

Физика и химия стекла, 2020, T. 46, № 2, стр. 213-216
Зарядовые состояния сорбционных центров в Матримиде
А. В. Петров 1, *, А. М. Тойкка 1
1 Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Университетская наб., 7/9, Россия
* E-mail: a.petrov@spbu.ru
Поступила в редакцию 11.11.19
После доработки 13.11.19
Принята к публикации 05.12.19
Аннотация
Рассмотрены особенности электронной структуры Матримида, важные для оценки сорбционной способности указанного полимера как мембранного материала. Методом теории функционала плотности рассчитаны зарядовые состояния сорбционных центров в Матримиде. Исследованы наиболее вероятные атомы полимера, ответственные за невалентные взаимодействия полярных молекул газов или жидкостей при прохождении через мембраны на основе данного полимера.
Матримид (Матримид®5218) является известным стеклообразным термопластом из класса полиимидов, обладающим достаточно высокой температурой стеклования (в форме полимерных пленок выше ~550 K) [1]. Это определило его широкое применение как мембранного материала для газоразделения и первапорации.
Известны, в частности, мембраны на основе Матримида для разделения промышленно значимых газов [2, 3], водно-органических и органических смесей [4, 5]. В указанных работах авторы ограничиваются преимущественно общей констатацией эффективности мембранного материала, без оценки сорбционных характеристик в ходе диффузии пенетранта в теле мембраны. Подобная информация существенна не только для анализа особенностей взаимодействия “полимер–пенетрант”, но и для прогнозирования хода мембранного разделения в целом, в частности, для оценки коэффициентов диффузии. В первую очередь, важна характеристика электростатических взаимодействий, которые вносят наибольший вклад в невалентные связи и, соответственно, влияют на сорбционные свойства. При прохождении молекул растворителя или газа через мембрану влияние активных сорбционных центров молекул полимера является одним из главных факторов, определяющих диффузию пенетрантов. С целью оценки этого влияния в нашей работе исследованы зарядовые состояния сорбционных центров в молекуле Матримида. Эти задачи решались с использованием различных функционалов, применяемых в методе ТФП. Влияние растворителя на величины зарядов оценивалось в рамках модели COSMOS.
В соответствии с проведенным анализом литературных данных, квантово-химические расчёты полиимидов, выполненные с различными приближениями, хорошо объясняют физико-химические свойства: ультрафиолетовые фотоэлектронные спектры [6], оптические абсорбционные спектры [7], фотоэмиссионные спектры [8], конформационные свойства [9], тонкая структура плотности состояний валентной зоны [10], адгезия меди и никеля [11], ИК-спектр [12], энергетика больших деформаций [13], диэлектрическая постоянная и энергетическая щель [14]. Однако, изучению зарядовых состояний атомов в полиимидах и, в частности, в Матримиде, практически не уделяется внимание. В то же время, такая информация представляется полезной и значимой при оценке механизмов сорбции полярных молекул в Матримиде в ходе мембранного разделения.
Атомы кислорода и азота в составе молекулы Матримида (детальный фрагмент молекулы представлен на рис. 1) являются сорбционными центрами: это связано с электростатическим взаимодействием с полярными молекулами диффундирующего флюида. Заряды на этих центрах формируются в результате распределения электронной плотности в молекуле Матримида. Неоднородность такого распределения оценивается по схеме Малликена [15] для относительного сравнения зарядовых состояний, выполненных в рамках одной методики.
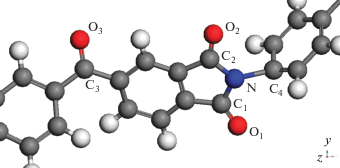
Для расчeтов электронной структуры мономера Матримида с полной оптимизацией геометрии применялся метод ТФП, реализованный на атомном базисе DNP (v. 4.4) с функционалами PBE [16], PW91 [17] и HCTH [18] в программе DMol3 [19] из программного пакета Materials Studio. Как видно из результатов расчётов в вакууме, представленных в таблице, максимальный отрицательный заряд имеют атомы кислорода O1 и O2, которые смещают на себя электронную плотность связанных с ними атомов углерода C1 и C2. Таким образом, атомы C1 и C2 имеют положительные заряды, близкие к атомам кислорода по абсолютной величине. Значительный отрицательный заряд имеет атом кислорода O3, который не компенсирует положительный заряд ближайшего атома углерода C3. Атом азота также имеет значительный отрицательный заряд, но меньший в сравнении с атомами кислорода.
Оценку взаимодействия полярных жидкостей с полимером проводили на основе континуальной модели окружающей среды COSMOS. Рассматривали конкретные жидкости – воду и метанол. Расчeты показали, что распределение электронной плотности, по сравнению с вакуумом, меняется таким образом, что отрицательные заряды атомов кислорода увеличиваются (по абсолютной величине), а заряд атома азота уменьшается (см. табл. 1). Наибольшие отрицательные заряды имеют атомы кислорода, что определяет их преимущественное влияние на сорбцию полярных молекул в ходе диффузии.
Сравнительный анализ применения различных функционалов обобщeнной градиентной аппроксимации отражает сохранение общих тенденций в расчетах зарядовых состояний сорбционных центров Матримида, как в вакууме, так и в полярных жидкостях, что говорит об устойчивости распределения электронной плотности в полимере. Полученные данные позволяют утверждать, что именно рассмотренные сорбционные центры должны оказывать существенное влияние на диффузию полярных флюидов в непористой полимерной мембране.
Работа выполнена при финансовой поддержке Российского научного фонда (РНФ), грант № 16-13-10164.
Таблица 1.
Заряды сорбционных центров в Матримиде (в единицах заряда электрона)
| Функционал | N | O1, 2 | O3 | C1, 2 | C3 | С4 |
|---|---|---|---|---|---|---|
| Вакуум | ||||||
| PBE | –0.303 | –0.386 –0.391 |
–0.380 | 0.388 0.386 |
0.343 | 0.195 |
| PW91 | –0.305 | –0.391 –0.387 |
–0.380 | 0.386 0.389 |
0.336 | 0.185 |
| HCTH | –0.337 | –0.393 –0.397 |
–0.380 | 0.394 0.388 |
0.318 | 0.212 |
| Вода | ||||||
| PBE | –0.289 | –0.442 –0.441 |
–0.435 | 0.408 0.405 |
0.361 | 0.179 |
| PW91 | –0.293 | –0.442 –0.440 |
–0.435 | 0.407 0.405 |
0.353 | 0.171 |
| HCTH | –0.317 | –0.451 –0.450 |
–0.436 | 0.413 0.407 |
0.340 | 0.194 |
| Метанол | ||||||
| PBE | –0.290 | –0.439 –0.440 |
–0.432 | 0.407 0.405 |
0.361 | 0.180 |
| PW91 | –0.294 | –0.439 –0.439 |
–0.433 | 0.407 0.404 |
0.353 | 0.172 |
| HCTH | –0.318 | –0.450 –0.448 |
–0.435 | 0.413 0.406 |
0.339 | 0.195 |
Список литературы
Konnertz N., Bohning M., Schonhals A. Dielectric investigations of nanocomposites based on Matrimid and Polyhedral Oligomeric Phenethyl-Silsesquioxanes (POSS) // Polymer. 2016. Vol. 90.P. 89. https://doi.org/10.1016/j.polymer.2016.02.060
Castro-Muñoz R., Martin-Gil V., Ahmad M.Z., Fíla V. Matrimid® 5218 in preparation of membranes for gas separation: Current state-of-the-art // Chem. Eng. Comm. 2018. V. 205(2). P. 161. https://doi.org/10.1080/00986445.2017.1378647
Aziz F., Ismail A.F. Preparation and characterization of cross-linked Matrimid® membranes using para-phenylenediamine for O2/N2 separation // Sep. Purif. Technol. 2010. V. 73. P. 421. https://doi.org/10.1016/j.seppur.2010.05.002
Castro-Muñoz R., Galiano F., Fíla V., Drioli E., Figoli A. Matrimid®5218 dense membrane for the separation of azeotropic MeOHMTBE mixtures by pervaporation // Sep. Purif. Technol. 2018. 199. P. 27. https://doi.org/10.1016/j.seppur.2018.01.045
Jiang L.Y., Chung T.-S., Rajagopalan R. Dehydration of alcohols by pervaporation through polyimide Matrimid asymmetric hollow fibers with various modifications // Chem. Eng. Sci. 2008. V. 63. P. 204. https://doi.org/10.1016/j.ces.2007.09.026
Brédas J.L., Clarke T. C. Electronic structure of polyimide // J. Chem. Phys. 1987. V. 86. P. 253. https://doi.org/10.1063/1.452615
LaFemina J.P., Arjavalingam G., Hougham G. Electronic structure and ultraviolet absorption spectrum of polyimide // J. Chem. Phys. 1989. V. 90. P. 5154. https://doi.org/10.1063/1.456558
Meyer III H.M., Wagner T.J., Weaver J.H., Feyereisen M.W., Almlof J. Photoemission, inverse photoemission, and ab initio SCF investigations of the electronic structure of polyimide // Chem. Phys. Lett. 1989. V. 164. P. 527. https://doi.org/10.1016/0009-2614(89)85251-0
Kafafi S.A., LaFemina J.P., Nauss J.L. Electronic Structure and Conformation of Polymers from Cluster Molecular Orbital and Molecular Mechanics Calculations: Polyimide // J. Am. Chem. Soc. 1990. V. 112. P. 8742. https://doi.org/10.1021/ja00180a015
Kowalczyk S.P., Stafström S., Brédas J.L., Salaneck W.R., Jordan-Sweet J.L. Electronic structure of polyimide and related monomers: Theory and experiment // Phys. Rev. B. V. 41. 1990. P. 1645. https://doi.org/10.1103/PhysRevB.41.1645
Zhang J., Sullivan M.B., Zheng J.W., Loh K.P., Wu P. Theoretical Study on Polyimide-Cu(100)/Ni(100) Adhesion // Chem. Mater. 2006. V. 18. P. 5312. https://doi.org/10.1021/cm052865n
Nishikawa Y., Nakano Y., Noda I. Two-Dimensional Correlation Analysis of Polyimide Films using Attenuated Total Reflection-Based Dynamic Compression Modulation Step-Scan Fourier Transform Infrared Spectroscopy // Appl. Spectroscopy. 2007. V. 61. P.873. https://doi.org/10.1366/000370207781540015
Fujinami A., Ogata S., Kimizuka H., Shibutani Y. The Energetics of Large Deformations of a Single Polyimide Molecular Chain: DFT and MO Calculations // Macromol. Theory Simul. 2008. V. 17. P. 488. https://doi.org/10.1002/mats.200800058
Ma R., Baldwin A.F., Wang C., Offenbach I., Cakmak M., Ramprasad R., Sotzing G.A. Rationally Designed Polyimides for High Energy Density Capacitor Applications // Appl. Mater. Interfaces. 2014. V. 6. P. 10445. https://doi.org/10.1021/am502002v
Mulliken R.S. Electronic population analysis on LCAO-MO molecular wave functions. I and Electronic population analysis on LCAO-MO molecular wave functions. II. Overlap populations, bond orders, and covalent bond energies // J. Chem. Phys., 1955. V. 23. P. 1833. https://doi.org/10.1063/1.1740588,10.1063/1.1740589
Perdew J.P., Burke K., Ernzerhof M. Generalized Gradient Approximation Made Simple // Phys. Rev. Lett., 1996. V. 77. P. 3865. https://doi.org/10.1103/PhysRevLett.77.3865
Perdew J.P., Wang Y. Generalized Gradient Approximation Made Simple // Phys. Rev. B. 1992. V. 45. P. 13244. https://doi.org/10.1103/PhysRevB.45.13244
Boese A.D., Handy N.C. A new parametrization of exchange–correlation generalized gradient approximation functionals // J. Chem. Phys. 2001. V. 114. P. 5497. https://doi.org/10.1063/1.1347371
Delley B. An all-electron numerical method for solving the local density functional for polyatomic molecules // J. Chem. Phys. V. 92. P. 508. https://doi.org/10.1063/1.458452
Дополнительные материалы отсутствуют.
Инструменты
Физика и химия стекла