

Журнал физической химии, 2022, T. 96, № 3, стр. 324-331
Закономерности деструкции порфиразинов в органических протоноакцепторных средах
a Ивановский государственный химико-технологический университет
Иваново, Россия
* E-mail: poa@isuct.ru
Поступила в редакцию 06.09.2021
После доработки 23.09.2021
Принята к публикации 25.09.2021
- EDN: GMZMQO
- DOI: 10.31857/S0044453722030207
Аннотация
Рассмотрены закономерности деструкции β-замещенных и β,β-аннелированных порфиразинов в присутствии циклических и ациклических азотсодержащих оснований в диметилсульфоксиде. Предложены и обоснованы возможные схемы механизмов. Показано влияние NH-кислотности порфиразинового макроцикла, пространственного строения азотсодержащего основания и его протоноакцепторной способности на кинетические параметры процесса. Обсуждены вопросы строения комплексов с переносом протонов в диметилсульфоксиде.
Порфиразины (Н2РА), благодаря разнообразным возможностям модификации их структуры, являются предметом все более пристального внимания исследователей, поскольку наличие наиболее полной информации о взаимосвязи строения и свойств Н2РА в различных химических процессах создает хорошую базу для использования результатов эксперимента в практических целях. В настоящее время порфиразины нашли применение в качестве жидкокристаллических, каталитических и сенсорных материалов, фотосенсибилизаторов синглетного кислорода, гасителей флуоресценции, нелинейной оптике и др. [1, 2]. Расширить спектр полезных свойств этого класса соединений возможно не только благодаря разработке новых эффективных методов синтеза, но и при всестороннем изучении устойчивости порфиразиновых молекул в различных условиях среды.
Кислотность порфиразинов
Под действием сильных оснований в среде полярных растворителей Н2РА подвергаются двухстадийной кислотной ионизации по внутрициклическим NH-связям с последовательным образованием моно- и дианионных форм [3]. При этом значения рKа1 и рKа2, часто полученные в различных условиях среды, достаточно сильно зависят от сольватационных эффектов, что затрудняет их использование для установления взаимосвязи между строением порфиразиновой молекулы и ее протонодонорной способностью. Наиболее достоверно эту зависимость отражает сравнение кислотных свойств Н2РА в газовой фазе [4].
Порфиразин (Н2Ра) обладает выраженной NH-кислотностью по сравнению с порфином (табл. 1) благодаря электроноакцепторному влиянию мезо-атомов азота. Введение в пиррольные кольца Н2Ра атомов галогенов приводит к значительному уменьшению величины рKа1 для тетрабромпорфиразина (Н2РаBr4) и тетрахлорпорфиразина (Н2РаCl4). Менее сильное влияние на кислотность молекулы оказывает β-фенильное замещение и расширение сопряженной π-системы макроцикла за счет тетрабензоаннелирования. Величины рKа1 для октафенилпорфиразина (Н2Ра(С6Н5)8) и фталоцианина (тетрабензопорфиразина) (Н2Рс) уменьшаются на 2 и 1 единицу соответственно по сравнению с Н2Ра. Напротив, аннелирование электронодефицитных пиразиновых циклов способствует значительному росту протонодонорной активности тетрапиразинопорфиразина (Н2Ра(Pyz)4). В ряду Н2Ра → Н2Рс → → Н2Ра(Pyz)4 значение рKа1 уменьшается на ∼6 единиц. Более сильное влияние на NH-кислотность оказывают аннелированные пятичленные гетероциклы. Для тетра(1,2,5-тиадиазоло)порфиразина (Н2Ра(SN2)4) и тетра(1,2,5-селенодиазоло)порфиразина (Н2Ра(SеN2)4) величины рKа1 имеют минимальные значения (табл. 1). Константы кислотной ионизации в газовой и жидкой фазах для окта(4-нитрофенил)порфиразина (Н2Ра(С6Н4NO2)8), тетра(4-трет-бутил)фталоцианина (Н2Рс(But)4), тетра(3-нитро-5-трет-бутил)фталоцианина (Н2Рс(3-NO2)4(But)4), тетра(4-нитро-5-трет-бутил)фта-лоцианина (Н2Рс(4-NO2)4(But)4), тетра(5-трет-бутилпиразино)порфиразина (Н2Ра(ButPyz)4), октаэтилтетрапиразинопорфиразина (Н2Ра(Et2Pyz)4) и октафенилтетрапиразинопорфиразина (Н2Ра(Ph2Pyz)4) отсутствуют:
Однако электронодонорные и электроноакцепторные заместители, находящиеся в их фенильных, бензольных и пиразиновых фрагментах, могут оказывать более или менее сильное влияние на кислотность порфиразинового макроцикла и, как следствие, на его устойчивость в органических протоноакцепторных средах.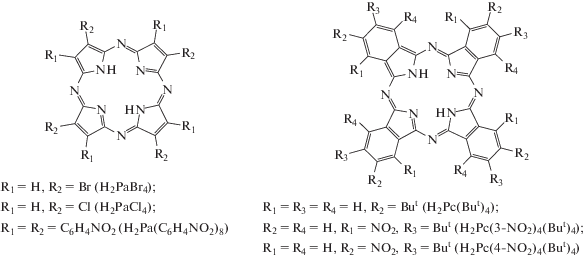

Таблица 1.
Константы кислотной ионизации порфина и порфиразинов по первой ступени (рKа1) в газовой фазе [4]
| Макроцикл | рKа1 |
|---|---|
| Н2Р | 22.35 |
| Н2Ра | 12.36 |
| Н2РаCl4 | 9.09 |
| Н2РаBr4 | 8.45 |
| Н2Ра(С6Н5)8 | 10.36 |
| Н2Рс | 11.23 |
| Н2Ра(Pyz)4 | 5.82 |
| Н2Ра(SN2)4 | 1.92 |
| Н2Ра(SеN2)4 | 1.60 |
Спектральная картина кислотно-основного взаимодействия порфиразинов с диметилсульфоксидом. Строение комплексов с переносом протонов
В инертном малополярном бензоле электронный спектр поглощения (ЭСП) Н2РаBr4, Н2РаCl4 и Н2Ра(С6Н4NO2)8 содержит в видимой области две расщепленные Qх- (λI) и Qy (λII)-составляющие Q-полосы, что соответствует D2h-симметрии π-хромофора молекулы [5, 6]. Аналогичная картина сохраняется для Н2Рс(3-NO2)4(But)4 [7], Н2Рс(4-NO2)4(But)4 [8], Н2Ра(ButPyz)4 [9], Н2Ра(Et2Pyz)4 [9], Н2Ра(Ph2Pyz)4 [10], Н2Ра(SN2)4 [11, 12] и Н2Ра(SеN2)4 [13, 14]. При замене бензола на слабоосновный диметилсульфоксид (DMSO) расщепление Q-полосы исчезает независимо от природы заместителя в макроцикле (рис. 1), а симметрия порфиразиновой молекулы в ходе кислотно-основного взаимодействия повышается от D2h до D4h. Исследованные порфиразины в присутствии DMSO проявляют свойства двухосновных NH-кислот и образуют устойчивые во времени комплексы с переносом протонов – Н2РА ⋅ 2DMSO. Исключение составляет тетра(4-трет-бутил)фталоцианин, ЭСП которого не претерпевает изменений при замене бензола на диметилсульфоксид [15].
Рис. 1.
Электронные спектры поглощения Н2Ра(SN2)4 в бензоле (1) и диметилсульфоксиде (2) при 298 K [11].

В комплексах Н2РА ⋅ 2DMSO протоны NH-групп, связанные с атомом кислорода молекул диметилсульфоксида и внутрициклическими атомами азота через водородные связи, располагаются над и под плоскостью макроцикла (рис. 2). Из-за пространственного экранирования молекулами DMSO четырех внутрициклических атомов азота они труднее вступают в реакции образования комплексов с солями металлов в отличие от молекулярных форм порфиразинов [16, 17]. В комплексах Н2РА ⋅ 2DMSO перенос протонов от кислоты к основанию, приводящий к образованию разделенных растворителем ионных пар с последующей их диссоциацией, представляется маловероятным [18, 19]. Кислотно-основное взаимодействие ограничивается либо стадией образования Н-комплекса – (PA)⋅⋅⋅(DMSOН)2, либо ионного комплекса, представляющего собой Н-связанную ионную пару – (PA)2–⋅⋅⋅(DMSOН)$_{2}^{ + }$ [5]. По мере увеличения протонодонорной способности порфиразина следует ожидать смещение кислотно-основного равновесия в сторону образования более полярной структуры:
(1)
$({\text{PA}})\cdot\cdot\cdot{{({\text{DMSOН}})}_{2}} \leftrightarrow {{({\text{PA}})}^{{2 - }}}\cdot\cdot\cdot({\text{DMSOН}})_{2}^{ + }.$
Спектральная картина деструкции порфиразинов в системе азотсодержащее основание-диметилсульфоксид
Введение в диметилсульфоксид достаточно слабых оснований (пиридина (Py), 2-метилпиридина (MePy)) не влияет на характер электронного спектра поглощения комплексов Н2РА ⋅ 2DMSO. В интервале концентраций $С_{{{\text{Py}}}}^{0}$ = $С_{{{\text{MePy}}}}^{0}$ = 0.31–9.93 моль/л он содержит нерасщепленную Q-полосу (рис. 1), интенсивность которой не претерпевает изменений в течение длительного промежутка времени при 323 K [6, 7, 9–14]. Качественно другая картина наблюдается при введении в DMSO добавок более сильных оснований – морфолина (Mor), пиперидина (Pip), н-бутиламина (BuNH2), трет-бутиламина и диэтиламина (Et2NH). В ЭСП Н2РА ⋅ 2DMSO с течением времени регистрируется уменьшение интенсивности Q-полосы (рис. 3–5) и полосы Соре, характеризующей наличие пиррольных фрагментов в макроцикле. Одновременно с этим наблюдается обесцвечивание раствора. Среди всех изученных Н2РА ⋅ 2DMSO только комплекс, образованный с участием тетра(4-нитро-5-трет-бутил)фталоцианина, не подвергается деструкции. На это указывает характер ЭСП Н2Рс(4-NO2)4(But)4 ⋅ 2DMSO, который остается без изменений в течение ∼37 ч при 323 K в DMSO с добавками 9.60 моль/л пиперидина [8], обладающего достаточно высокой протонодонорной способностью. Напротив, комплексы Н2РаBr4 ⋅ 2DMSO и Н2РаCl4 ⋅ 2DMSO в системе DMSO-пиперидин разрушаются предельно быстро, со скоростями, не позволяющими измерить их обычными кинетическими методами [6].
Рис. 3.
Изменение электронного спектра поглощения Н2Рс(3-NO2)4(But)4 ⋅ 2DMSO в системе н-бутиламин–DMSO в течение 45 мин при СоBuNH2 = = 5.05 моль/л, Т = 338 K [7].

Рис. 4.
Изменение электронного спектра поглощения Н2Ра(ButPyz)4 ⋅ 2DMSO в системе пиперидин–DMSO в течение 50 мин при СоPip = 9.96 моль/л, Т = = 323 K [9].

Рис. 5.
Изменение электронного спектра поглощения Н2Ра(SеN2)4 ⋅ 2DMSO в системе пиперидин–DMSO в течение 40 мин при СоPip = 2.02 моль/л, Т = 298 K [13].

Кинетические особенности деструкции порфиразинов в системе азотсодержащее основание-диметилсульфоксид
В зависимости от природы азотсодержащего основания и особенностей геометрического строения Н2РА ⋅ 2DMSO деструкция комплексов с переносом протонов может осуществляться по различным альтернативным механизмам.
В системе н-бутиламин (диэтиламин)-DMSO процесс деструкции Н2РаBr4 ⋅ 2DMSO [6] описывается суммарным кинетическим уравнением третьего порядка
(2)
$ - d{{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{PaB}}{{{\text{r}}}_{{\text{4}}}}\, \cdot \,{\text{2DMSO}}}}}{\text{/}}d\tau = k{{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{PaB}}{{{\text{r}}}_{{\text{4}}}}\, \cdot \,{\text{2DMSO}}}}}C_{{\text{B}}}^{2},$Лимитирующей стадией в этом случае является не тримолекулярный процесс, а бимолекулярное взаимодействие между комплексом с переносом протонов и Н-связанными димерными молекулами н-бутиламина (диэтиламина), которые образуются за счет NH-связей одной и неподеленной электронной пары атома азота другой молекулы [20]:
(4)
${\text{В}} + {\text{В}}\;\overset {{{K}_{{\text{p}}}}} \longleftrightarrow \;{\text{В}} \cdots {\text{В}}{\text{.}}$Благодаря более выраженной протоноакцепторной способности азотсодержащие основания вступают в конкурентную реакцию за протон и вытесняют молекулы DMSO:
(5)
$\begin{gathered} {{{\text{H}}}_{{\text{2}}}}{\text{PaB}}{{{\text{r}}}_{4}} \cdot 2{\text{DMSO}} + {\text{В}} \cdots {\text{В}}\;\xrightarrow{{{{k}_{1}}}} \\ \xrightarrow{{{{k}_{1}}}}\;{{({\text{PaB}}{{{\text{r}}}_{{\text{4}}}})}^{{2 - }}} + 2{{({\text{НВ}})}^{ + }} + 2{\text{DMSO}}. \\ \end{gathered} $Если принять во внимание, что значения Kр для первичных и вторичных ациклических аминов в DMSO отсутствуют, то нельзя полностью исключить возможность протекания деструкции в две стадии с k1 ≈ k2:
(6)
$\begin{gathered} {{{\text{H}}}_{{\text{2}}}}{\text{PaB}}{{{\text{r}}}_{{\text{4}}}} \cdot {\text{2DMSO}} + В\;\xrightarrow{{{{k}_{{\text{1}}}}}} \\ \xrightarrow{{{{k}_{{\text{1}}}}}}\;{{({\text{HPaB}}{{{\text{r}}}_{{\text{4}}}} \cdot {\text{DMSO}})}^{ - }} + {\text{Н}}{{{\text{В}}}^{ + }} + {\text{DMSO}}, \\ \end{gathered} $(7)
$\begin{gathered} {{({\text{HPaB}}{{{\text{r}}}_{{\text{4}}}} \cdot {\text{DMSO}})}^{ - }} + В\;\xrightarrow{{{{k}_{2}}}} \\ \xrightarrow{{{{k}_{2}}}}\;{{({\text{PaB}}{{{\text{r}}}_{{\text{4}}}})}^{{2 - }}} + {\text{Н}}{{{\text{В}}}^{ + }} + {\text{DMSO}}. \\ \end{gathered} $Напротив, деструкция Н2Ра(С6Н4NO2)8 ⋅ ⋅ 2DMSO [6] под влиянием BuNH2 (Et2NH) в DMSO описывается уравнением
(8)
$\begin{gathered} - d{{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{Pa(}}{{{\text{С}}}_{{\text{6}}}}{{{\text{Н}}}_{{\text{4}}}}{\text{N}}{{{\text{O}}}_{{\text{2}}}}{{{\text{)}}}_{{\text{8}}}}\, \cdot \,{\text{2DMSO}}}}}{\text{/}}d\tau = \\ = k{{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{Pa(}}{{{\text{С}}}_{{\text{6}}}}{{{\text{Н}}}_{{\text{4}}}}{\text{N}}{{{\text{O}}}_{{\text{2}}}}{{{\text{)}}}_{{\text{8}}}}\, \cdot \,{\text{2DMSO}}}}}{{C}_{{\text{B}}}}. \\ \end{gathered} $(9)
$\begin{gathered} {{{\text{Н}}}_{{\text{2}}}}{\text{Ра(}}{{{\text{С}}}_{{\text{6}}}}{{{\text{Н}}}_{{\text{4}}}}{\text{N}}{{{\text{O}}}_{{\text{2}}}}{{{\text{)}}}_{{\text{8}}}} \cdot {\text{2DMSO}} + {\text{В}}\;\xrightarrow{{{{k}_{{\text{1}}}}}} \\ \xrightarrow{{{{k}_{{\text{1}}}}}}\;{{({\text{НРа(}}{{{\text{С}}}_{{\text{6}}}}{{{\text{Н}}}_{{\text{4}}}}{\text{N}}{{{\text{O}}}_{{\text{2}}}}{{{\text{)}}}_{{\text{8}}}} \cdot {\text{DMSO}})}^{ - }} + \\ + \;{\text{Н}}{{{\text{В}}}^{ + }} + {\text{DMSO}}, \\ \end{gathered} $(10)
$\begin{gathered} {{({\text{НРа(}}{{{\text{С}}}_{{\text{6}}}}{{{\text{Н}}}_{{\text{4}}}}{\text{N}}{{{\text{O}}}_{{\text{2}}}}{{{\text{)}}}_{{\text{8}}}} \cdot {\text{DMSO}})}^{ - }} + {\text{В}}\;\xrightarrow{{{{k}_{2}}}} \\ \xrightarrow{{{{k}_{2}}}}\;{{({\text{Ра(}}{{{\text{С}}}_{{\text{6}}}}{{{\text{Н}}}_{{\text{4}}}}{\text{N}}{{{\text{O}}}_{{\text{2}}}}{{{\text{)}}}_{{\text{8}}}} \cdot {\text{DMSO}})}^{{2 - }}} + \\ + \;{\text{Н}}{{{\text{В}}}^{ + }} + {\text{DMSO}}. \\ \end{gathered} $Делокализованная π-связь по внутреннему 16-членному контуру (C8N8) и увеличение числа π-электронов в ароматической системе за счет мезо-атомов азота, а также ее расширение в результате аннелирования четырех бензольных (пиразиновых, тиадиазольных, селенодиазольных) колец способствует экранированию атомами и π-электронами протонов NH-групп в комплексах Н2РА ⋅ ⋅ 2DMSO. Наряду с этим, алкильные (фенильные, нитрофенильные) заместители создают дополнительные пространственные затруднения для благоприятного контакта молекул-партнеров. В результате этого процесс деструкции Н2РА ⋅ 2DMSO характеризуется низкими значениями констант скорости и достаточно высокими значениями энергии активации (табл. 2, 3).
Таблица 2.
Кинетические параметры деструкции комплексов с переносом протонов галогензамещенных порфиразинов в системе азотсодержащее основание–DMSO [6]
| Основание (В) | $k_{{\text{H}}}^{{298}}$ × 104, с–1 |
k298 × 103, л2/(моль2с) |
Еа, кДж/моль |
|---|---|---|---|
| Н2РаBr4 ⋅ 2DMSO | |||
| н-Бутиламин | 19.30 | 7.20 | 10 |
| Диэтиламин | 3.00 | 1.04 | 28 |
| Н2РаCl4 ⋅ 2DMSO | |||
| н-Бутиламин | 30.25 | 10.54 | 10 |
| Диэтиламин | 3.20 | 1.15 | 27 |
Таблица 3.
Кинетические параметры деструкции комплексов с переносом протонов β-замещенных и β,β-аннелированных порфиразинов в системе азотсодержащее основание-DMSO [6, 7, 9–14]
| Основание (В) | $k_{{\text{H}}}^{{298}}$ × 104, С–1 |
k298 × 105, л/(мольс) |
Еа, кДж/моль |
|---|---|---|---|
| Н2Ра(С6Н4NO2)8 ⋅ 2DMSO | |||
| н-Бутиламин | 10.20 | 210 | 29 |
| Диэтиламин | 0.38 | 7 | 51 |
| Н2Рс(3-NO2)4(But)4 ⋅ 2DMSO | |||
| Морфолин | устойчив | ||
| Пиперидин | 0.04 | 0.04 | 67 |
| н-Бутиламин | 0.02 | 0.02 | 57 |
| Диэтиламин | устойчив | ||
| Н2Ра(ButPyz)4 ⋅ 2DMSO | |||
| Морфолин | 3.00 | 3.00 | 94 |
| Пиперидин | 4.00 | 6.50 | 76 |
| н-Бутиламин | 6.30 | 9.00 | 64 |
| Диэтиламин | устойчив | ||
| Н2Ра(Et2Pyz)4 ⋅ 2DMSO | |||
| Морфолин | 4.50 | 4.00 | 85 |
| Пиперидин | 6.00 | 10.00 | 62 |
| н-Бутиламин | 4.50 | 8.00 | 68 |
| Диэтиламин | устойчив | ||
| Н2Ра(Ph2Pyz)4 ⋅ 2DMSO | |||
| Морфолин | 2.58 | 7.50 | 47 |
| Пиперидин | 3.14 | 23.20 | 44 |
| н-Бутиламин | 1.70 | 24.80 | 28 |
| Диэтиламин | устойчив | ||
| Н2Ра(SN2)4 ⋅ 2DMSO | |||
| Морфолин | 0.83 | 59.60 | 61 |
| Пиперидин | 35.10 | 161 | 48 |
| н-Бутиламин | 2.90 | 242 | 53 |
| трет-Бутиламин | 0.48 | 2.00 | 55 |
| Диэтиламин | устойчив | ||
| Триэтиламин | устойчив | ||
| Н2Ра(SеN2)4 ⋅ 2DMSO | |||
| Морфолин | 1.43 | 102 | 27 |
| Пиперидин | 6.20 | 520 | 37 |
| н-Бутиламин | 2.75 | 550 | 39 |
| трет-Бутиламин | 2.83 | 6.20 | 28 |
| Диэтиламин | 0.50 | 4.00 | 32 |
| Триэтиламин | устойчив | ||
Как уже упоминалось ранее, среди всех изученных порфиразинов образование комплекса Н2Рс(But)4 ⋅ 2DMSO в диметилсульфоксиде не наблюдается. Введение нитрогрупп в бензольные кольца тетра(4-трет-бутил)фталоцианина повышает протонодонорную способность макроцикла [22] и благоприятствует образованию комплексов с переносом протонов. Однако, несмотря на структурную близость, комплекс Н2Рс(4-NO2)4(But)4 ⋅ 2DMSO не подвергается деструкции в сильноосновной полярной среде – BuNH2 (Et2NH)-DMSO в отличие от Н2Рс(3-NO2)4(But)4 ⋅ ⋅ 2DMSO [8]. Электроноакцепторные нитрогруппы, находящиеся в четвертом положении бензольного кольца, оказывают менее сильное влияние на кислотные свойства Н2Рс(4-NO2)4(But)4 [22], в результате чего при его взаимодействии с диметилсульфоксидом равновесие (1) ограничивается стадией образования малополярного Н-комплекса, который не вступает в конкурентную реакцию за протон.
Комплексы с переносом протонов тетрапиразинопорфиазинов в отличие от комплексов фталоцианинов гораздо легче подвергаются распаду с течением времени. Так, при переходе от Н2Рс(3-NO2)4(But)4 ⋅ 2DMSO к Н2Ра(ButPyz)4 ⋅ 2DMSO скорость деструкции в присутствии н-бутиламина и пиперидина, судя по величинам k298, возрастает в ∼450 и 160 раз соответственно (табл. 3). При этом дестабилизация комплекса Н2Ра(ButPyz)4 ⋅ ⋅ 2DMSO наблюдается и в менее сильной протоноакцепторной среде (морфолин-DMSO). Замена трет-бутильных групп в пиразиновых фрагментах макроцикла на этильные не оказывает существенного влияния на значения k298 и Еа. Напротив, при переходе от близких по устойчивости Н2Ра(ButPyz)4 ⋅ 2DMSO и Н2Ра(Et2Pyz)4 ⋅ ⋅ 2DMSO к Н2Ра(Ph2Pyz)4 ⋅ 2DMSO скорость деструкции в системе Mor (BuNH2, Et2NH)-DMSO возрастает на фоне уменьшения энергии активации процесса (табл. 3). Фенильные заместители в Н2Ра(Ph2Pyz)4 ⋅ 2DMSO оказывают менее сильное дестабилизирующее влияние на комплекс, чем п-нитрофенильные заместители в Н2Ра(С6Н4NO2)8 ⋅ 2DMSO. Так, под влиянием н-бутиламина значение константы скорости деструкции для Н2Ра(С6Н4NO2)8 ⋅ 2DMSO в диметилсульфоксиде возрастает в ∼8 раз. В системе BuNH2-DMSO распад комплексов Н2Ра(С6Н4NO2)8 ⋅ 2DMSO и Н2Ра(SN2)4 ⋅ 2DMSO характеризуется близкими значениями k298, в то время как в системе Et2NH-DMSO комплекс, образованный с участием тетра(1,2,5-тиадиазоло)порфиразина, не подвергается распаду (табл. 3). Минимальной кинетической устойчивостью в системе азотсодержащее основание-DMSO обладает комплекс Н2Ра(SеN2)4 ⋅ 2DMSO. При переходе от Н2Ра(SN2)4 ⋅ ⋅ 2DMSO к структурно близкому Н2Ра(SеN2)4 ⋅ ⋅ 2DMSO скорость деструкции в присутствии BuNH2, ButNH2, Mor и Pip возрастает в ∼2–3 раза, а Eа процесса уменьшается (табл. 3).
Потеря кинетической устойчивости комплексов с переносом протонов в ряду Н2Рс(But)4 ⋅ ⋅ 2DMSO → Н2Рс(4-NO2)4(But)4 ⋅ 2DMSO → → Н2Рс(3-NO2)4(But)4 ⋅ 2DMSO → Н2Ра(ButPyz)4 ⋅ ⋅ 2DMSO ≈ Н2Ра(Et2Pyz)4 ⋅ 2DMSO → Н2Ра (Ph2Pyz)4 ⋅ 2DMSO → Н2Ра(SN2)4 ⋅ 2DMSO → → Н2Ра(С6Н4NO2)8 ⋅ 2DMSO → Н2Ра(SеN2)4 ⋅ ⋅ 2DMSO связана с увеличением NH-кислотности макроцикла и смещением равновесия (1) в сторону образования более полярного ионного комплекса, который в результате протекания конкурентной реакции за протон (9), (10) переходит в лабильный порфиразиновый дианион, подвергающийся дальнейшей деструкции.
Тетрагалогензамещенные порфиразины обладают близкой протонодонорной способностью (табл. 1), поскольку влияние атомов галогена на кислотные NH-центры передается с полуизолированных Сβ = Сβ-связей по индуктивному (–I) эффекту и за счет эффекта n, π-сопряжения с макроциклом (+М-эффект). –I-эффект при переходе от брома к хлору увеличивается, что способствует росту полярности NH-связей. Действие мезомерного (+М) эффекта, возрастая в том же порядке, напротив способствует уменьшению подвижности протонов NH-групп. В результате электронные эффекты атомов галогенов нивелируют кислотность Н2РаBr4 и Н2РаCl4, а их комплексы с диметилсульфоксидом подвергаются распаду в системе BuNH2 (Et2NH)-DMSO с близкими значениями k298 и Еа процесса (табл. 2).
Достаточно сильное влияние на кинетические параметры деструкции оказывает пространственное строение азотсодержащего основания и его протоноакцепторная способность. Среди циклических оснований максимальная скорость распада Н2Ра(SеN2)4 ⋅ 2DMSO наблюдается в присутствии пиперидина (${\text{p}}K_{{\text{a}}}^{{298}}$ = 11.23 [23]), который является сильным акцептором протона и имеет стерически доступный атом азота в составе молекулы [24]. Введение в пиперидиновый цикл гетероатома кислорода не влияет на пространственное строение амина [25], однако приводит к понижению ${\text{p}}K_{{\text{a}}}^{{298}}$ на ∼2.5 единицы. В результате этого при переходе от пиперидина к морфолину (${\text{p}}K_{{\text{a}}}^{{298}}$ = 8.50 [23]) значение k298 уменьшается в ∼5 раз на фоне незначительного изменения Еа процесса (табл. 3). Уменьшение ${\text{p}}K_{{\text{a}}}^{{298}}$ оснований на ∼6 единиц в ряду Pip → Mor → Py приводит к дальнейшему росту стабильности комплекса Н2Ра(SеN2)4 ⋅ 2DMSO. В системе Py-DMSO он не подвергается деструкции, поскольку пониженная протоноакцепторная способность пиридина (${\text{p}}K_{{\text{a}}}^{{298}}$ = 5.23 [23]) не позволяет ему конкурировать с молекулой DMSO за протон. Аналогичное изменение устойчивости наблюдается для комплексов Н2Ра(SN2)4 ⋅ 2DMSO, Н2Ра(Ph2Pyz)4 ⋅ ⋅ 2DMSO, Н2Ра(Et2Pyz)4 ⋅ 2DMSO, Н2Ра(ButPyz)4 ⋅ ⋅ 2DMSO и Н2Рс(3-NO2)4(But)4 ⋅ 2DMSO (табл. 3).
Объемные алкильные заместители, связанные с атомом азота в основании, также противодействуют процессу деструкции комплексов с переносом протонов, поскольку затрудняет благоприятный контакт взаимодействующих молекул в ходе протекания конкурентной реакции за протон (9), (10). Так, замена н-бутиламина (${\text{p}}K_{{\text{a}}}^{{298}}$ = 10.60 [23]) на близкий по основности трет-бутиламин (${\text{p}}K_{{\text{a}}}^{{298}}$ = 10.68 [23]) приводит к уменьшению значений k298 более, чем в 80 и 120 раз для Н2Ра(SеN2)4 ⋅ 2DMSO и Н2Ра(SN2)4 ⋅ 2DMSO соответственно (табл. 3). Наряду с разветвлением углеводородной цепи, дестабилизации комплексов с переносом протонов противодействует увеличение числа алкильных заместителей в амине. Так, для Н2Ра(SеN2)4 ⋅ 2DMSO и Н2Ра(С6Н4NO2)8 ⋅ ⋅ 2DMSO скорость деструкции под влиянием диэтиламина (${\text{p}}K_{{\text{a}}}^{{298}}$ = 10.84 [23]), судя по значениям k298 (табл. 3), уменьшается в ∼130 и 30 раз соответственно по сравнению с н-бутиламином. Напротив, комплексы образованные с участием тетра (1,2,5-тиадиазоло)порфиразина, тетра(3-нитро-5-трет-бутил)фталоцианина и замещенных тетрапиразинопорфиразина, в системе Et2NH-DMSO не разрушаются с течением времени. Аналогичной устойчивостью обладают комплексы Н2Ра(SN2)4 и Н2Ра(SеN2)4 с диметилсульфоксидом в присутствии триэтиламина (${\text{p}}K_{{\text{a}}}^{{298}}$ = 10.75 [23]) (табл. 3).
Работа выполнена при поддержке гранта ФГБОУ ВО “Ивановский государственный химико-технологический университет” (13-ISUCT-21).
Список литературы
Novakova V., Donzello M.P., Ercolani C. et al. // Coord. Chem. Rev. 2018. V. 361. № 4. P. 1.
Сесслер Дж.Л., Гейл Ф.А., Вон-Сеоб Хо. Химия анионных рецепторов. М.: Красанд, 2011. 456 с.
Stuzhin P.A. // J. Porphyrins Phthalocyanines. 1999. V. 3. № 6–7. P. 500.
Stuzhin P.A. // Ibid. 2003. V. 7. № 12. P. 813.
Петров О.А. // Журн. физ. химии. 2021. Т. 95. № 4. С. 549.
Petrov O.A. // Russ. J. Phys. Chem. 2000. V. 74. Suppl. 3. P. S454.
Петров О.А., Осипова Г.В., Кузмина Е.Л. // Журн. общ. химии. 2011. Т. 81. Вып. 6. С. 1018.
Петров О.А., Осипова Г.В., Майзлиш В.Е., Родионов А.В. // Изв. вузов. Химия и хим. технология. 2014. Т. 57. Вып. 6. С. 54.
Осипова Г.В., Петров О.А., Ефимова С.В. // Журн. общ. химии. 2013. Т. 83. Вып. 3. С. 510.
Петров О.А., Стужин П А., Иванова Ю.Б. // Журн. физ. химии. 2008. Т. 82. № 2. С. 266.
Петров О.А., Киселев А.Н., Сырбу С.А. // Журн. общ. химии. 2014. Т. 84. Вып. 9. С. 1492.
Петров О.А., Киселев А.Н., Сырбу С.А. // Росс. хим. журнал. 2016. Т. 60. № 2. С. 89.
Петров О.А., Киселев А.Н., Телецкий З.А., Беляева А.О. // Журн. общ. химии. 2019. Т. 89. № 3. С. 400.
Петров О.А., Аганичева К.А., Гамов Г.А., Киселев А.Н. // Журн. физ. химии. 2020. Т. 94. № 9. С. 1379.
Кузмина Е.Л., Петров О.А. // Журн. общ. химии. 2012. Т. 82. Вып. 5. С. 850.
Петров О.А., Хелевина О.Г., Чижова Н.В. // Координац. химия. 1997. Т. 23. № 2. С. 143.
Kokareva E.A., Petrov O.A., Khelevina O.G. // Macroheterocycles. 2009. V. 2. № 2. P. 157.
Молекулярные взаимодействия / Под ред. Г. Ратайчака, У. Орвилл-Томаса. М.: Мир, 1984. 599 с.
Zundell G. Hydrogen Bonds with large Proton Polarizability and Transfer Processes in Electrochemistry and Biology / Ed. by I. Prigogin, S.F. Rise. N.Y.: Willy and Sons. Inc. 2000. 217 p.
Райхардт К. Растворители и эффекты среды в органической химии. М.: Мир, 1991. 764 с.
Stuzhin P., Khelevina O., Berezin B. // Phthalocyanines: Properties and Applications. N.Y.: VCH Publ. Inc., 1996. V. 4. P. 23.
Петров О.А., Кузмина Е.Л., Майзлиш В.Е., Родионов А.В. // Журн. физ. химии. 2014. Т. 88. № 1. С. 11.
The Handbook of Chemistry and Physics / Ed. by W.M. Haynes. Boca Raton, London, N.Y.: Taylor and Francis, 2013. 2668 p.
Anet F.A.L., Yavari I. // J. Amer. Chem. Soc. 1977. V. 99. P. 2794.
Blackburne I.D., Katritzky A.R., Takeuchi Y. // Accounts. Chem. Res. 1975. V. 8. № 9. P. 300.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии