

Физика и химия стекла, 2022, T. 48, № 3, стр. 272-282
Влияние воды на зарождение кристаллов пирофосфата олова в оловоцинковофосфатном стекле
Г. А. Сычева 1, *, Т. Г. Костырева 1
1 Институт химии силикатов им. И.В. Гребенщикова РАН
199034 Санкт-Петербург, наб. Макарова, 2, Россия
* E-mail: sycheva_galina@mail.ru
Поступила в редакцию 22.07.2021
После доработки 31.12.2021
Принята к публикации 07.02.2022
- EDN: VYJMOK
- DOI: 10.31857/S0132665122030106
Аннотация
Синтезированы стекла в системе SnO–ZnO–P2O5. Проведено определение химического состава стекла с помощью мокрой химии с применением комплексонометрического, гравиметрического и редокс методов. Методом проявления изучено зарождение кристаллов пирофосфата олова в оловоцинковофосфатном стекле. Определены параметры зарождения кристаллов пирофосфата олова: стационарная скорость зарождения кристаллов и время нестационарного зарождения. Установлено влияние воды, примесей WO3 и MoO3 и рентгеновского излучения на параметры зарождения.
ВВЕДЕНИЕ
Стекла и стеклокристаллические материалы, содержащие элементы переменной валентности, играют важную роль в науке и технике. Многие из них обладают уникальными оптическими и электрическими свойствами, улучшенными прочностными характеристиками, химической и коррозионной стойкостью, повышенной каталитической активностью и т.д. Обоснованием технологии стеклокристаллических материалов является изучение закономерностей гомогенного и гетерогенного зарождения кристаллов в стеклах различного химического состава в зависимости от времени и температуры [1]. Влиять на зарождение кристаллов можно путем введения в стекло нуклеационных агентов, снижающих поверхностное натяжение σ на границе расплав–зародыш. Из экспериментальных исследований Аппена [2] известно, что такие оксиды, как WО3, V2О5, МоО3, обладают сильным эффектом снижения поверхностного натяжения щелочных силикатных расплавов, что может привести к изменению значения σ на границе зародыш–стекло. Влиянию на кристаллизацию литиевосиликатных стекол пентаоксидов фосфора, ванадия и элементов VВ и VIB подгрупп таблицы Менделеева посвящена работа [3]. Действие концентрации тербия на свойства фосфатного стекла изучено в [4]. Фосфатные стекла еще в начале 60-х гг. ХХ в. привлекли внимание исследователей как возможные матрицы для захоронения радиоактивных (РАО) и высокоактивных (ВАО) отходов. Их преимуществами были признаны низкая температура плавления и высокая растворимость химических соединений, находящихся в составе отходов [5–7]. В настоящей работе исследовано влияние воды, примесей WO3 и MoO3 и рентгеновского излучения на параметры зарождения кристаллов пирофосфата олова.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Характеристика стекол исходных составов. Стекла синтезировали из фосфорной кислоты (Н3РО4), углекислого цинка и диоксида олова. МоО3 вводили в шихту в виде молибденовой кислоты [8], а WО3 — в виде моновольфрамовой кислоты. Все реактивы имели марку “чда”. Шихту высушивали и затем перемешивали в шаровой мельнице в течение 10 ч. Варку стекол проводили в платиновом тигле при температуре 1170°С в течение 2 ч с автоматическим перемешиванием 30 мин. Стекло вырабатывали быстрым охлаждением расплава на массивную металлическую изложницу.
Химический анализ стекол. При проведении химического анализа использовали классические методы анализа: комплексонометрический, гравиметрический и редокс методы [9–11]. Предварительно было установлено, что данные стекла при нагревании легко растворяются в хлористоводородной кислоте, а олово полностью окисляется до Sn(IV). Для определения содержания компонентов навеску образца растворяли в 10 мл HCl и полученный раствор осторожно (во избежание полимеризации олова) разбавляли в мерной колбе на 250 мл 0.5 н раствором HCl. В аликвотных порциях этого раствора определяли валовое содержание олова, фосфора и цинка. Для определения олова использовали комплексонометрический метод титрования [9]. Предварительные испытания показали, что в присутствии фосфора, для полного протекания реакции комплексонообразования необходимо кипячение анализируемой аликвотной порции (аликвотная часть раствора – объем раствора, номинально необходимый для проведения анализа по конкретной методике, кратный объему раствора) с избытком комплексона III (другие названия: трилон Б, хелатон III – динатриевая соль этилендиаминтетрауксусной кислоты, представляющая собой белый кристаллический порошок или кристаллы белого цвета, хорошо растворимые в воде и щелочах и очень слабо растворяется в спирте, образующие устойчивые комплексные соединения с большинством катионов). Цинк титровали комплексонoм III при рН 10 [10]. Точность метода ±0.5% отн.
При определении фосфора использовали метод количественного химического анализа, основанный на точном измерении массы вещества, – гравиметрический метод [11].
Свойства стекол в значительной мере зависят от степени окисления элементов, входящих в состав материалов. При разработке метода анализа валентного состояния компонентов в первую очередь приходится решать вопрос о выборе растворителя и устойчивости той или иной степени окисления элемента. В тех случаях, когда определяемая валентная форма неустойчива при вскрытии навески пробы, растворение проводят в присутствии другого элемента, устойчивого в этих условиях и способного эквивалентно восстановить или окислить определяемый элемент. При этом реакция должна протекать количественно и быть избирательной по отношению к определяемому компоненту. Скорость реакции должна быть достаточно высокой по сравнению со скоростью протекания возможных побочных процессов.
Двухвалентное олово является сильным восстановителем, его растворы легко окисляются на воздухе. Известно, что олово эквивалентно окисляется трeхвалентным железом, при этом обе валентные формы железа достаточно устойчивы в условиях кислотного разложения материалов. В связи с этим для селективного определения SnO была разработана следующая методика. В коническую колбочку помещали 1 г углекислого натрия (для создания инертной атмосферы), затем вносили навеску стекла, добавляли 5 мл ~ 0.012 М раствора трехвалентного железа и 10 мл HCl (1 : 1). Колбочку закрывали конусной воронкой и нагревали в течение 5 мин. Не удаляя воронки, охлаждали, ополаскивали воронку водой, добавляли 5 мл (1 : 1), 1 мл 0.01% раствора дифениламиносульфоната натрия и титровали раствором бихромата калия до появления фиолетовой окраски. Содержание двухвалентного олова рассчитывали по количеству образовавшегося Fe(II). Предлагаемая методика очень проста в исполнении, не требует специального оборудования (в т. ч. баллона с инертным газом). На одно определение затрачивается 15–20 мин. Точность метода ±0.3 отн. %. Составы исследованных стекол приведены в табл. 1.
Таблица 1.
Составы стекол, мол. %
| Номер образца |
Оксид | Цвет стекла |
|||||
|---|---|---|---|---|---|---|---|
| SnO | SnO2 | P2O5 | ZnO | WO3* | MoO3* | ||
| по анализу | по анализу | по анализу | по синтезу сверх 100 | ||||
| 1 | 45.0 | 1.5 | 33.5 | 20.0 | – | Бесцветное | |
| 2 | 43.2 | 3.5 | 33.2 | 20.1 | 0.5 | – | Голубое |
| 3 | 43.6 | 3.1 | 33.1 | 20.2 | 2.0 | – | Синее |
| 4 | 42.6 | 3.6 | 33.3 | 20.5 | 0.5 | Бежевое | |
| 5 | 43.3 | 2.5 | 33.5 | 20.7 | – | 2.0 | Коричневое |
Кинетика кристаллизации исследуемых стекол. Поверхностное зарождение. Объемное зарождение кристаллов наблюдали только в стекле 1. В составах 2–5 зарождение кристаллов происходило только с поверхности образцов. При температурах 280–470°С во всех изученных стеклах, согласно рентгенофазовому анализу (РФА), зарождались и росли кристаллы пирофосфата олова. Был использован дифрактометр ДРОН-2, излучение СuKα, условия съемки: Сu-антикатод, Ni-фильтр, 30 кВ, 20 мА. Индицирование рентгенограмм выполнялось при помощи программы PDWin 3.0, разработанной НПП “Буревестник”.
Рентгенограммы исходных (до термообработки) стекол без примесей и с примесями WО3 и МоО3 приведены на рис. 1а. На рис. 1б представлены дифрактограммы стекла 2 (табл. 1) на разных стадиях термообработки.
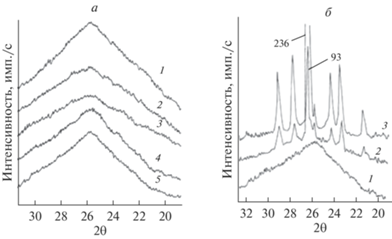
Введение примесей как WО3, так и МоО3 препятствует объемному зарождения кристаллов. Кристаллизация в стеклах 2–5 идет только с поверхности образцов. На рис. 2 приведен внешний вид кристаллической корочки пирофосфата олова в образце после термообработки при 400°С в течение 456 ч.
Рис. 2.
Внешний вид кристаллической корочки пирофосфата олова в образце после термообработки при 400°С в течение 456 ч.

Скорость роста кристаллической корки в стекле без примесей (1) при Т = 400°С равна 17.4 × 10–5 мм/мин, при Т = 450°С – 43.4 × 10–5 мм/мин.
Кинетика кристаллизации исследуемых стекол. Объемное зарождение. В стекле 1 кристаллы пирофосфата олова зарождались не только на поверхности стекла и росли внутрь образца в виде игл, но и в объеме. Для определения числа зародившихся кристаллов в объеме использовали метод проявления, предложенный Тамманом при исследовании кристаллизации органических веществ и получивший развитие в исследованиях гомогенного и гетерогенного зарождения кристаллов в стеклах [12]. Суть метода заключается в выращивании при температуре проявления Тпр до видимых размеров кристаллов, зародившихся при более низкой температуре. Тпр выбирается такой, чтобы скорость роста при ней была бы достаточно высокой, а скоростью зарождения можно было бы пренебречь. В качестве Тпр выбрали 420°С. При Тпр кристаллы пирофосфата олова в стекле без примесей росли в виде сферолитов. Образцы стекла просматривали на микроскопе “Jenaval” в проходящем свете. Стационарную скорость зарождения Icт(Т) рассчитывали как тангенс угла наклона линейного участка зависимости n(t), индукционный период tинд определяли как точку пересечения его продолжения с осью времени. Максимальная скорость зарождения 15.6 × 10–4 мм–3 мин–1 наблюдалась при Т = 400°С.
Результаты количественного рентгенофазового анализа. В силу отсутствия объемного зарождения кристаллов в стеклах с примесями WО3 и МоО3 и малых значений скорости зарождения пирофосфата олова процесс кристаллизации в стеклах с примесями изучали с помощью рентгенофазового количественного анализа [13]. Оценку количества образовавшейся кристаллической фазы проводили, используя эталон (максимально закристаллизованный при данной температуре образец) и данные о высоте самого яркого дифракционного пика, определяемого для Sn2P2О7 при угле рассеяния 2θ = 26°. В качестве величины, характеризующей степень закристаллизованности, использовали отношение α = V(Т, t)/Vmax, где V(Т, t) – объем образовавшейся фазы пирофосфата олова при температуре Т в момент времени t, а Vmax – объем, соответствующий полной длительности эксперимента [14].
На рис. 3 приведены зависимости степени закристаллизованности α от количества примеси WО3 и МоО3 при изотермической выдержке в течение фиксированного времени при температурах 400 и 450°С (рис. 3).
Рис. 3.
Зависимость степени закристаллизованности α от количества примеси в стекле, выдержанном при 400°С. Время выдержки, ч: – 144 (I), 456 (II), 620 (III), 933 (IV). Примеси WО3 (1 – кружочки) и МоО3 (2 – треугольники).

Из рис. 3 видно, что α уменьшается с увеличением количества примеси WО3 и МоО3. Для 400°С при количестве примеси RО3, равном 1.6–1.65 мол. %, степень закристаллизованности α одинакова независимо от вида примеси.
В табл. 2 приведены значения степени закристаллизованности α для различных температур и времен термообработок стекол 1–5. Из табл. 2 видно, что α максимальна для стекла без примесей и уменьшается с увеличением количества примесей во всем температурном интервале.
Таблица 2.
Степень закристаллизованности α для различных температур и времени термообработки стекла
| Т, °С | t, ч | α для стекол | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| 390 | 144 | 0.030 | 0.025 | 0.020 | 0.015 | 0.010 |
| 456 | 0.260 | 0.100 | 0.040 | 0.020 | 0.015 | |
| 620 | 0.990 | 0.300 | 0.080 | 0.050 | 0.020 | |
| 933 | 1.000 | 0.620 | 0.260 | 0.110 | 0.025 | |
| 400 | 20 | 0.010 | 0 | 0 | 0 | 0 |
| 150 | 0.013 | 0 | 0 | 0 | 0 | |
| 180 | 0.015 | 0.010 | 0 | 0 | 0 | |
| 300 | 0.100 | 0.030 | 0.020 | 0 | 0 | |
| 450 | 0.250 | 0.085 | 0.070 | 0.050 | 0 | |
| 620 | 0.980 | 0.220 | 0.130 | 0.080 | 0.015 | |
| 960 | 1.000 | 0.750 | 0.270 | 0.120 | 0.040 | |
| 450 | 40 | 1.000 | 0.400 | 0.200 | 0.100 | 0.020 |
| 140 | 1.000 | 0.800 | 0.400 | 0.250 | 0.150 | |
| 190 | 1.000 | 1.000 | 0.500 | 0.350 | 0.180 | |
| 470 | 1 | 0.200 | 0.006 | 0 | 0 | 0 |
| 2 | 0.340 | 0.130 | 0.020 | 0 | 0 | |
| 3 | 0.530 | 0.180 | 0.080 | 0.020 | 0 | |
| 10 | 0.770 | 0.540 | 0.420 | 0.180 | 0.100 | |
| 17.5 | 1.000 | 0.920 | 0.830 | 0.360 | 0.220 | |
| 500 | 1 | 0.500 | 0.400 | 0.300 | 0.200 | 0.100 |
| 1.5 | 0.750 | 0.650 | 0.500 | 0.300 | 0.200 | |
| 2 | 1.000 | 0.950 | 0.900 | 0.850 | 0.800 | |
Действие рентгеновского облучения на зарождение кристаллов пирофосфата олова. При изучении эффекта уменьшения степени закристаллизованности от времени изотермической выдержки при температуре 500°С обнаружено, что этот эффект более сильно проявился для образца стекла 1, подвергшегося рентгеновскому облучению. При визуальном осмотре образцов обнаружено, что участки стекла, не подвергшиеся облучению, закристаллизовались, в то время как облученная часть образцов оставалась прозрачной после выдержки образцов при 400°С в течение 2300 ч и проявлении при 420°С в течение 10 мин. Оцененная скорость зарождения кристаллов в облученной зоне на один порядок ниже по сравнению со скоростью в необлученной зоне (рис. 4). В данном случае следует говорить о подавлении центров кристаллизации излучением, т.е. об аморфизирующем действии рентгеновского излучения на зарождение кристаллов.
Рис. 4.
Зависимость числа n проявленных кристаллов пирофосфата олова от глубины образца необлученного (1) и облученного в течение 10 мин (2). Кривая 3 иллюстрирует ослабление излучения СuKα при прохождении через образец стекла 1, Δ = 0.013 мм – слой половинного ослабления излучения.

Таблица 3.
Зависимость содержания Н2О, мол. %, от количества водяного пара, продуваемого через расплав за единицу времени
| Н2О, мл/ч | 30 | 100 | 100 | 125 | 125 | 200 | 200 | 250 |
|---|---|---|---|---|---|---|---|---|
| Н2О, мол. % | 0.26 | 0.33 | 0.33 | 0.37 | 0.45 | 0.45 | 0.47 | 0.89 |
Аналогичное явление было обнаружено и при изучении зарождения кристаллов в натриевоцинковофосфатном стекле в системе Na2O–ZnO–P2O5 и SnO–ZnO–P2O5 в стекле близкого состава.11
Влияние воды на зарождение кристаллов пирофосфата олова. Определение содержания воды в пирофосфатных стеклах с помощью химического анализа. Определение содержания воды в твердых телах, являющееся важным для практики, продолжает оставаться одной из наиболее сложных проблем аналитической химии. Это обусловлено многообразием способов, которыми вода может быть связана с матрицей исследуемого вещества. А именно, вода может быть химически связана с матрицей, удерживаться мощными адсорбционными силами, или быть “свободной”. Независимо от выбранного аналитического метода определить, в какой конкретной форме находится вода в исследуемом веществе, трудно. Для фосфатных стекол используется метод, основанный на термическом разложении образца с последующим кулонометрическим измерением содержания воды. В наших экспериментах использовали автоматический кулонометрическиий измеритель влажности газа “Байкал-3” с непрерывной регистрацией. Действие этого прибора основано на адсорбции водяного пара фосфорным ангидридом с последующим электролитическим разложением адсорбированной воды. Индикатором служит стеклянная трубка, содержащая спиральные платиновые электроды, с напыленной между ними пленкой пятиокиси фосфора. Напряжение источника постоянного тока, присоединенного к электродам, выше потенциала разложения воды. Ток электролиза, пропорциональный количеству разлагаемой воды, регистрируется самописцем. Установка включает высокотемпературную электрическую печь, трубку из кварцевого стекла, измеритель влажности газа “Байкал-3”, снабженный самописцем, и осушительную систему, использующую концентрированную серную кислоту. Вода, выделенная из исследуемого образца при термообработке, переносится потоком гелия в измеритель влажности и подвергается электролизу. Содержание воды в исследуемом образце рассчитывается по площади под пиком, регистрируемым самописцем. Используя построенные калибровочные кривые с помощью веществ с известным содержанием воды (например, кристаллогидрата хлорида бария), получаем значения содержания воды в образце стекла. Интервал измерений составляет 10–500 микрограммов H2O, что соответствует содержанию воды 0.01–0.95 в 100-граммовом образце. Через 2 ч плавки при 1170°С в расплаве остается ~0.26 мас. % Н2О, через 19 ч – 0.13%. Полностью удалить воду из расплава не удается. Подъем температуры при этом производится медленно во избежание вспучивания шихты. Фриттированную таким образом шихту измельчали и загружали в тигель для плавки. Плавку шихты производили при возможно быстром подъеме температуры. Длительный процесс плавки шихты, а также выдержка расплава в печи более 2 ч приводили к интенсивному улетучиванию компонентов стекломассы, особенно оксида фосфора.
Для дальнейших исследований были подготовлены образцы стекла 1 (табл. 1) 45.0SnO∙1.5SnO2∙33.5P2O5∙20.0ZnO. Воду вводили в образцы путем продувания водяного пара через расплав стекла (барботированием расплава). Расплавы стекол отливали в форму размерами 5 × 25 × 25 мм, что позволяло получать достаточно большие отливки для приготовления образцов как для исследования кристаллизационных свойств, так и для химического анализа. Для снятия термического напряжения проводили отжиг стекол. Режимы насыщения водой полученных образцов отличались друг от друга количеством водяного пара, продуваемого через расплав за единицу времени. Это количество находилось в пределах от 30 дo 250 мл воды в ч.
Под микроскопом были обнаружены как полые пузыри, так и пузыри, захватившие в процессе барботирования расплава различные атмосферные газы – активные пузыри. Для того чтобы иметь возможность выяснить влияние воды на зарождение кристаллов пирофосфата олова, мы прибегли к некоторым методическим приемам [15, 16]. Синтез стекол провели в кварцевых тиглях. Кварцевый тигель с имеющимся в нем стеклорасплавом из варочной печи помещали в муфельную печь для отжига с целью снятия имеющихся в стекле напряжений, чтобы предотвратить растрескивание образцов стекол в процессе последующей механической обработки стекла. Из охлажденного тигля затем алмазной пилой вырезали образец в форме параллелепипеда высотой 110 мм. После этого проводили подготовку образцов для изучения образовавшейся в них пузырьковой структуры.
Из образца данного синтеза были получены 14 пластин с расстоянием между ними 4 мм. В результате механической обработки были получены пластины стекла толщиной 4 мм, пригодные для изучения образовавшейся в них пузырьковой структуры. Первый слой в отличие от всех остальных шлифовали и полировали только с нижней стороны. Верхняя сторона слоя прозрачна. Она образуется в процессе синтеза стекла и представляет собой границу между стеклом и атмосферой воздуха. В этом образце, помимо пузырей, находящихся внутри объема образца, можно наблюдать единичные пузыри, вышедшие на поверхность стекла. В табл. 4 приведены положение середины i-го (li) слоя, глубины залегания слоев (hi) и плотности числа пузырьков Ni·мм–3 в каждом hi слое образца, синтезированного при температуре 1170°С в течение 2 ч. Отсчет глубины залегания слоя производили от поверхностного слоя стекла вглубь образца. Порядок числа пузырей в образцах, синтезированных в конкретных температурно-временных условиях, воспроизводили от опыта к опыту.
Таблица 4.
Характеристика слоев стекла для подсчета пузырьковой структуры после синтеза при Т = 1170°С, t = 2 ч
| № i | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| li, мм | 2 | 10 | 18 | 26 | 34 | 42 | 50 | 58 | 66 | 74 | 82 | 90 | 98 | 106 |
| hi, мм | 0–4 | 8–12 | 16–20 | 24–28 | 32–36 | 40–44 | 48–52 | 56–60 | 64–68 | 72–76 | 80–84 | 88–92 | 96–100 | 104–108 |
Затем с помощью оптического микроскопа JENAVAL в проходящем свете определили размеры всех пузырей в конкретном i-м слое образца и построили распределения пузырей по размерам. На рис. 5 представлены распределения пузырей по размерам для стекла 1, синтезированного при температуре 1170°С в течение 2 ч. Величина N на рис. 5 соответствует доле пузырьков, приходящейся на интервал, расположенный вблизи данного размера. Распределения, приведенные на рис. 5 имеют вид несимметричного максимума. Несимметричность максимума по данным [15] обуславливается однонаправленным процессом формирования пузырьковой структуры, в основном только их слиянием. Как видно из рис. 5, максимальное количество пузырьков наблюдали в нижних слоях стекла, с увеличением подъема слоя количество пузырей уменьшается, а их размеры увеличиваются.
Рис. 5.
Гистограммы распределения пузырей по размерам для образцов стекла, синтезированного при 1170°С в течение 2 ч для различных значений глубины залегания слоя от поверхности вглубь образца hi, (мм): 0–4 (а), 8–12 (б), 16–20 (в), 24–28 (г), 32–36 (д), 40–44 (е), 48–52 (ж); суммарное распределение числа пузырьков – 1 и распределение активных (содержащих воду) пузырьков – 2 (з).

На рис. 5з приведены гистограммы пузырей, образовавшихся в стекле 1. Гистограмма 1 представляет собой суммарное распределение пузырей по размерам в объеме параллелепипеда, а гистограмма 2 представляет собой суммарное распределение “активных” пузырей.22
ЗАКЛЮЧЕНИЕ
Исследовано влияние воды, примесей WO3 и MoO3 и рентгеновского излучения на зарождение кристаллов пирофосфата олова в системе SnO–SnO2–ZnO–P2O5. Показано, что в стекле без примесей зарождение кристаллов происходит в объеме. Определены параметры объемного зарождения кристаллов пирофосфата олова: температурные зависимости стационарной скорости зарождения и времени нестационарного зарождения. Под действием облучения стационарная скорость зарождения уменьшается на порядок. Введение примесей препятствует объемному зарождению кристаллов. Кристаллизация идет только с поверхности образцов стекол. При малых временах выдержки существует определенное количество примеси (1.65 мол. %), при котором степень закристаллизованности не зависит от вида примеси. Облучение оказывает ингибирующее (уменьшающее кристаллизационную активность) влияние облучения на зарождение кристаллов пирофосфата олова. Барботирование расплава стекла парами воды приводит к увеличению скорости зарождения пирофосфата олова за счет появления катализированного парами воды дополнительного числа активных пузырей.
Работа выполнена в рамках государственного задания ИХС РАН при поддержке Минобрнауки России (тема № АААА-А19-119022290088-8).
КОНФЛИКТ ИНТЕРЕСОВ
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Gurzow I., Toshev S. The kinetics of nucleation and the formation of glass-ceramic materials // Advances in nucleation and crystallization in glasses. Columbus, Ohio: Amer. Ceram. soc., 1971. 290 p.
Аппен А.А. Химия стекла. Л.: Химия, 1974. 351 с.
Harper H., McMillan P.V. The formation of glass-ceramic microstructures // Phys. Chem. Glasses. 1972. V. 13. № 4. P. 97–101.
Babkina A., Kulpina E., Sgibnev Y., Fedorov Y., Starobov A., Palashov O., Niconorov N., Ignatiev A., Zyryanova K., Oreshkina K., Zhizhin E., Pudikov D. Terbium concentration effect on magneto-optical properties of ternary phosphate glass // Optical Materials. 2020. V. 100. P. 109692.
Брежнева Н.Е., Озиранер С.Г., Минаев А.А., Кузнецов Д.Г. Свойства фосфатных и силикатных стекол для отверждения радиоактивных отходов // Management of Radioactive Wastes from the Nuclear Fuel Cycle. Vienna: IAEA. 1976. V. 2. P. 85–94.
Sales B.C., Boatner L.A. Lead-Iron Phosphate Glass // Radioactive Waste Forms for the Future. Amsterdam: Elsevier Science Publishers B.Y., 1988. P. 193–231.
Stefanovsky S.V., Ivanov I.A., Gulin A.N. Aluminophosphate Glasses with High Sulfate Content // Mat. Res. Soc. Symp. Proc. 1995. V. 353. P. 101–106.
Schlesinger M.E., Lynch D.C. Effect of VB and VIB oxides on nucleation parameters in lithium disilicate glass // J. Non-Crystalline Solids. 1989. V. 108. № 3. P. 237–248.
Пирютко М.М., Костырева Т.Г. Изучение поведения олова в условиях его определения в минеральных объектах // Журн. аналитической химии. 1982. Т. 37. № 9. С. 1644–1647.
Schwarzenbach G., Flaschka H. The complexometric Titration. Stuttgart: Ferdinand Enke Verlag. 1965. 360 p.
Пирютко М.М., Голубева Л.Г. Определение фосфора в силикатах фосфорномолибденовохинолиновым методом // Заводская лаборатория. 1966. Т. 32. № 8. С. 927–928.
Тамман Г. Стеклообразное состояние. М., Л.: ОНТИ Главная редакция общетехнической литературы. 1953. 136с.
Китайгородский И.И., Павлушкин Н.М., Ходаковская Р.Я. Исследование возможности применения количественного рентгенофазового анализа к стеклокристаллическим материалам // Изв. АН СССР. Неорган. Матер. 1966. Т. 2. №4. С. 726–737.
Зевин Л.С., Завьялова Л.Л. Количественный рентгенографический фазовый анализ. М.: Недра. 1974. 184 с.
Васнецова В.Б., Шардаков Н.Т., Кудяков В.Я., Дерябин В.А. Формирование пузырьковой структуры в пленках стеклообразующих расплавов // Физика и химия стекла. 1999. Т. 25. № 3. С. 373–377.
Дополнительные материалы отсутствуют.
Инструменты
Физика и химия стекла