте.

Рис. 2.
Изменения транзитной разностной оптической плотности этанольных растворов смеси t-AB/c-AB = 75%/25% под действием импульсов света с λех = = 435 нм на длинах волн λpr: 1 – 360; 2, 5 – 415; 3, 6 – 467, 4 – 563 нм. Пояснения в тексте.

Завершая описание результатов эксперимента [6], истолкованных с традиционным отнесением Vis-полос изомеров AB к n → π*-переходам, следует отметить, что выводы [6] неадекватны реальной модели фотоизомеризации c-AB. Между тем, таковую можно построить на основе экспериментальных данных [6] с учетом раскрытой в [20] роли e-поляризованных таутомеров (схема 1) в фотохимии t-AB .
Природа Vis-поглощения c-AB в интервале 450–750 нм
Представленная на рис. 1 кривая 1 характеризует Vis-поглощение с λm = 550 и 650 нм. Для выяснения его природы на рис. 1 приведены спектры 3–6, полученные для красителя t-аминоазобензола [41] методом пикосекундной спектроскопии и объясненные в [42]. Согласно [42], основным состоянием красителя являются ридимеры t-AAB2, чье Vis-поглощение обеспечивают катионы PhAT. Возбуждение принадлежащих t-AAB2 катионов PhAT ведет к расщеплению t-AAB2 и образованию плоских поляризованных мономеров t-AAB с бинарными катионами PhAT, заряды которых уравновешиваются отрицательными зарядами $R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ [42]:

Плоские t-мономеры Ph+N$_{{ \bullet + }}^{{ + \bullet }}$–N$_{{ \bullet + }}^{{ \bullet + }}$Pћ+N$^{{ \bullet \bullet }}$R2 с бинарными катионами PhAT имеют полосу Vis-поглощения при λmax = 625–650 нм (рис. 1, кривая 3), связанную с механизмом e-резонанса по Симпсону [13, 15, 21]. (Сходные Vis-полосы имеют дикатионы Ph+HN+–N+HPh+ дипротонированного азобензола [19].) Сильное кулоновское отталкивание заряженных частей дикатионов Ph+N$_{{ \bullet + }}^{{ + \bullet }}$–N$_{{ \bullet + }}^{{ \bullet + }}$Pћ+N$^{{ \bullet \bullet }}$R2 и наличие в них торсионных колебаний вокруг связи N–N приводят к быстрой потере (с τ = 0.2 пс) плоской структуры и замене полосы с λmax = 625–650 нм на полосу с λmax = 550 нм [42]. При этом на некоторое время появляется промежуточное планарное состояние (intermediate state, с-A${\text{AB}}_{{{\text{ims}}}}^{*}$), предшествующее устойчивой форме с-AAB [42]. Реализация кратковременной планарности у с-A${\text{AB}}_{{{\text{ims}}}}^{*}$ сочетается с появлением Vis-полосы при λmax = 520–550 нм по механизму Симпсона [15, 21] (рис. 1, спектры 4, 5), тогда как спектр 6 (рис. 1) в основном принадлежит молекулам c-AAB.
На рис. 1 полоса 1 с λm = 550 и 650 нм по своей форме схожа с транзитным спектром мономерного дикатиона Ph+N$_{{ \bullet + }}^{{ + \bullet }}$–N$_{{ \bullet + }}^{{ \bullet + }}$Pћ+N$^{{ \bullet \bullet }}$R2 (рис. 1, кривая 4), в котором нет π-сопряжения между аминогруппой –N$^{{ \bullet \bullet }}$R2 и соседним фениленовым кольцом катиона PhAT. Это сходство позволяет заключить, что полоса 1 (рис. 1) принадлежит не расходующемуся в c → t-изомеризации “n,π*”-состоянию молекул с-AB, а суперпозиции сигналов от возникших из с-AB* e-возбужденных дикатионов в планарной конформации с-${\text{AB}}_{{{\text{ims}}}}^{*}$ (подобной с-A${\text{AB}}_{{{\text{ims}}}}^{*}$ с раздвинутыми ph-кольцами) и планарной конформации t-AB$_{1}^{*}$ (Ph+N$_{{ \bullet + }}^{{ + \bullet }}$–N$_{{ \bullet + }}^{{ \bullet + }}$Pћ+). Можно заметить, что максимальная оптическая плотность mΔDm = 15 на спектральной полосе 1 (рис. 1, λm = 550 нм td = 100 фс) весьма близка к mΔDm = 16 на кривой 4 (рис. 2, λpr = = 563 нм). В то же время нулевая точка mΔD0 моноэкспоненциального участка кривой 4 (с константой τ = 170 фс) находится практически на максимуме mΔDm = 16. Отсюда возникает мысль, что быстрый (с τ = 170 фс) спад сигнала с λpr = = 563 нм от начальной точки (mΔD0 ≈ mΔDm) характеризует не изомеризацию с-AB в “n,π*”-состоянии, как полагают в [6], а релаксацию образовавшегося из с-${\text{AB}}_{{{\text{ims}}}}^{*}$ возбужденного состояния t-AB$_{1}^{*}$. Важно, что максимумы всех кинетических кривых 1–4 (рис. 2) достигаются одновременно, а начальные стадии спадов от точек mΔD0 ≈ mΔDm подчиняются, согласно [6], моноэкспоненциальной кинетике (τ = 170 фс) с максимальной начальной скоростью. По этой причине все моноэкспоненциальные стадии кривых на рис. 2, скорее всего, отражают не изомеризацию молекул с-AB, а релаксацию транс-дикатиона t-AB$_{1}^{*}$ (Ph+N$_{{ \bullet + }}^{{ + \bullet }}$–N$_{{ \bullet + }}^{{ \bullet + }}$Pћ+), на котором сложились энергия возбуждения с λex = 435 нм и энергия, запасенная в c-AB.
Кинетика спада фотовозбужденных молекул c-AB
Возникшая для трактовки [6] альтернатива с интермедиатами с-${\text{AB}}_{{{\text{ims}}}}^{*}$ и t-AB$_{1}^{*}$ ставит вопрос о реальной кинетике c → t-изомеризации. В этой связи следует обратить внимание на то, что на кривых 1–4 (рис. 2) даны последовательности импульсов с λpr в зонах отрицательных значений td, осуществленные без импульсов возбуждения (λex = 435 нм) для обозначения осей абсцисс. Они заканчиваются в моменты включения импульсов с λex = 435 нм, значительно опережая “нулевые” моменты (mΔD0 ≈ mΔDm) кривых спада с τ = 170 фс. Более того, на кривых 2, 3 (рис. 2, λpr = 415 и 467 нм) имеются слабые отрицательные сигналы (– mΔD), опережающие моменты подъема сигналов еще в области отрицательных значений td. Этим слабым сигналам в [6] не придали значения, но они обозначают начало расходования молекул c-AB (по разностности mDtd,c-AB – mD0,c-AB < 0). Начальные участки расходования c-AB быстро маскируются растущими сигналами промежуточных продуктов релаксации от молекул с-${\text{AB}}_{{{\text{ims}}}}^{*}$ и t-AB$_{1}^{*}$. Разностную кинетическую кривую спада молекул c-AB можно рассчитать с учетом величины интервала значений Δtd на кривых 2, 3 (рис. 2) от момента включения импульсов (td0) с λex = = 435 нм до максимальных значений сигналов mΔD0 ≈ mΔDm (с tdm), при которых рост сигналов заканчивается вследствие практически полного исчезновения c-AB. Момент включения импульсов с λex = 435 нм (это начальная точка сканирования разностных сигналов) наблюдается на кривых 2, 3 с λpr = 415, 467 нм (рис. 2) в отрицательной области td. Оценка интервала (td0,tdm) дает Δt ≈ 0.33 пс.
Для распадающегося по моноэкспоненциальному закону соединения обычно полагают, что его практически полный расход реализуется за время, равное десяти периодам полураспада, и в данном случае это 10T0.5 = 0.33 пс. Отсюда следуют равенства T0.5 = 0.033 пс, τc-AB* = 0.033/ln 2 = = 0.047 пс и формула спада сигнала c-AB при c → → t-фотоизомеризации: mΔD = (– mΔDmin)(1 – ‒ exp(– t/0.047).
Рассчитанные по этой формуле для λpr = 415 и 467 нм (–mΔDmin= – 4) кривые 5, 6 (рис. 2) свидетельствуют, что изомеризация c-AB* → t-AB$_{1}^{*}$ идет значительно быстрее (τc-AB* = 47 фс), чем последующая релаксация с-${\text{AB}}_{{{\text{ims}}}}^{*}$ и t-AB$_{1}^{*}$ (τ = 170 фс).
Механизм индуцированных с λex = 435 нм превращений c-AB
Учитывая, что импульсное возбуждение с λex = = 435 нм вызывает в c-AB π → π*-перенос электрона в катионе PhAT с ВЗМО на НСМО, можно представить последующую цепочку e-конфигураций до состояния t-AB$_{1}^{*}$ в виде схемы 3:

Согласно схеме 3, возбужденный в c-AB* на НСМО катиона PhAT (E+,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ + hvVis → E+*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$) электрон быстро переходит с НСМО на пустую $p_{{\text{z}}}^{ + }$-орбиталь атома N в состояние $p_{{\text{z}}}^{1}$ и затем взаимодействует с pz-электроном углерода ph-кольца, образуя e-конфигурацию $E_{1}^{{ + *}}$,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ с двойной связью C=N. Отдав на образование C=N из своего e-секстета два π-электрона, ph-кольцо преобразуется в катион типа бензония, имеющего полосу поглощения c λm ≈ 400 нм [20]. В бензонии заряд локализуется в кольце, не изменяя состояния орбитали $R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ и компенсации зарядов на правом азоте ${\text{N}}_{{ + \bullet }}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ e-конфигурации $E_{1}^{{ + *}}$,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$. Далее один из $p_{{\text{z}}}^{2}$-электронов этого азота ${\text{N}}_{{ + \bullet }}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ переходит на $R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$-орбиталь, создавая интермедиат $E_{1}^{{ + + *}}$,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$. В нем происходит e-обмен, в котором $R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$-орбиталь отдает электрон (переходя в $R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$) на аннигиляцию заряда бензония, превращая его в свободный радикал, захватывающий электрон из π-связи C=N с восстановлением ph-кольца при локализации другого π-электрона на орбиталиь $p_{{\text{z}}}^{1}$ азота. Затем сама $R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$-орбиталь захватывает электрон из e-пары (:sp2) того же азота, возвращаясь в $R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ и завершая чередование франк-кондоновских (FC) e-конфигураций состоянием $E_{2}^{{ + + *}}$,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$. В состоянии $E_{2}^{{ + + *}}$,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ катионы PhAT испытывают значительное кулоновского отталкивание друг от друга при практической потере π-связи в группе ${\text{N}}_{{ + \bullet }}^{{ + \bullet }}$–${\text{N}}_{{ + \bullet }}^{{ + \bullet }}$. В итоге оба заряженных ph-кольца раздвигаются в противоположные стороны: $E_{2}^{{ + + *}}$,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ → с-${\text{AB}}_{{{\text{ims}}}}^{*}$, создавая неустойчивую планарную конформацию с-ABims, обладающую Vis-полосой с λm = 550 нм (рис. 1, спектр 1).
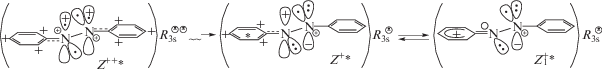
При этом идет суммирование энергии фотовозбуждения (λex = 435 нм, 65.75 ккал/моль) и энергетического избытка c-AB над t-AB (0.7 эВ, 16.14 ккал/моль [8]) до величины Q = 81.89 ккал/моль. А оставшееся в с-${\text{AB}}_{{{\text{ims}}}}^{*}$ взаимное отталкивание групп ph+ и их торсионные движения вокруг связи 2${\text{N}}_{{ + \bullet }}^{{ + \bullet }}$–${\text{N}}_{{ + \bullet }}^{{ + \bullet }}$ обеспечивают изомеризацию (с τ = 170 фс) в планарную e-конфигурацию Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ с Vis-полосой при λm = 650 нм (рис. 1, спектр 1).
В [6] быстрый подъем и последующий спад сигналов на кривых 1–4 (рис. 2) отнесены к мнимому n,π*-состоянию E*, обеспечивающему c → t -изомеризацию с константой τ = 170 фс. Но реально, источниками этих сигналов служат возбужденные состояния → с-${\text{AB}}_{{{\text{ims}}}}^{*}$ и → t-AB$_{1}^{*}$ с энергией Q = 81.89 ккал/моль, которая, согласно формуле Q = 28600/λ (нм) [43], соответствует энергии с λ = 348 нм, достаточной для образования t-AB$_{1}^{*}$ (Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$). С труктура t-AB$_{1}^{*}$ (Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$) сохраняет планарную конформацию в течение определенного времени, и в ней наступает чередование e-конфигураций, которое должно идти с той же скоростью, что и в FC-состоянии (τi$ \ll $ τ = = 170 фс). (Об этом свидетельствует весьма низкая интенсивность полосы с λm = 650 нм относительно полосы с λm = 550 нм (рис. 1, спектр 1)). Поэтому быстрые спады сигналов (рис. 2, кривые 1–4) от возникающих в t-AB$_{1}^{*}$ e-конфигураций лимитируются скоростью предшествующей стадии изомеризации с-${\text{AB}}_{{{\text{ims}}}}^{*}$ ~→ t-AB$_{1}^{*}$ (схема 4) и регистрируются с константами τ = 170 фс.
Релаксационные превращения молекул t-AB$_{1}^{*}$
Как отмечалось, спад полосы с λpr = 563 нм (τ = 170 фс; рис. 1, спектр 1; рис. 2, кривая 4) до ≈80% от начального значения mΔD0 ≈ mΔDm [6] относится к изомеризации с-${\text{AB}}_{{{\text{ims}}}}^{*}$ в t-AB$_{1}^{*}$ (Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$, схема 3). Последующую стадию медленного затухания сигнала с λpr = 563 нм (рис. 2, кривая 4, td > 0.5 пс) можно связать с небольшим вкладом актов релаксации (Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$) в колебательно возбужденное состояние (Z++V,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$), передающее v-энергию молекулам среды. Спад сигнала с λpr = 360 нм (τ = 170 пс) до ≈50% от значения mΔD0 (рис. 2, кривая 1) можно отнести к рекомбинации двух пар зарядов в t-AB$_{1}^{*}$ с образованием нейтрального и поляризованного таутомеров с v-возбужденными химическими связями: Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ → → (ZV) → ZV,$R_{{{\text{3s}}}}^{0}$ ↔ Z+V,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$. Возникающие при этом v-таутомеры в итоге приходят к равновесию по схеме 1. А быстрые спады сигналов с λpr = 415 и 467 нм (рис. 2, кривые 2, 3) можно связать с последовательностью e-конфигураций, рассмотренной в [20] (схема 4).
Согласно схеме (4), в t-AB$_{1}^{*}$ (Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$) сначала электрон с орбитали $R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ рекомбинирует с зарядом sp2-орбитали атома ${{{\text{N}}}_{{ \bullet + }}}$. Потеряв заряд, sp2-орбиталь обеспечивает перенос электрона с орбитали pz своего азота на орбиталь pz соседнего азота. При этом появляется e-конфигурация Z+*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ с возбужденным катионом PhAT, получившим электрон на НСМО. Этот электрон быстро переходит на опустевшую $p_{{\text{z}}}^{ + }$-орбиталь атома N, а возникающая $p_{{\text{z}}}^{1}$-орбиталь взаимодействует с $p_{{\text{z}}}^{1}$-орбиталью атома С ph-кольца, образуя $Z_{1}^{{ + *}}$,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ с двойной связью C=N и катионом типа бензония с λm ≈ 400 нм (эта трансформация соответствует переходу, указанному в схеме 3).
e-Конфигурация $Z_{1}^{{ + *}}$,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ находится в обратимом равновесии с Z+*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ [20], но у нее нет основного состояния, и релаксация ее энергии возбуждения идет через e-конфигурацию Z+*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ с катионом PhAT и Vis-полосой при λm ≈ 440 нм [20]. Поэтому спад сигнала с λpr = 415 (рис. 2, кривая 2) идет сразу со спадом сигнала катиона PhAT. Зондирующие импульсы с λpr = 467 нм (рис. 2, кривая 3) попадали на склон Vis-полосы катиона PhAT (λm ≈ 440 нм), однако они позволили установить спад с константой τ = 170 фс.
В целом, интермедиат Z+*,$R_{{{\text{3s}}}}^{ \bullet }$ с энергией фотовозбуждения от e-конфигурации Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ релаксирует по ряду направлений. Часть его энергии расходуется на рекомбинацию зарядов с образованием колебательно возбужденной пары из нейтрального и поляризованного таутомеров: Z+*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ → ZV,$R_{{{\text{3s}}}}^{0}$ ↔ Z+V,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$. Этот канал релаксации придает сложный вид кинетике спада сигнала с λpr = 360 нм (рис. 2, кривая 1). Сначала сигнал с λpr = 360 нм идет от t-ABV, возникающего первоначально за счет рекомбинации двух пар зарядов: Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ → (ZV) → ZV,$R_{{{\text{3s}}}}^{0}$ ↔ Z+V,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$, он спадает с константой τ = 170 фс до практической остановки при значении ≈0.5 mΔD0. В это время и на последующем этапе медленной релаксации до td = 100 фс, импульсы с λpr = 360 нм зондируют UV-полосу S0-состояния таутомера ZV,$R_{{{\text{3s}}}}^{0}$, которая не только снижает интенсивность, но и гипсохромно смещается к λm = 320 нм. Наблюдаемую задержку v-релаксации (не обсужденную в [6]) можно связать с тем, что на сигнал от первоначального канала релаксации Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ → (ZV) → ZV, $R_{{{\text{3s}}}}^{0}$ ↔ Z+V,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ на некоторое время налагается сигнал от дополнительного канала Z+*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ → ZV,$R_{{{\text{3s}}}}^{0}$ ↔ Z+V,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$. В кончном итоге, v-энергия релаксирует на молекулы растворителя [20] и наступает таутомерное равновесие Z+,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ ↔ Z,$R_{{{\text{3s}}}}^{0}$ (схема 1).
Рассмотренные выше превращения молекул t-AB$_{1}^{*}$ (Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$) и Z+*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ ответственны за расход основного количества энергии возбуждения c-AB. Весьма незначительное расходование энергии идет путем флуоресценции катионов PhAT, спектр которой зеркально симметричен спектру Vis-поглощения катионов PhAT (ее квантовый выход Ф ≈ 1 × 10– 6 [44]). В [44] эта флуоресценция была отнесена к эмиссии c-AB*, однако, в соответствии со схемами 3 и 4, ее источником должны быть катионы PhAT* e-конфигурации Z+*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$, возникшей из t-AB$_{1}^{*}$ (Z++*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$). В другом второстепенном канале релаксации образуется малое количество c-AB из-за выделения энергии рекомбинации зарядов в Z+*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$, идущей на поддержание торсионных колебаний ph-колец (Z+*,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ ~→ ~→ E+,$R_{{{\text{3s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ ↔ E,$R_{{{\text{3s}}}}^{0}$) [20]. Об этом свидетельствует слабый подъем сигнала с λpr = 467 нм (рис. 2, кривая 3) при td > 0.5 пс. Без указанной энергетической подпидки спад разностного сигнала с λpr = = 467 нм катиона PhAT шел бы до равновесных таутомеров c-AB (схема 1) в сответствии с кривой 6 (рис. 2).
Влияние природы растворителя на квантовые выходы фотоизомеризации
Согласно работе [38], квантовые выходы (Ф) c ↔ t-фотоизомеризации AB под действием Vis-света с λex = 439 нм изменяются при замене растворителя – гептана – на смесь “80% вода/20% этанол“: происходит увеличение Фt→c от = 0.25 до 0.35 и снижение Фс→t от 0.56 до 0.41 [38]. Это можно связать с тем, что изомеризация в том и другом направлениях идет через один и тот же планарный интермедиат с-${\text{AB}}_{{{\text{ims}}}}^{*}$:
Таким образом, следует отметить высокое значение экспериментальных данных [6], на основе которых теперь создана качественная модель фотоизомеризации c-AB. Кроме того, дополнительно к выводам работы [20] показано, что проводимые до настоящего времени теоретические вариации с молекулами c-AB и t-AB без учета роли 3s-орбиталей азогрупп и e-таутомеров с катионами PhAT ведут лишь к виртуальным моделям фотоизомеризации, не давая желательного адекватного описания соответствующих механизмов.
Список литературы
Google. https://ru.wikipedia.org/wiki/Азобензол. Оптические свойства.
Birnbaum P.P., Linford J.H., Style D.W.G. // Trans. Far. Soc. 1954. V. 50. P. 735.
Birnbaum P.P., Style D.W.G. // Ibid. 1954. V. 50. P. 1192.
Sidman J.W. // J. Chem. Revs. 1958. V. 58. № 4. P. 689.
Jaffe H.H., Yeh Si-Jung, Gardner R. // J. Mol. Spectroskopy, 1958. V. 2. № 1–6. P. 120.
Nägele T., Hoche R., Zinth W., Wachtveitl J. // Chem. Phys. Lett. 1997. V 272. № 5, 6. P. 489.
Böckmann M., Doltsinis N.L., Dominik M. // J. Phys. Chem. A .2010. V. 114. № 2. P. 745.
Pederzoli M., Pittner J., Barbatti M., Lischka H. // Ibid. 2011. V. 115. № 41. P. 11136.
Harabuchi Yu., Ishii M., Nakayama A., Noro T., Taketsugu T. // J. Chem. Phys. 2013. V. 138. № 6. P. 4305.
Neukirch A.J., Shamberger L.C., Abad E., Haycock et al. // J. Chem. Theory and Computation. 2014. V. 10. № 1. P. 14.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2015. Т. 89. № 11. С. 1773.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 8. С. 1251.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 10. С. 1552.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 2. С. 313.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 7. С. 1111.
Михеев Ю.А., Ершов Ю.А. // Там же. 2020. Т. 94. № 1. С. 143.
Михеев Ю.А., Ершов Ю.А. // Там же. 2020. Т. 94. № 8. С. 1269.
Михеев Ю.А., Ершов Ю.А. // Там же. 2020. Т. 94. № 11. С. 1706.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Там же. 2015. Т. 89. № 2. С. 243.
Михеев Ю.А. // Там же. 2021. Т. 95. № 11. С. 1771.
Robin B., Simpson W.T. // J. Chem. Phys. 1962. V. 36. № 3. P. 580.
Robertson J.M. // J. Chem. Soc. 1939. P. 232.
Hampson G.C., Robertson J.M. // Ibid. 1941. P. 409.
Tsui T., Takashima H., Takeuchi H., Egava T., Konaka Sh. // J. Phys. Chem. A. 2001. V. 105. № 41. P. 9347.
Клар Э. Полициклические углеводороды. Т. 1. М.: Химия, 1971.
Hartley G.S., Le Fèvre R.J.W. // J. Chem. Soc. 1939. P. 531.
Brealey G.J., Kasha M. // J. Amer. Chem. Soc. 1955. V. 77. № 17. P. 4462.
Beveridge D.L., Jaffe H.H. // Ibid. 1966. V. 88. № 9. P. 1948.
Gerson F., Heilbronner E., van Veen A., Wepster B.M. // Helvet. Chim. Acta. 1960. V. 43. № 7. P. 1889.
Mason S.F. // J. Chem. Soc. 1959. P. 1240.
Коулсон Ч. Валентность. М.: Мир, 1965. 426 с.
Калверт Дж., Питтс Дж. Фотохимия. М.: Мир, 1968. 671 с.
Хигаси К., Баба Х., Рембаум А. Квантовая органическая химия. М.: Мир, 1967. 379 с.
Крамаренко В.Ф., Туркевич Б.М. Анализ ядохимикатов. М.: Химия, 1978. С. 248.
Чукин Г.Д. Строение оксида алюминия и катализаторов гидрообессеривания. Механизмы реакций. М.: Типография Паладин, ООО “Принта”. 2010. 288 с. (chem.msu.su>rus/books/chukin – 2010/chukin – 2010.pdf).
Jacquemin D., Preat J., Perpète E.A. et al. // Intern. J. Quantum Chem. 2011. V. 111. № 15. P. 4224.
Zapata F., Fernandez-Gonzalez M.A., Rivero D. et al. // J. Chem. Theory Comput. 2014. V. 10. P. 312.
Bortolus P., Monti S. // J. Phys. Chem. 1979. V. 83. № 6. P. 648.
Rau H., Lüddecke E. // J. Am. Chem. Soc. 1982. V. 104. № 6. P. 1616.
Rau H. // J. Photochem. 1984. V. 26. № 2–3. P. 221.
Hirose Ya., Yui H., Sawada Ts. // J. Phys. Chem. A. 2002. V. 106. № 13. P. 3067.
Михеев Ю.А., Ершов Ю.А. // Журн. физ. химии. 2019. Т. 93. № 6. С. 946.
Теренин А.Н. Фотоника молекул красителей и родственных органических соединений. Л.: Наука, 1967. С. 17, 18.
Stuart Ch.M., Frontiera R.R., Mathies R.A. // J. Phys. Chem. A. 2007. V. 111. № 48. P. 12072.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии