

Журнал физической химии, 2022, T. 96, № 5, стр. 739-746
О хемилюминесценции люминола, инициируемой ионами железа(II)
Ю. Б. Цаплев a, *, А. В. Трофимов a, b, **
a Институт биохимической физики им. Н.М. Эмануэля РАН
119334 Москва, Россия
b Московский физико-технический институт
141701 Долгопрудный, Московской области, Россия
* E-mail: tsap_04@mail.ru
** E-mail: avt_2003@mail.ru
Поступила в редакцию 26.10.2021
После доработки 26.10.2021
Принята к публикации 28.10.2021
- EDN: AUPOHJ
- DOI: 10.31857/S0044453722050296
Аннотация
Определены квантовые выходы по люминолу и FeII в хемилюминесцентной реакции люминола, инициированной растворами солей FeII. На основании значений этих величин сделан вывод, что инициация хемилюминесценции вызвана не супероксид-анионом, а более эффективным первичным окислителем. При исследовании тушения хемилюминесценции под действием диметилсульфоксида показано, что гидроксильный радикал также никак не может претендовать на роль первичного окислителя. Установлено, что влияние на хемилюминесценцию хелатирования FeII трилоном-Б не связано с потерей FeIIEDTA инициирующего действия в хемилюминесценции люминола, а обусловлено быстрым превращением в FeIIIEDTA в ходе приготовления смеси FeII с трилоном-Б.
Исследование хемилюминесценции, сопровождающей окисление различных субстратов в щелочной среде в присутствии солей FeII и растворенного кислорода, было начато в работах Михельсона и др. [1–4]. В этих работах среди субстратов окисления – люциферин двухстворчатого моллюска Pholas dactylus (камнеточец обыкновенный) [1, 2], лофин и люминол [3, 4]. При изучении влияния каталазы и супероксиддисмутазы на хемилюминесценцию в системе FeII/O2/люминол выявлено образование и участие в хемилюминесценции супероксид-аниона (${\text{O}}_{2}^{ - }$). Михельсон предположил, что хемилюминесценция люминола происходит в ходе его окисления супероксидом.
Позднее хемилюминесценция люминола под действием супероксид-аниона (как единственного окислителя) была исследована в работе [5]. Было установлено, что квантовый выход хемилюминесценции небольшой, он изменяется от 3 × 10–6 до 6 × 10–6 фотон на молекулу супероксида при изменении pH от 9 до 11. В этой хемилюминесцентной реакции ни растворенный кислород, ни H2O2 не влияют существенно на кинетику и выход света, что является важным отличием от хемилюминесценции в системе FeII/O2/люминол. Квантовый выход хемилюминесценции в системе FeII/O2/люминол не измерен до настоящего времени.
Хемилюминесценция люминола в щелочной среде, инициированная добавкой растворов солей FeII, была применена для высокочувствительного определения железа(II) в воде [6]. При наблюдении хемилюминесценции пробу вводили в проточный реактор непрерывного смешивания. Максимальный хемилюминесцентный отклик регистрировался при pH ⁓ 11. Хемилюминесценция инициируется также катионами CoII, CuII и MnII, но ее интенсивность составляет соответственно 0.3–2, 0.02–0.3 и ⁓0.003% от интенсивности в присутствии FeII [7]. Инструментарий, обеспечивающий хемилюминесцентное определение железа, со времен работы [6] претерпел ряд изменений [8–10]. Сейчас используется проточно-инжекционный вариант введения пробы. В этом случае регистрируемый сигнал сложным образом зависит как от кинетических параметров хемилюминесценции, так и от инструментальных параметров [11], а определение квантового выхода хемилюминесценции в этих условиях, как правило, невозможно. Интересно отметить, что авторы работы [6] отводили ионам FeII роль катализатора окисления люминола кислородом, но, как справедливо замечено в [12], приписывание FeII каталитической роли в этой реакции не только неверно по существу, но и затушевывает незнание действительных механизмов реакции.
Хемилюминесценция люминола возникает в реакциях со многими реагентами. Нитью Ариадны, которая позволяет не заблудиться в лабиринтах хемилюминесценции люминола, служат результаты, полученные Мереньи и его соавт. в исследованиях 1980–1990 годов [см. 13, 14 и ссылки там]. В этих работах была установлена схема световых и темновых превращений люминола после его одноэлектронного окисления. В конкретных случаях инициирования хемилюминесценции требуется установить первичный окислитель люминола и пути перевода кислорода в реакционные формы. Для хемилюминесценции люминола, инициируемой солями FeII, это не сделано до настоящего времени.
С другой стороны, ответ на вопрос о природе первичного окислителя в системе FeII/O2 представляет интерес для понимания механизма окисления липидов, инициированного катионами железа [15–17]. Окисление и липидов в микросомах, и люминола в хемилюминесцентной реакции может быть основано на схожих молекулярных процессах взаимодействия кислорода с железом (II), несмотря на различие условий (нейтральная среда в одном случае и сильнощелочная в другом). Судьба катионов Fe2+ в водных средах (вне связи с хемилюминесценцией и биохимией) – еще одна проблема, которой занимаются, главным образом, геохимики [9, 12, 18–20]. Кроме того, окисление Fe(II) кислородом – важный процесс в технологии производства пигментов и при водоподготовке [21–23], а взаимодействие ионов железа и молекулярного кислорода – центральная проблема для катализаторов на основе оксидов и гидроксидов железа [24–26].
В водных растворах катионы ${\text{Fe}}_{{{\text{aq}}}}^{{2 + }}$ и гидроксиды: FeOH+, Fe(OH)2, Fe(OH)$_{3}^{ - }$ подвергаются окислению растворенным кислородом. Исследования этой реакции были выполнены при pH < 9, т.е. вне области максимального инициирующего эффекта железа(II) в хемилюминесцентной реакции. Константы скорости окисления железа(II) возрастают в ряду ${\text{Fe}}_{{{\text{aq}}}}^{{2 + }}$ ≪ FeOH+ ≪ Fe(OH)2, увеличиваясь на 5 порядков при переходе от одного члена ряда к другому [18]. В области pH 8–10 константа скорости первого порядка для окисления железа(II) d(ln[Fe(II)]/dt) принимает максимальное значение, равное 0.05 с–1 (в морской воде соленостью 3.2% при температуре 15–25°C и парциальном давлении O2 1 бар) [20]. Константа скорости понижается до 0.01 с–1 при парциальном давлении O2 0.2 бар, время полупревращения в этом случае составляет 70 с, что в ⁓100 раз превышает длительность наблюдаемой хемилюминесценции.
В области pH > 10.8 превалирующей формой железа(II) является Fe(OH)$_{3}^{ - }$ [18]. Можно предполагать, что скорость окисления при больших pH определяется скоростью окисления кислородом именно этой формы:
(1)
${\text{Fe}}({\text{OH}})_{3}^{ - } + {{{\text{O}}}_{2}} \leftrightarrow {\text{Fe}}{{({\text{OH}})}_{3}} + {\text{O}}_{2}^{ - }.$Задачи настоящего исследования – определить частные квантовые выходы хемилюминесценции по железу и люминолу; изучить влияние комплексообразования на выход света в реакции; изучить эффекты диметилсульфоксида (ДМСО), считающегося нередко специфическим перехватчиком гидроксильных радикалов, на наблюдаемую хемилюминесценцию.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали следующие реактивы: NaOH, КОН “х.ч.”, люминол (95%, AppliChem), FeSO4 ⋅ 5H2O (“ч.д.а.”), FeCl3 ⋅ 6H2O (“ч”). Люминол (L) для использования в работе очищали по методике, описанной в [29].
Кинетику хемилюминесценции регистрировали посредством двухканального хемилюминометра на основе фотоприемных модулей H7360-1 и H7360-2 (Hamamatsu, Япония) и одноканального хемилюминометра на основе модуля H7360-1 со счетчиками импульсов CNT-202 (Спецприбор, Беларусь) и компьютера. Двухканальная люминометрическая система регистрировала хемилюминесценцию в двух спектральных каналах: в полосе 430–490 нм и в полосе 480–560 нм. Схема двухканального хемилюминометра описана в [30].
Спектральное отношение при двухканальной спектрально-кинетической регистрации хемилюминесценции для конкретного эмиттера хемилюминесценции и конкретного регистрирующего прибора – вполне определенная величина, равная: $\frac{{\int {S(\lambda ){{P}_{{\text{B}}}}(\lambda ){{F}_{{\text{B}}}}(\lambda )d\lambda } }}{{\int {S(\lambda ){{P}_{{\text{G}}}}(\lambda ){{F}_{{\text{G}}}}(\lambda )d\lambda } }}$, где S(λ) – спектр испускания эмиттера, а PB(λ), PG(λ) и FB(λ), FG(λ) – спектральные чувствительности фотоприемников и спектры пропускания светофильтров в B- и G-каналах регистрации соответственно. Для двухканального люминометра, используемого в нашей лаборатории, спектральное отношение равно 1.9 ± 0.2 при регистрации хемилюминесценции люминола в присутствии гемина и пероксида водорода.
Квантометрическую калибровку хемилюминометров выполняли, используя “люминольный стандарт” по Ли и Селигеру [31], при величине квантового выхода хемилюминесценции по люминолу 0.012.
Реакторами для проведения реакции служили полипропиленовые микропробирки объемом 2 мл, в которых смешивали растворы реагентов. Для смешивания реагентов порцию раствора FeSO4 инжектировали в щелочной раствор люминола посредством светоизолированного дозатора.
Исходные растворы FII (FeSO4, FeCl2) и их последующие разведения подкисляли соляной кислотой до pH 3 с целью предотвращения гидролиза.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Общая характеристика хемилюминесценции
Хемилюминесценция, наблюдаемая при смешивании щелочного раствора люминола с раствором соли FeII, является люминольной хемилюминесценцией. Об этом свидетельствует спектр испускаемого света, соответствующий спектру хемилюминесценции люминола в других системах, в частности тех, в которых Ли и Селигер измеряли квантовый выход хемилюминесценции люминола (люминол/H2O2/гемин, люминол/H2O2/пероксидаза при pH > 12). На нашем двухканальном люминометре это видно в каждом опыте, поскольку отношение выхода света в синем и зеленом спектральных каналах во всех опытах постоянно и равно 1.9 ± 0.2.
Хемилюминесцентная реакция протекает быстро. Половина света испускается за 0.3–2 с после смешивания реагентов, а вся реакция заканчивается за время от 1 до нескольких секунд.
Зависимости выхода света (светосуммы Σ) от концентрации люминола (рис. 1) и FeII (рис. 2) однотипны: Σ линейно увеличивается с ростом концентрации, а затем перестает расти. Верхний концентрационный предел линейной зависимости для Σ([L]) соответствует концентрации [L] ⁓ ⁓ 0.3 мкМ, а для зависимости Σ([FeII]) соответствует концентрации [FeII] = 3 мкМ. Время испускания половины светосуммы (t0.5) не зависит от концентрации люминола, для данных, представленных на рис. 1, оно равно 1.3 ± 0.2 с. Зависимость t0.5 от концентрации FeII показана на рис. 2.
Рис. 1.
Зависимости выхода света от концентрации люминола. Условия: концентрация NaOH 0.8 мМ, FeSO4 – 30 мкМ, объем реакционной смеси 0.5 мл.

Рис. 2.
Концентрационные зависимости выхода света (1) и времени испускания половины светосуммы (2) от концентрации FeII. Условия: концентрация NaOH – 0.8 мМ, люминола – 4 мкМ. Объем реакционной смеси 0.5 мл.

Линейные участки зависимостей Σ([L]) и Σ([FeII]) позволяют найти частные квантовые выходы света по люминолу и FeII. Из данных, представленных на рис. 1 и 2, частные квантовые выходы света по люминолу, ∂Σ([L])/∂[L] = $\Phi _{{\text{L}}}^{{{\text{app}}}}$ = = (9 ± 1) × 10–5 Эйнштейн/моль, и по FeII, ∂Σ([FeII])/∂[FeII] = ΦFe = (1.2 ± 0.2) × 10–5 Эйнштейн/моль. В случае люминола в обозначение $\Phi _{{\text{L}}}^{{{\text{app}}}}$ добавлен индекс “app”, для того, чтобы подчеркнуть, что это кажущаяся величина, поскольку нет полного расходования люминола в реакции. Даже при начальной концентрации люминола 1 нМ и избытке FeII над люминолом в 30000 раз нет полного расходования люминола, что проявляется в возобновлении хемилюминесценции при повторном добавлении порции раствора FeII.
Величины и $\Phi _{{\text{L}}}^{{{\text{app}}}}$, и ΦFe значительно превышают квантовый выход хемилюминесценции в системе люминол/супероксид, определенный в работе [5], что указывает на образование в исследуемой реакции первичного окислителя (POx) люминола, значительно более эффективного, чем супероксид.
Для полного “сжигания” люминола требуется добавление нескольких порций раствора FeII. Суммированием Σ в серии таких добавлений при концентрации люминола 0.5 нМ был получен квантовый выход по люминолу (ΦL), равный 2 × 10–3 Эйнштейн/моль. Такая величина типична для хемилюминесценции люминола под действием окислителя в отсутствие пероксида водорода в концентрации более 0.1 мМ [31], например, для ХЛ под действием NaOCl.
Уравнение хемилюминесцентной реакции, учитывающее различие ΦFe и $\Phi _{{\text{L}}}^{{{\text{app}}}}$, имеет вид:
В отдельных опытах мы установили, что в щелочном растворе катионы Fe2+ быстро теряют способность инициировать хемилюминесценцию люминола. Спустя 10 с пребывания в 1 мМ растворе щелочи инициирующая способность пропадает практически полностью. Таким образом, частицы POx, способные инициировать хемилюминесценцию люминола, появляются при попадании катионов Fe2+ в щелочной раствор и существуют непродолжительное время, длительность которого сравнима с длительностью наблюдаемой хемилюминесценции.
Верхний концентрационный предел линейной зависимости Σ([L]) (рис. 1) можно объяснить тем, что при концентрации люминола более 0.3 мкМ все активные частицы, образующиеся из FeII, перехватываются люминолом до их гибели. Пусть время жизни частиц POx равно τ, тогда в присутствии люминола возникает конкуренция между взаимодействием POx с люминолом с бимолекулярной константой скорости kPOx и процессом их гибели. В этом случае выход света при концентрации люминола, равной [L], пропорционален величине kPOx[L]/(1/τ + kPOx[L]).
Существование верхнего концентрационного предела у линейной зависимости Σ([FeII]) было бы легко объяснить тем, что при [FeII] > 3 мкМ весь имеющийся в реакционной смеси люминол окисляется, однако этого, как уже подчеркнуто выше, не происходит. Верхний концентрационный предел не связан также и с перегрузкой фотоприемника, поскольку при работе с высокоинтенсивными сигналами между фотоприемником и реакционным сосудом помещали нейтральный светофильтр, который ослаблял световой поток. Верхний концентрационный предел не обусловлен и ограничением реакции каким-либо другим реагентом, например, растворенным кислородом или щелочью, поскольку их концентрация много больше концентраций FeII или люминола.
Возможной причиной существования верхнего концентрационного предела линейной зависимости Σ([FeII]) при условии отсутствия полного окисления люминола является взаимодействие POx c FeII:
Влияние ДМСО
Специфичность взаимодействия ДМСО с гидроксильными радикалами в настоящее время возможна, вероятно, только в ряду активных форм кислорода. Установлено, что даже в реакции Фентона, в которой, как известно, при низких pH генерируется гидроксилный радикал, в нейтральных средах первичным окислителем ДМСО служит не гидроксильный радикал, а формы железа(IV) [32].
ДМСО – конкурентный тушитель хемилюминесценции люминола, инициируемой FeII (рис. 3). Концентрация ДМСО, при которой выход света уменьшается в 2 раза, зависит от концентрации люминола. Величина [ДМСО]0.5 увеличивается от 7.3 мкМ до 3.3 мМ при увеличении начальной концентрации люминола от 0.01 до 10 мкМ, что свидетельствует о наличии конкуренции между люминолом и ДМСО за первичный окислитель (POx). Конкуренция между реакциями (L + POx) с константой скорости kLum, и (ДМСО + POx) с константой скорости kDMSO, при kDMSO[ДМСО]0.5 = = kLum[L] приводит к уменьшению выхода света в 2 раза, при этом [ДМСО]0.5/[L] = kLum/kDMSO.
Рис. 3.
Влияние ДМСО на выход света. Условия: концентрация люминола равна 0.01 (1) и 10 (2) мкМ, [Fe(II)] = 30 (1) и 0.1 мкМ(2), концентрация NaOH – 0.8 мМ.

Из данных, представленных на рис. 3, следует, что отношение kLum/kDMSO равно 300–700, т.е. много больше 1. Это справедливо и для растворов, в которых определенно образуются коллоидные формы гидроксидов железа (линия 1, концентрация железа(II) – 30 мкМ), и в растворах, в которых нет образования коллоидных форм (линия 2, концентрация железа(II) – 0.1 мкМ). В коллоидообразующих растворах отношение kLum/kDMSO возрастает. Причина этого, вероятно, в том, что взаимодействие аниона люминола и первичного окислителя происходит на поверхности коллоидной частицы по месту образования первичного окислителя. Однако и в растворах с низкой концентрацией железа(II) отношение kLum/kDMSO > 300.
Гидроксильный радикал часто выдвигается на роль первичного окислителя в рассматриваемой хемилюминесцентной реакции. Константа скорости реакции ДМСО + ${\text{H}}{{{\text{O}}}^{ \bullet }}$ равна 5 × 109 М–1 с–1 [33], а константа скорости реакции аниона люминола с ${\text{H}}{{{\text{O}}}^{ \bullet }}$ равна 8.7 × 109 М–1 с–1 [34], и, значит, отношение констант скоростей ⁓2, что ≪300. Из чего следует, что гидроксильный радикал никак не может быть первичным окислителем в рассматриваемой хемилюминесцентной реакции.
Влияние ЭДТА
Хемилюминесценцию люминола, инициируемую FeII, наблюдают при смешивании щелочного раствора люминола с раствором соли FeII, поэтому влияние ЭДТА на хемилюминесцентную реакцию можно изучать двумя способами: либо вводя ЭДТА в раствор c FeII, либо – в раствор c люминолом. Первый способ используют при апробации методик обнаружения железа в природных водах, имитируя наличие в пробе агентов, хелатирующих железо. Второй способ не исследован. Мы выполнили такое исследование, и оказалось, что влияние ЭДТА на хемилюминесценцию в этих случаях разное.
Введении ЭДТА в раствор c FeII при отношении [ЭДТА]/[Fe(II)] > 1 снижает на несколько порядков количества света, испускаемого в реакции с щелочным раствором люминола. На рис. 4a показаны кинетические кривые хемилюминесценции, наблюдаемой при некоторых значениях этого отношения. Там же на вставке показана зависимость светосуммы от отношения [ЭДТА]/ [Fe(II)]. Тушащее действие хелатирующих агентов таких, как этилендиамин, глицин, тартрат, 2.4-пентандион отмечено в [6], а для ЭДТА – в [35], и было интерпретировано [6, 12, 35] как исчезновение каталитического действия FeII после хелатирования.
Рис. 4.
Кинетические профили ХЛ и зависимость светосуммы от отношения [ЭДТА]/[Fe(II)] при добавлении 50 мкл смеси растворов FeSO4 и трилона-Б к 450 мкл раствора люминола (0.11 мкМ) в 1.8 мМ KOH (а) и при добавлении 50 мкл раствора FeSO4 к 450 мкл раствора люминола и трилона-Б в KOH (б). Числа при кинетических кривых обозначают величину отношения [ЭДТА]/[Fe(II)], при котором получена данная кривая. Во всех опытах концентрация FeSO4 1 мкМ.
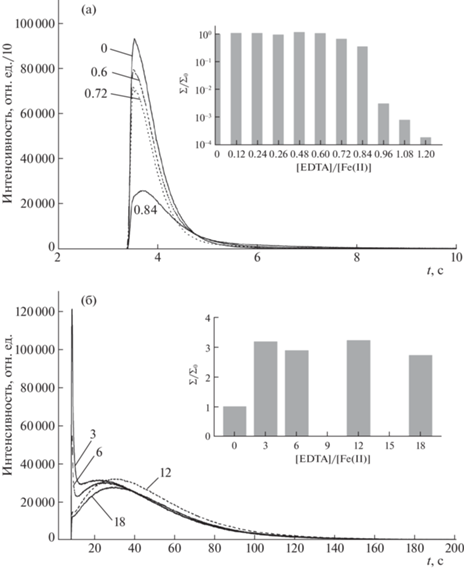
Такая интерпретация находится в явном противоречии с результатами, полученными при втором способе введения ЭДТА в систему, которые показаны на рис. 4б. В этом случае кинетика свечения по мере увеличения отношения [ЭДТА]/[Fe(II)] меняется от импульсной на более плавную. Длительность свечения возрастает до 100 с. Максимальная интенсивность хемилюминесценции уменьшается более чем в 30 раз, но выход света в реакции не только не уменьшается, а возрастает. При 12-кратном превышении концентрации ЭДТА над концентрацией Fe(II) на кинетических кривых хемилюминесценции все еще заметен импульсный выброс, наблюдаемый сразу после смешивания реагентов, но при большем отношении [ЭДТА]/[Fe(II)] он уже отсутствует.
Естественно задать вопрос: в чем причина разительного отличия эффектов ЭДТА в разных схемах его введения в реакционную смесь? Прежде всего, нужно признать, что полное тушение хемилюминесценции при отношении [ЭДТА]/[FeII] > 1 в первом случае не связано с отсутствием у FeIIEDTA инициирующего действия в хемилюминесценции люминола. Действительная причина тушения состоит в отсутствии самого FeIIEDTA в растворе. При добавлении ЭДТА к раствору соли FeII происходят быстрое образование комплекса FeIIEDTA и его окисление кислородом в FeIIIEDTA. Реакция (${\text{F}}{{{\text{e}}}^{{{\text{II}}}}}{\text{EDTA}}\;\xrightarrow{{{\text{ }}{{{\text{O}}}_{{\text{2}}}}}}\;{\text{F}}{{{\text{e}}}^{{{\text{III}}}}}{\text{EDTA}}$) подробно исследована в [36, 37]. Время полуреакции при pH 2–7 составляет всего несколько секунд, поэтому за время приготовления смеси FeII с ЭДТА реакция успевает закончиться. При нейтральных pH реакция протекает по нерадикальному механизму с восстановлением O2 до H2O2. Пероксид водорода нетрудно обнаружить по хемилюминесценции люминола в присутствии гемина [38]. И, действительно, добавление смеси растворов FeII и ЭДТА к раствору люминола и гемина в щелочной среде инициирует хемилюминесценцию, кинетический профиль которой схож с профилем хемилюминесценции в присутствии H2O2.
Во второй схеме введения ЭДТА ион железа поступает в реакционную смесь в форме ${\text{Fe}}_{{{\text{aq}}}}^{{2 + }}$, поэтому в данном случае конкурентно образуются и гидроксокомплексы железа(II), и комплексы FeIIЭДТА. Выход гидроксокомплексов железа(II) снижается с увеличением концентрации ЭДТА. Но и при высокой концентрации ЭДТА происходит инициирование хемилюминесценции, причем с большим выходом света, и, следовательно, ни о какой потери инициирующего действия FeIIEDTA не может быть и речи.
Железо(II) окисляется растворенным кислородом независимо от состава образующихся комплексов, однако лабильные интермедиаты окисления разные. Очевидно, что быстропротекающая импульсная хемилюминесценция инициируется первичным окислителем, который образуется при окислении гидроксокомплексов железа(II). Возможно, что первичным окислителем люминола являются лабильные формы железа(IV), но не гидроксильный радикал или супероксид-анион.
Список литературы
Henry J.P., Isambert M.F., Michelson A.M. // Biochim. Biophys. Acta. Bioenergetics. 1970. V. 205. № 3. P. 437. https://doi.org/10.1016/0005-2728(70)90109-X
Henry J.P., Michelson A.M. // Ibid. 1970. V. 205. № 3. P. 451. https://doi.org/10.1016/0005-2728(70)90110-6
Michelson A.M. // Biochimie. 1973. V. 55. № 4. P. 465. https://doi.org/10.1016/S0300-9084(73)80213-5
Michelson A.M. // Ibid. 1973. V. 55. № 8. P. 925. https://doi.org/10.1016/S0300-9084(73)80170-1
Merenyi G., Lind J., Eriksen T.E. // Photochem. Photobiol. 1985. V. 41. № 2. P. 203. https://doi.org/10.1111/j.1751-1097.1985.tb03472.x
Seitz W.R., Hercules D.M. // Anal. Chem. 1972. V. 44. № 13. P. 2143. https://doi.org/10.1021/ac60321a020
Klopf L.L., Nieman T.A. // Ibid. 1983. V. 55. № 7. P. 1080. https://doi.org/10.1021/ac00258a023
O'Sullivan D.W., Hanson A.K. (Jr.), Kester D.R. // Mar. Chem. 1995. V. 49. № 1. P. 65. https://doi.org/10.1016/0304-4203(94)00046-G
King D.W., Lounsbury H.A., Millero F.J. // Environ. Sci. Technol. 1995. V. 29. № 3. P. 818. https://doi.org/10.1021/es00003a033
Hopwood M.J., Birchill A.J., Gledhill M. et al. // Front. Mar. Sci. 2017. V. 4. P. 192. https://doi.org/10.3389/fmars.2017.00192
Цаплев Ю.Б. // Журн. аналит. химии. 2004. Т. 59. № 2. С. 182. Tsaplev Y.B. // J. Anal. Chem. 2004. V. 59. № 2. P. 160.
Rose A.L., Waite T.D. // Anal. Chem. 2001. V. 73. № 24. P. 5909. https://doi.org/10.1021/ac015547q
Merenyi G., Lind J., Shen X. et al. // J. Phys. Chem. 1990. V. 94. № 2. P. 748. https://doi.org/10.1021/j100365a043
Merenyi G., Lind J., Eriksen T.E. // J. Biolum. Chemilum. 1990. V. 5. № 1. P. 53. https://doi.org/10.1002/bio.1170050111
Владимиров Ю.А., Арчаков А.И. Перекисное окисление липидов в биологических мембранах. М.: Наука, 1972. 252 с.
Welch K.D., Davis T.Z., Aust S.D. // Arch. Biochem. Biophys. 2002. V. 397. № 2. P. 360. https://doi.org/10.1006/abbi.2001.2694
Welch K.D., Davis T.Z., Van Eden M.E. et al. // Free Rad. Biol. Med. 2020. V. 32. № 7. P. 577. https://doi.org/10.1016/S0891-5849(02)00760-8
Morgan B., Lahav O. // Chemosphere. 2007. V. 68. № 11. P. 2080. https://doi.org/10.1016/j.chemosphere.2007.02.015
Jones A.M., Griffin P.J., Waite T.D. // Geochim. Cosmochim. Acta. 2015. V. 160. P. 117. https://doi.org/10.1016/j.gca.2015.03.026
Roekens E.J., Van Grieken R. // Mar. Chem. 1983. V. 13. № 3. P. 195. https://doi.org/10.1016/0304-4203(83)90014-2
Prasad T.P., Ramasastry V.V. // J. Appl. Chem. Biotech. 1974. V. 24. № 12. P. 769. https://doi.org/10.1002/jctb.5020241208
Prasad T.P., Ramasastry V.V. // Ibid. 1977. V. 27. № 2. P. 409. https://doi.org/10.1002/jctb.5020270207
Охлопкова Л.Б., Кузнецова Н.И. //Кинетика и катализ. 2008. Т. 49. № 1. С. 38. Okhlopkova L.B., Kuznetsova N.I. // Kinet. Catal. 2008. V. 49. № 1. P. 34. https://doi.org/10.1134/S0023158408010059
Сычев А.Я., Исак В.Г. // Успехи химии. 1995. Т. 64. № 12. С. 1183. Sychev A.Y., Isak V.G. // Rus. Chem. Rev. 1995. V. 64. № 12. P. 1105. https://doi.org/10.1070/RC1995v064n12ABEH000195
Shubin A.A., Ruzankin S.P., Zilberberg I.L., Parmon V.N. // Chem. Phys. Lett. 2015. V. 640. P. 94. https://doi.org/10.1016/j.cplett.2015.10.016
Чикунов А.С., Таран О.П., Шубин А.А. et al. // Кинетика и катализ. 2018. Т. 59. № 1. С. 29. Chikunov A.S., Taran O.P., Shubin A.A. et al. // Kinet. Catal. 2018. V. 59. № 1. P. 23. https://doi.org/10.1134/S0023158418010032
Лилич Л.С., Хрипун М.К. Растворы в неорганической химии. Л.: Изд. ЛГУ, 1984. 160 с.
Armstrong D.A., Huie R.E., Koppenol W.H. et al. // Pure Appl. Chem. 2015. V. 87. № 11–12. P. 1139. https://doi.org/10.1515/irm-2013-0005
Воробьева Т.П., Козлов Ю.Н., Колтыпин Ю.В. et al. // Изв. АН СССР. Сер. хим. 1976. № 10. С. 2187.
Цаплев Ю.Б., Васильев Р.Ф., Кънчева В.Д., Трофимов А.В. // Хим. физика. 2020. Т. 39. № 6. С. 7. Tsaplev Y.B., Vasil’ev R.F., Kancheva V.D., Trofimov A.V. // Russ. J. Phys. Chem. В. 2020. V. 14. № 3. P. 431. https://doi.org/10.1134/S1990793120030252
Lee J., Seliger H.H. // Photochem. Photobiol. 1972. V. 15. № 2. P. 227. https://doi.org/10.1111/j.1751-1097.1972.tb06241.x
Bataineh H., Pestovsky O., Bakac A. // Chem. Sci. 2012. V. 3. № 5. P. 1594. https://doi.org/10.1039/C2SC20099F
Czapski G. // Israel J. Chem. 1984. V. 24. № 1. P. 29. https://doi.org/10.1002/ijch.198400005
Baxendale J.H. // J. Chem. Soc. Faraday Trans. 1. 1973. V. 69. № 9. P. 1665. https://doi.org/10.1039/F19736901665
Bottu G. // J. Biolum. Chemilum. 1991. V. 6. № 3. P. 147. https://doi.org/10.1002/bio.1170060303
Пурмаль А.П., Скурлатов Ю.И., Травин С.О. // Изв. АН СССР. Сер. хим. 1980. № 3. С. 492. Purmal’ A.P., Skurlatov Y.I., Travin S.O. // Russ. Chem. Bull. 1980. V. 29. P. 315. https://doi.org/10.1007/BF00949607
Травин С.О., Скурлатов Ю.И. // Журн. физ. химии. 1981. Т. 55. № 6. С. 1453. Travin S.O., Skurlatov Y.L. // Russ. J. Phys. Chem. 1981. V. 55. P. 815.
Цаплев Ю.Б. // Журн. аналит. химии. 2012. Т. 67. № 6. С. 564. Tsaplev Y.B. // J. Anal. Chem. 2012. V. 67. № 6. P. 506. https://doi.org/10.1134/S1061934812040028
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии