

Физика плазмы, 2020, T. 46, № 9, стр. 783-829
Спектральные методы контроля примесей, их потоков и локализации в неравновесной низкотемпературной плазме пониженного давления
А. В. Бернацкий a, *, И. В. Кочетов a, b, **, В. Н. Очкин a, ***
a Физический институт им. П.Н. Лебедева РАН
Москва, Россия
b “Троицкий институт инновационных и термоядерных исследований”
Москва, Россия
* E-mail: bernatskiyav@lebedev.ru
** E-mail: kochet@triniti.ru
*** E-mail: ochkin@sci.lebedev.ru
Поступила в редакцию 28.02.2020
После доработки 26.03.2020
Принята к публикации 26.03.2020
Аннотация
Обсуждается состояние исследований процессов с участием малых фракций частиц, образующихся в результате химических превращений исходного газа либо появившихся извне как примеси. Рассматриваются различные высокочувствительные методы измерений малых концентраций частиц. Основное внимание уделяется спектральным методам, как классическим, так и лазерным. Выделяются основные современные области применений неравновесной плазмы, особенности и достижения используемых в них диагностических средств. Акцент делается на спектроскопические исследования чистоты плазмы в низкотемпературных пристеночных слоях термоядерных реакторов. В этом случае особое значение принимает вопрос о проникновении молекул воды в плазменную камеру из контуров охлаждения. Ввиду ограниченной доступности к реакторам развиваются новые дистанционные версии спектральных эмиссионных методов, ориентированных на диагностику пристеночной плазмы ИТЭР. Обсуждается новый метод мультиспектральной оптической актинометрии, в котором роль актинометров могут выполнять, в том числе, промежуточные химически нестабильные частицы. Это расширяет круг возможностей для количественных измерений концентраций частиц различных сортов, повышает чувствительность. Для рассматриваемых объектов найдены эффективные актинометрические пары, например, атомы водорода и дейтерия. Схемы проработаны на лабораторных установках, развиты кинетические модели плазмохимических процессов. Масштабирование результатов модельных экспериментов позволяет прогнозировать измерение потоков примесей на уровне (10–9–10–13) Па · м3 с–1 с локализацией источников, определяемой пространственным разрешением оптической системы.
1. ВВЕДЕНИЕ
По оценке, более 90% известного в природе вещества находится в состоянии плазмы [1], являясь, естественно, одним из самых распространенных и традиционных объектов исследований и применений. Значительная доля приходится на низкотемпературную плазму (НТП) с частичной ионизацией. Долгое время основные применения связывались и продолжают связываться с нагревом при горении или прохождении тока через газовые среды (световые источники, резка, сварка, обработка, закалка материалов, анализ состава вещества и пр.). В последние десятилетия центр тяжести заметно сместился в те области применений, где плазменные эффекты не ограничиваются тепловыми и проявляют селективные свойства. Это стимулировано задачами интерпретации и формирования линейчатых спектров, развития лазерной техники, реализации направленных химических реакций и потребовало более детального описания состояний плазмы [2, 3].
Формы и проявления НТП классифицируются, в первую очередь, по отношению к состоянию ее равновесия [4–6]. При термодинамическом равновесии (ТР) плазма замкнута, однородна и единственный параметр – температура – полностью описывает ее свойства, включая состав и распределения энергий нейтральных и заряженных химических частиц вещества. В этом состоянии она, однако, практически не встречается, в основном из-за нарушения условия замкнутости. Различные приближения к ТР описываются моделями: локального (ЛТР) и частично локального (ЧЛТР) термодинамического равновесия, корональной (МКР), столкновительно-радиационной (СРМ) и др. [4–8]. В них обычно сохраняется основной параметр – температура Т, хотя бы в отношении отдельных сортов частиц, их состояний, степеней свободы и пр.: кинетическая (поступательная, газовая) для нейтральных частиц Tt, электронная Te, колебательная TV, вращательная TR, температура возбуждения электронных уровней Texc, ионизационная Ti. Несмотря на условность, такие модели полезны, поскольку, в частности, позволяют рассматривать определенные больцман-максвелловские ансамбли частиц с помощью парциальных температур, хотя для плазмы в целом таких ансамблей может быть и несколько. Подобные неравновесные модели рассматривают распределение частиц по энергиям, но о предложениях относительно моделей химического состава, в какой-то мере восполняющих детально-равновесные аррениусовы правила, не известно.
Наряду с развитием аналитических моделей, в современной практике основным становится кинетический подход с возможно полным учетом частиц всех сортов, их состояний и взаимодействий. Трудность связана с большим числом процессов. В книге [9] описана концепция ограничения числа реакций по принципу исключения менее вероятных и значимых. Привлечение, начиная с 1970-х годов, вычислительных средств со специальными пакетами программ и накопление экспериментальных данных поддерживают такую логику. Проблема концепции, однако, состоит в выработке критериев полноты отбора процессов из числа таковых в реальном объекте. Описание кинетики в форме балансных уравнений предполагает также наличие информации о скоростях процессов, если и известных, то с погрешностями, и часто трудно оценить надежность результатов расчета в целом. Например, в обзоре [10] рассматривается детальная схема уровневой кинетики для интерпретации спектра излучения разряда в азоте при пониженном давлении. Используются данные о скоростях реакций с участием более 500 уровней. При этом число связанных состояний только в N2 порядка 2 × 104, и полноту описания состояния плазмы оценить сложно. Важны сведения о химическом составе плазмы, который существенно отличается от газов, из которых она образована.
В данной статье мы рассмотрим задачи, связанные с малыми составляющими в неравновесной плазме. В дополнение к указанным трудностям, некоторые могут возникать не только из-за химических превращений исходных газов, но и за счет их конечной чистоты, выделений из конструкционных элементов, появления извне через дефекты стенок. В этих случаях важен вопрос не только о наличии и количестве частиц, но и об источниках, их локализации, потоках эмитированных частиц и их превращениях. При анализе процессов и, как упоминалось, вынужденном ограничении их числа, иногда используется соображение, что некоторые допустимо исключить, если на промежуточных этапах число реагирующих частиц в них оказывается малым. Известно, однако, много подтвержденных на практике случаев, когда частицы в малых количествах играют важную роль. Например, радикалы ОН (~1013 см–3) замедляют разложение молекул СО2 в разряде, и это сказывается на работе СО2 лазеров [11]. В СО лазерах передача энергии от колебательно-возбужденных молекул СО к электронно-возбужденным молекулам CN (~1012 см–3) приводит к радиационному тушению лазерных уровней [12], наличие атомов кислорода О (~1014 см–3) влияет на цепочку превращений рабочих молекул СО [12, 13], а горение атомов углерода С (~(1013–1014) cм–3) дает вклад в заселение лазерных уровней [14]. Появление малых (~1010 см–3) количеств молекул Н2О в пристеночном слое плазменного крупного ядерного реактора нарушает режим его работы [15].
Во многих случаях именно малые составляющие и радикалы, в частности, определяют результаты целевых применений плазмы. Атомы F, Cl (~(1010–1014) см–3) и радикалы HFx, HClx (~(109–1012) cм–3) играют ключевую роль в технологиях изготовления элементов электроники [16–20]. Малым фракциям радикалов углеводородов принадлежит решающая роль в различных химических производствах и экологии [21]. Соотношение количеств радикалов С, Н, СF3 в плазме, зависящее от состава газа и энерговклада, влияет на морфологию роста нанотрубок, графена [22], алмазоподобных пленок [23].
Как правило, в стационарной плазме самостоятельного разряда отступления от равновесия возрастают с понижением давления плазмообразующих газов из-за замедления релаксационных процессов при относительно невысокой кинетической температуре нейтральной составляющей в объеме и участия в реакциях на поверхности. В импульсном режиме, однако, глубокие нарушения равновесия также могут наблюдаться и при атмосферных, и при более высоких давлениях. Это используется, в частности, в мощных импульсных газовых лазерах с планарными разрядами и поперечном пробое. Нередко необходимый результат достигается тем, что в плазме происходит возбуждение и наработка частиц, которые потом вносятся в отделенную от плазмы среду. Из наиболее известных примеров можно отметить создание первых мощных молекулярных лазеров при смешении колебательно-возбужденных в ВЧ-разряде молекул N2 с потоком молекул СО2, СО [11]. В йодно-кислородных лазерах генерация возникает при смешении паров йода с потоком метастабильного кислорода О2(1Δ) из газового разряда, и в этом случае важную роль играют приемы для управления энергией электронов [24]. Большой интерес в последние годы проявляется к плазменным объектам, в которых газ непрерывно прокачивается под повышенным давлением через разряд малых размеров (типично – миллиметры) и последующим расширением в сопле или свободном пространстве. При этом на расстоянии в несколько сантиметров струя имеет кинетическую температуру, близкую к комнатной, но ее состав радикально отличен от исходного, содержит колебательно- и электронно-возбужденные частицы, радикалы. В таком состоянии струи обладают выраженными бактерицидными свойствами, изучаются и применяются в интересах медико-биологических технологий и обработке пищевых продуктов с минимальными тепловыми эффектами [25–27]. В процедурах типа травления материалов расширение происходит в вакуум. Для некоторых других применений в обработке поверхностей вклад энергии в разряд увеличивают, а неравновесная плазменная струя на тех же стадиях истечения имеет кинетическую температуру ~1500 К [28]. Подобных практически важных результатов известно много, и число их растет.
Даже из таких отдельных примеров ясно, что при разработке методов как измерений, так и моделирования состава неравновесной плазмы различных объектов и решаемых с их помощью задач вряд ли можно и сейчас и, по крайней мере, в близком будущем, рассчитывать на универсальный ответ. Ясно также, что логика развития физики и техники неравновесной плазмы не дает альтернативы в пользу расчетов или измерений, они должны быть дополняющими.
Экспериментальные методы исследований малых составляющих должны иметь высокую чувствительность детектирования. В одной из последних книг по спектральным исследованиям плазмы [4] описаны различные методы измерений состава и свойств частиц. Однако арсенал этих средств, с одной стороны, постоянно пополняется, и, с другой стороны, уже известные методы адаптируются к новым условиям их применения. На эти, не возмущающие плазму методы и их применения и делается акцент в настоящем обзоре.
Мы учитываем, что интерес к проблеме чрезвычайно велик и что выполненные в последние примерно 20 лет в этом направлении работы трудно осветить достаточно полно и, тем более, проанализировать их компактно. Отдельным циклам, связанным с заметными продвижениями в выделенных вопросах, посвящены специальные обзоры. Часть из них упомянута, другие будут указаны в последующих разделах статьи. Это касается и появления новых объектов, и достигнутых с их помощью результатов научно-технического характера. Общим для них, однако, является то, что объекты относительно компактны, как, например, разряды для травления поверхностей основ больших электронных схем [19], синтеза наноструктур [22], электронно-пучковые установки [29], “холодные” плазменные струи [25] и др. Непосредственно на них и проводятся исследования в целях оптимизации технологий. Ряд обзоров посвящен новым методам диагностики, в т.ч. возможностям применения лазерной техники [30–33].
Помимо ряда общих вопросов техники исследований, мы описываем состояние проблемы химического состава неравновесной пристеночной зоны плазменных энергетических реакторов. Обычно это крупные охлаждаемые установки с большими нагрузками на оболочку рабочей камеры, и проблемы вакуумной изоляции, возможного поступления примесей, их контроля и локализации источников стоят остро. На это, исходя из опыта работы реакторов предыдущих поколений, особое внимание обращается в проекте создаваемого наиболее крупного (~1000 м3) реактора И-ТЭР [15]. Проблема обсуждается уже более 10 лет и включает разработку физически и технически адекватных методов контроля, их апробацию на модельных установках и описание физико-химических процессов в целях масштабирования результатов.
2. ОСОБЕННОСТИ ПРИМЕНЕНИЙ МЕТОДОВ, СВЯЗАННЫХ С ОТБОРОМ ПРОБ, ДЛЯ ИЗУЧЕНИЯ ПЛАЗМОХИМИЧЕСКИХ ПРЕВРАЩЕНИЙ
В 1960-х годах начались работы по мощным газоразрядным CO2, CO, N2O лазерам, исследовались механизмы их действия и возможности создания приборов с длительным сроком службы в условиях протекания химических реакций. Это стимулировало развертывание систематических работ по неравновесной плазмохимии. Начали создаваться приемлемые для этого количественные методы, и предприняты попытки использовать опыт и приборы для газового анализа на основе хроматографии, масс-спектрометрии, ЭПР, ИК-анализа. Они предполагали работу с газами, образовавшимися после воздействия плазмы, – отбор проб, анализ потока газа, прошедшего через плазму или после выключения разряда. В простейших версиях производился отбор проб газа в съемные ампулы на разрядной трубке с последующим анализом на удаленных приборах [34–36]. В развитие этого были предложены методы, когда аналитические приборы интегрировались с плазменными объектами. На рис. 1 показаны эскизы некоторых из первых таких схем.
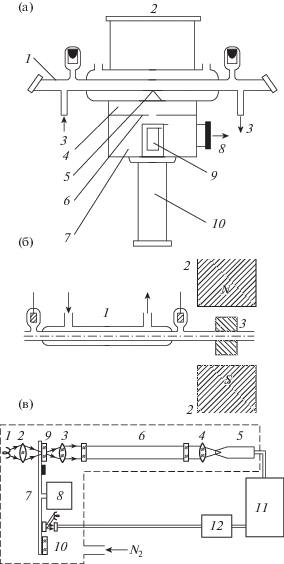
На рис. 1а приведен фрагмент схемы с масс-спектрометром. Здесь 1 – охлаждаемая разрядная трубка, проходящая через камеру прибора, откачиваемого через апертуру 2. Напуск и откачка лазерной смеси газов проводилась через каналы 3. Первая камера дифференциальной откачки спектрометра 4 сообщалась с внутренним объемом трубки через малое отверстие диафрагмы 5. Диафрагма 6 отделяла первую камеру от камеры анализатора с откачкой через порт 8, ионизационной камерой 9 и датчиком квадрупольного масс-спектрометра 10. Особого внимания в этом методе требует организация отбора пробы через диафрагму 5. Это должно быть малое отверстие в тонкой пленке, выбор размеров и материалов связан с минимизацией искажений состава частиц, проходящих через него, зависит от условий в плазме и обсуждался в работе [37].
На рис. 1б дана схема анализа состава потока газа, прошедшего через разряд 1 с помощью ЭПР спектрометра. Указаны полюса магнита 2 и СВЧ‑резонатор. На рис. 2в показана принципиальная схема оптического газоанализатора. ИК-излучение от теплового источника 1 линзы 2–4 коллимируют и фокусируют на детектор 5 после прохождения газовой среды в кювете 6. Излучение модулируется диском 7, вращающимся мотором 8. Два отверстия на диске закрыты интерференционными фильтрами 9, 10. Полоса пропускания одного из фильтров соответствует полосе поглощения анализируемых молекул, полоса другого лежит в окне прозрачности. Поглощение и концентрации молекул определялись синхронным детектированием с помощью электроники 11, 12 при опорном сигнале с фотосопротивлением. Быстродействие установок а), в) около 10 с, время пролета частиц от плазмы до СВЧ-резонатора в установке б) варьировалось от 0.2 с до 5 с.
Рис. 2.
Изменение концентрации СО2 в разряде в смеси СО2–N2–He. 1 и 4 – измерения на газоанализаторе (рис. 1в [39]), 2 – данные с удаленным масс-спектрометрическим анализом проб [36], 3 – измерения при удаленном хроматографическом анализе [35]. На врезке – начальный участок зависимости 4 при кратковременном воздействии разряда. В случае 4 электроды из Ni, в случае 1 – из Pt.
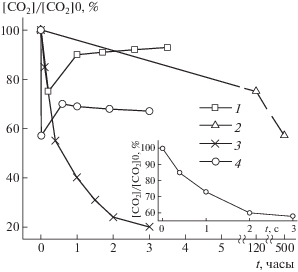
Важным результатом этих ранних экспериментов с интегрированными схемами стало то, что удалось разделить превращения, протекающие в разных фазах воздействия неравновесной плазмы. Было показано, что в случаях СО2 и СО лазеров стационарный состав частиц устанавливается достаточно быстро, за времена порядка секунд, а более медленные смещения этого динамического равновесия связаны с влиянием поверхностей: адсорбцией частиц, десорбцией примесей, каталитическими процессами. На рис. 2 приведены примеры динамики концентрации СО2 при измерениях различными методами.
Это определило стратегию развития технологий создания отпаянных СО2 и СО лазеров с большим сроком службы без смены наполнения активных элементов. Напротив, в случае N2O лазеров уже на первом этапе динамическое равновесие достигается при полном распаде рабочих молекул N2O, и создание таких отпаянных систем невозможно.
В последующий период и до настоящего времени, как говорилось во Введении, исследования состава плазмы значительно расширялись, но анализ с отбором проб не потерял актуальности. Принципы измерений, заложенные в приведенных на рис. 1 схемах, постоянно используются и модифицируются.
Как наиболее универсальные, по отношению к сорту и заряду частиц, получили развитие методы масс-спектрометрии неравновесной плазмы. Хотя прототип (рис. 1) и методология в целом сохраняются, проводится их усовершенствование и устранение недостатков, отмечавшихся уже на начальном этапе применений. Так, принцип формирования молекулярного пучка с помощью системы двух диафрагм в современных установках реализуется с помощью трех диафрагм (скиммеров), трехступенчатой дифференциальной откачки и модуляции пучка [40]. Это позволяет в большей степени исключить протекание вторичных реакций на пути от границы плазмы до ионизационной камеры спектрометра, отмечавшееся при работе со схемой рис. 1а [37]. За такой модификацией закрепилось название масс-спектрометрии молекулярных пучков (molecular beam mass spectrometry – MBMS). Другое предложение направлено на минимизацию влияния осколочных частиц в ионизационной камере. Эта трудность также отмечалась в [37] и связана с тем, что появление в ионном масс-спектре какой-либо частицы может связываться либо с прямой, либо с диссоциативной ионизацией. Второй процесс имеет более высокий порог. Предложение сводится к тому, чтобы измерять интенсивность ионного пика для массы (M/Z) соответствующей частицы в зависимости от энергии электронов в ионизационной камере. Появляющиеся особенности позволяют разделить вклады процессов. Аналогичные особенности могут быть зафиксированы, если диссоциативная ионизация происходит с участием метастабильных молекул, это понижает порог, и их концентрация может быть определена. Такая методика теперь называется масс-спектрометрией с контролем порогов ионизации (threshold ionization mass spectrometry – TIMS) [41]. В ряде работ используются оба подхода [17]. Абсолютная калибровка концентраций nX измеряемых частиц X в масс-спектрометрических измерениях может проводиться при наличии в составе плазмообразующих газов атомов инертных газов А с известной концентрацией nA, которые не участвуют в реакциях,
(1)
${{n}_{{\text{X}}}} = {{n}_{{\text{A}}}}{{I}_{{\text{X}}}}{{\sigma }_{{\text{A}}}}{\text{/}}{{I}_{{\text{A}}}}{{\sigma }_{{\text{X}}}},$При отключении электронного пучка или при его энергии ниже минимального порога ионизации могут фиксироваться ионы плазмы. Это также демонстрировалось в работе [42], в которой описан ионный состав плазмы СO- и N2O-лазеров. В частности, обнаружен гомологический ряд ${{{\text{C}}}_{k}}{\text{O}}_{l}^{ + }$ при l = 2, 2 ≤ k ≤ 10. При этом об абсолютных концентрациях ионов говорить по-прежнему проблематично из-за сложности калибровок.
Одной из центральных остается проблема стыковки масс-спектрометра с реальным плазменным объектом в сочетании с необходимостью отбора вещества при минимальных искажениях его состава. Эти обстоятельства отличают данную технику от также современных коммерчески доступных масс-спектрометрических комплексов для определения элементного состава образцов, включающих атомизаторы, роль которых играет равновесная плазма (пламя, индуктивно-связанная плазма с высокой газовой температурой).
Хотя применимость техники ЭПР ограничена парамагнитными в основном электронном состоянии частицами, схема (рис. 1б) используется для диагностики плазменных потоков и определения содержания в них не только атомов кислорода (как в работе [13]), но и других радикалов [43]. При необходимости исследовать взаимодействие этих радикалов с различными средами методика ЭПР дополняется техникой спиновых ловушек [44].
Техника абсорбционной спектроскопии ИК высокой чувствительности, реализованная по схеме рис. 1в, также была модифицирована. В работе [38] в схеме рис. 1в в качестве источника использовалась сборка металл–керамика, и измерения проводились после выключения разряда либо при непрерывной прокачке от внешнего разряда. В работе [37] для целей пирометрии плазмы источником 1 служила нагреваемая многополостная графитовая конструкция со спектром черного тела при температурах до 2500 К. При таких температурах, превышающих молекулярные колебательные температуры, вклад собственного ИК-излучения разряда в общую интенсивность мал, и концентрации молекул могут измеряться не только после выключения разряда, но и непосредственно в плазме. Впоследствии с появлением частотно-перестраиваемых лазеров методы ИК-анализа были переведены на новую базу, о чем более подробно будет сказано в следующих разделах.
Резюмируя, можно сказать, что методы исследования состава неравновесной плазмы с помощью отбора проб, пройдя довольно долгий путь, достаточно хорошо проработаны до уровня текущих потребностей с применением специальных масс-спектрометров, ЭПР- и ИК-анализа. Характерная чувствительность ~(1012–1013) см–3. Общим для этих методов является то, что локализация измерений практически невозможна. Сведения о составе плазмы, в зависимости от условий, относятся либо к усредненным из-за диффузии значениям, либо к окрестности места отбора вещества (в схеме рис. 1а).
3. ИССЛЕДОВАНИЯ СОСТАВА ПЛАЗМЫ МЕТОДАМИ КЛАССИЧЕСКОЙ ОПТИЧЕСКОЙ СПЕКТРОСКОПИИ
Эти методы в настоящее время широко развиты и применяются в разных целях. Для плазмы они, в частности, дают возможность проводить исследования химического состава непосредственно в объекте без существенных возмущений. В отсутствие равновесия, однако, такие измерения также не являются прямыми, поскольку дают информацию о выделенном энергетическом состоянии или их группе. Это приводит к необходимости установления связи заселенностей уровней с полной концентрацией исходя из физики процессов и, часто, к использованию нескольких независимых методов. Методы разделяются на эмиссионные (излучения), абсорбционные (поглощения) и рассеяния. Кратко остановимся на основных.
3.1. Спонтанное излучение плазмы
Интенсивность линии IXul излучения частиц Х при переходе u–l связана с измеряемой концентрацией NX при переходе u–l на частоте νX оптически тонкого однородного слоя соотношением [4]
(2)
${{I}_{{Xul}}} = {{C}_{X}} \cdot h{{\nu }_{{Xul}}} \cdot {{A}_{{Xul}}}{{k}_{{Xu}}}{{N}_{X}}{{\left( {{{Q}_{{Xu}}} + {{{\left( {{{\tau }_{{Xu}}}} \right)}}^{{ - 1}}}} \right)}^{{ - 1}}},$Технически это один из самых простых методов определения концентраций частиц. Коэффициенты СХ определяются для конкретной установки, величины AXul и QXu для большого числа частиц, переходов и состояний доступны в базах спектроскопических данных, как и величины τXu, учитывающие факторы ветвления радиационных переходов. Абсолютные интенсивности IXul измеряются по эталонной лампе со сплошным спектром и известной яркостной температурой. Это также хорошо отработанная процедура [45].
Трудности связаны с отсутствием равновесия в рассматриваемом объекте. Заселенности уровней, в т.ч. излучающего уровня u, могут определяться различными механизмами с участием других уровней i с заселенностями NXi и скоростями kXiu. Иногда, с учетом опыта предыдущих исследований и оценок их соответствия условиям проводимых измерений делаются упрощающие предположения. Принимается, что подавляющее число частиц находится в основном электронном состоянии ΣNXi = NX. Если, исходя из вида спектра, выбрать наиболее сильные оптические переходы с рассматриваемого возбуждаемого и излучающего состояния, то (τXu)–1= AXul. В случае электронно-возбужденных молекул вклад в QXu могут давать переходы в другие отталкивательные состояния (предиссоциация). Если это исключается структурой термов или рассматриваются атомы с излучающим состоянием, удаленным от границы сплошного спектра, то частоты QXu могут быть приписаны к тушащим столкновениям с другими частицами Y c концентрациями NY и между собой с константами скоростей qXY, qXX:
где $v$, σq – скорости частиц и сечения тушения. Такие допущения действительно часто оправдываются и заметно упрощают использование (2). Принимается также, что возбуждение уровней происходит прямым электронным ударом и скорость возбуждения в (2) зависит от концентрации электронов ne и их распределения f по энергиям ε(5)
$\left\{ \begin{gathered} {{k}_{{Xu}}} = \sqrt {\frac{2}{{{{m}_{e}}}}} {{n}_{e}}\int {\sigma (\varepsilon )} f(\varepsilon )\sqrt \varepsilon d\varepsilon , \hfill \\ \int {f(\varepsilon )d\varepsilon } = 1, \hfill \\ \end{gathered} \right.$Этот простой метод, напротив, является одним из наиболее востребованных в количественных измерениях. С точки зрения обсуждаемой темы – определения концентраций частиц – он часто используется как сопровождающий иные методы, не связанные со столь серьезными предпосылками и ограничениями. За редкими исключениями, практически все высокочувствительные спектральные методы, как отмечалось, дают информацию о заселенности выделенного энергетического уровня частицы. Они ориентированы на то, чтобы этот уровень относился к основному электронному (иногда – метастабильному, если интересует его заселенность) состоянию. Для атомов в большинстве случаев это выполняется. Любой спектральный метод при анализе результатов для неравновесной плазмы требует дополнительной информации. Важно знание температуры нейтрального газа. Спонтанное излучение при низких плотностях дает нужную информацию из допплеровского уширения [4]. Эту же информацию можно получить и из распределения интенсивностей во вращательной структуре электронно-колебательных полос [4, 220]. Из распределения интенсивностей в колебательной структуре полос переходов между электронно-возбужденными состояниями можно восстановить колебательные температуры двухатомных молекул в основных электронных состояниях. С их помощью находятся полные внутренние статистические суммы молекул, необходимые для применения любых спектральных методов измерения их концентраций.
3.2. Актинометрия спонтанного излучения плазмы
Указанные трудности эмиссионных методов, связанные с необходимостью знания параметров электронной составляющей плазмы, могут быть существенно уменьшены при применении метода оптической актинометрии (ОА) [4, 46]. Метод ОА не использует абсолютные оптические измерения и основан на сопоставлении интенсивностей спектров двух частиц различного сорта. Если концентрацию одних частиц (Х) следует определить, а концентрация других (А) известна, то частица А называется актинометром. Если записать выражения (2) для каждой из частиц и составить их отношение, то концентрация NX равна
(6)
${{N}_{X}} = {{N}_{A}}\frac{{{{I}_{{Xul}}}}}{{{{I}_{{Aul}}}}}\frac{{{{\lambda }_{{Xul}}}}}{{{{\lambda }_{{Aul}}}}}\frac{{{{C}_{A}}}}{{{{C}_{X}}}}\frac{{{{k}_{{Au}}}}}{{{{k}_{{Xu}}}}}\frac{{{{A}_{{Aul}}}}}{{{{A}_{{Xul}}}}}\frac{{{{Q}_{{Xu}}} + {{A}_{{Xul}}}}}{{{{Q}_{{Au}}} + {{A}_{{Aul}}}}}.$Концентрация электронов в (6) не входит, а вместо абсолютных скоростей возбуждения излучающих состояний входит их отношение. Привлекательность ОА метода связана именно с этим последним обстоятельством; важно, чтобы удалось выбрать пары сечений с близкими значениями порогов εt и схожими энергетическими зависимостями. Подобие форм сечений при близких порогах не случайно, поскольку в борновском приближении они связаны с силой осциллятора fgu оптически разрешенного перехода между основным g и возбуждаемым u состояниями [4]:
(7)
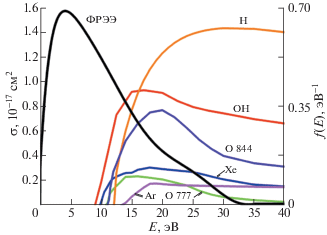
$\sigma (\varepsilon ) = 4\pi a_{0}^{2}{{\left( {\frac{{{\text{Ry}}}}{{{{\varepsilon }_{t}}}}} \right)}^{2}}\frac{{{{\varepsilon }_{t}}}}{\varepsilon }\ln \left( {\frac{\varepsilon }{{{{\varepsilon }_{t}}}}} \right){{f}_{{gu}}},$Примеры вида сечений, используемых при измерении концентраций молекул Н2О и ее фрагментов в плазме, приведены на рис. 3 [49]. Там же показан вид функции распределения энергий электронов (ФРЭЭ), измеренной зондовым методом в разряде с полым катодом в смеси He + + Xe + Ar + H2O при давлении 0.8 мбар [49]. Парциальное давление смеси инертных газов He : Xe : Ar = 99 : 1 : 1 – 0.6 мбар; паров воды – 0.2 мбар. Ток разряда 280 мА.
Рис. 3.
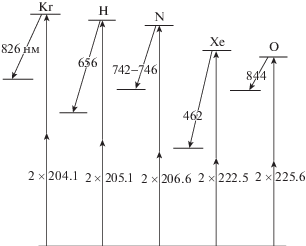
Сечения возбуждения атомов, диссоциации молекулы H2O и ФРЭЭ [49]: OH-диссоциативное возбуждение H2O; Xe 823 нм; O 777 нм и 844 нм; H 656 нм; Ar 751 нм.
Таким образом, актинометрический подход полностью исключает влияние на измерения концентраций частиц электронной плотности ne и сводит влияние f(ε) к сравнительно небольшим оцениваемым поправкам. С учетом этих факторов искомая концентрация частиц определяется простой формулой
(9)
$N_{X}^{\Gamma } = {{N}_{A}}\frac{{{{I}_{{Xul}}}}}{{{{I}_{{Aul}}}}}\frac{{{{\lambda }_{{Xul}}}}}{{{{\lambda }_{{Aul}}}}}\frac{{{{C}_{A}}}}{{{{C}_{X}}}}\Gamma (1 + {{\delta }_{X}})\frac{{{{A}_{{Aul}}}}}{{{{A}_{{Xul}}}}}\frac{{{{Q}_{{Xu}}} + {{A}_{{Xul}}}}}{{{{Q}_{{Au}}} + {{A}_{{Aul}}}}}.$В табл. 1 приведены Х и А, подобранные с учетом замечаний о сечениях возбуждения в работах различных авторов. Там же указаны комбинации длин волн в этих парах. В приведенных ссылках содержатся данные о сечениях возбуждения Х и А.
Таблица 1.
Апробированные актинометрические пары
| X | A | Ссылка | X | A | Ссылка | ||||
|---|---|---|---|---|---|---|---|---|---|
| λ, нм | λ, нм | λ, нм | λ, нм | ||||||
| O | 777.2 | Ar | 750.4 | [17, 50–54] | Cl | 754.7 | N2 | 337.1 | [80] |
| 751.5 | [47, 49, 56–58] | He | 388.8 | [80] | |||||
| 811.5 | [59] | Ar | 750.4 | [80] | |||||
| Xe | 823.2 | [47, 49, 55–58] | 837.6 | Ar | 826.4 | [81] | |||
| Kr | 758.7 | [52] | CCl | 277.8 | N2 | 337.1 | [80] | ||
| 811.3 | [52] | He | 388.8 | [80] | |||||
| 844.6 | Ar | 750.4 | [17, 54, 60–68] | Ar | 750.4 | [80] | |||
| 751.5 | [49, 58] | H2O | 306–320 | Xe | 823.2 | [49, 55–58, 72, 82–86] | |||
| 811.5 | [51, 54, 59, 69] | 306–320 | Ar | 751.5 | [49, 58] | ||||
| Xe | 823.2 | [49, 55–58] | OH | 306–320 | Xe | 823.2 | [49, 58] | ||
| H | 486.1 | N2 | 380.5 | [70] | 306.4 | 834.7 | [66, 67] | ||
| 656.3 | [70] | O2 | 760.5 | Ar | 750.4 | [64] | |||
| Ar | 750.4 | [66, 67, 71] | N2 | 337.1 | Ar | 750.4 | [79] | ||
| 751.5 | [47, 49, 55–58, 72] | 380.5 | [53] | ||||||
| 811.5 | [63] | CF | 202.4 | Ar | 750.4 | [87] | |||
| Xe | 823.2 | [47, 49, 58] | 202.6 | Xe | 834.7 | [66, 67] | |||
| Kr | 427.4 | [73] | 207.8 | Ar | 750.4 | [53, 78] | |||
| 431.9 | [73] | CF2 | 251.9 | Ar | 750.4 | [87] | |||
| 432.0 | [73] | 276.6 | [53, 78] | ||||||
| 557.0 | [73] | CH | 431.3 | N2 | 380.5 | [70] | |||
| 587.1 | [73] | 431.3 | Xe | 834.7 | [66, 67] | ||||
| D | 656.1 | [55–58] | CO | 217.3 | Ar | 750.4 | [53] | ||
| N | 744.2 | Ar | 750.4 | [59, 74] | 297.7 | [67, 88] | |||
| 746.8 | [59, 74] | Xe | 834.7 | [66, 67, 88] | |||||
| 821.6 | [71] | CO+ | 180–315 | Ar | 750.4 | [53] | |||
| 811.5 | [17, 59, 63] | CO2 | 218–439 | Ar | 750.4 | [53] | |||
| F | 685.6 | Ar | 750.4 | [17, 75] | ${\text{CO}}_{2}^{ + }$ | 289.0 | Xe | 834.7 | [53, 66, 67] |
| 703.7 | [17, 18, 53, 66, 67, 75–79] | HF | 255.0 | Xe | 834.7 | [67] | |||
| C | 247.7 | Ar | 750.4 | [66] | HF+ | 390.0 | Xe | 834.7 | [67] |
| Xe | 834.7 | [67] | |||||||
В табл. 2 собраны данные о частицах и оптических переходах, потенциально пригодных для составления актинометрических пар с информацией о пороговых значениях сечений возбуждения различных термов, обозначениях и конфигурации термов. Для тяжелых инертных газов указаны по 2 вида используемых в литературе обозначений (из-за промежуточного типа связи), коэффициенты A Эйнштейна и времена жизни τu верхних уровней переходов.
Таблица 2.
Некоторые параметры частиц для составления актинометрических пар. Данные из базы [89] для атомов и из [4, 90] для молекул. Обозначения в тесте
| λul, нм | Eu, эВ | u состояние; терм; J | l состояние; терм; J | Aul × 107, с–1 | τu, нс | |
|---|---|---|---|---|---|---|
| Ar | 750.4 | 13.48 | 3s23p5($^{2}P_{{1/2}}^{{\text{o}}}$)4p; 2[1/2]; 0 | 3s23p5($^{2}P_{{1/2}}^{{\text{o}}}$)4s; 2[1/2]o; 1 | 4.5 | 22 |
| 751.5 | 13.27 | 3s23p5($^{2}P_{{3/2}}^{{\text{o}}}$)4p; 2[1/2]; 0 | 3s23p5($^{2}P_{{3/2}}^{{\text{o}}}$)4s; 2[3/2]o; 1 | 4.0 | 25 | |
| 811.5 | 13.08 | 3s23p5($^{2}P_{{3/2}}^{{\text{o}}}$)4p; 2[5/2]; 3 | 3s23p5($^{2}P_{{3/2}}^{{\text{o}}}$)4s; 2[3/2]o; 2 | 3.3 | 30 | |
| 826.4 | 13.32 | 3s23p5($^{2}P_{{1/2}}^{{\text{o}}}$)4p; 2[1/2]; 1 | 3s23p5($^{2}P_{{1/2}}^{{\text{o}}}$)4s; 2[1/2]o; 1 | 1.53 | 28* | |
| Xe | 823.2 | 9.82 | 5p5($^{2}P_{{3/2}}^{{\text{o}}}$)6p; 2[3/2]; 2 | 5p5($^{2}P_{{3/2}}^{{\text{o}}}$)6s; 2[3/2]o; 2 | 2.86 | 27* |
| 834.7 | 11.05 | 5p5($^{2}P_{{1/2}}^{{\text{o}}}$)6p; 2[3/2]; 2 | 5p5($^{2}P_{{1/2}}^{{\text{o}}}$)6s; 2[1/2]o; 2 | 4.2 | 22* | |
| Kr | 427.4 | 12.82 | 4s24p5($^{2}P_{{3/2}}^{{\text{o}}}$)6p; 2[3/2]; 2 | 4s24p5($^{2}P_{{3/2}}^{{\text{o}}}$)5s; 2[3/2]; 2 | 0.199 | 414* |
| 431.9 | 12.79 | 4s24p5($^{2}P_{{3/2}}^{{\text{o}}}$)6p; 2[5/2]; 2 | 4s24p5($^{2}P_{{3/2}}^{{\text{o}}}$)5s; 2[3/2]o; 2 | 5.13 × 10–2 | 772* | |
| 432.0 | 12.78 | 4s24p5($^{2}P_{{3/2}}^{{\text{o}}}$)6p; 2[5/2]; 3 | 4s24p5(2P°3/2)5s; 2[3/2]o; 2 | 0.125 | 800 | |
| 557.0 | 12.14 | 4s24p5($^{2}P_{{1/2}}^{{\text{o}}}$)5p; 2[1/2]; 1 | 4s24p5($^{2}P_{{3/2}}^{{\text{o}}}$)5s; 2[3/2]o; 2 | 9.8 × 10–2 | 28* | |
| 587.1 | 12.14 | 4s24p5($^{2}P_{{1/2}}^{{\text{o}}}$)5p; 2[3/2]; 2 | 4s24p5($^{2}P_{{3/2}}^{{\text{o}}}$)5s; 2[3/2]; 1 | 7.1 × 10–2 | 28* | |
| 758.7 | 11.67 | 4s24p5($^{2}P_{{3/2}}^{{\text{o}}}$)5p; 2[1/2]; 0 | 4s24p5($^{2}P_{{3/2}}^{{\text{o}}}$)5s; 2[3/2]o; 1 | 4.31 | 23 | |
| 811.3 | 11.44 | 4s24p5($^{2}P_{{3/2}}^{{\text{o}}}$)5p; 2[5/2]; 3 | 4s24p5($^{2}P_{{3/2}}^{{\text{o}}}$)5s; 2[3/2]o; 2 | 3.61 | 27 | |
| He | 388.9 | 23.0 | 1s3p; 3Po; 1 | 1s2s; 3S; 1 | 0.947 | 105 |
| 388.9 | 23.0 | 1s3p; 3Po; 2 | 1s2s; 3S; 1 | 0.947 | 105 | |
| O | 777.2 | 10.74 | 2s22p3(4S°)3p; 5P; 3 | 2s22p3(4S°)3s; 5So; 2 | 3.69 | 27 |
| 777.4 | 10.74 | 2s22p3(4S°)3p; 5P; 2 | 2s22p3(4S°)3s; 5So; 2 | 3.69 | 27 | |
| 777.5 | 10.74 | 2s22p3(4S°)3p; 5P; 1 | 2s22p3(4S°)3s; 5So; 2 | 3.69 | 27 | |
| 844.6 | 10.99 | 2s22p3(4S°)3p; 3P; 0 | 2s22p3(4S°)3s; 3So; 1 | 3.22 | 31 | |
| 844.6 | 10.99 | 2s22p3(4S°)3p; 3P; 2 | 2s22p3(4S°)3s; 3So; 1 | 3.22 | 31 | |
| 844.7 | 10.99 | 2s22p3(4S°)3p; 3P; 1 | 2s22p3(4S°)3s; 3So; 1 | 3.22 | 31 | |
| H | 486.1 | 12.74 | 4p; 2Po; 3/2 | 2s; 2S; 1/2 | 0.967 | 103 |
| 486.1 | 12.74 | 4p; 2Po; 1/2 | 2s; 2S; 1/2 | 0.967 | 103 | |
| 656.3 | 12.09 | 3d; 2D; 3/2 | 2p; 2Po; 1/2 | 5.39 | 18 | |
| 656.3 | 12.09 | 3p; 2Po; 3/2 | 2s; 2S; 1/2 | 2.24 | 45 | |
| 656.3 | 12.09 | 3p; 2Po; 1/2 | 2s; 2S; 1/2 | 2.24 | 45 | |
| 656.3 | 12.09 | 3d; 2D; 5/2 | 2p; 2Po; 3/2 | 6.47 | 15 | |
| N | 744.2 | 11.99 | 2s22p2(3P)3p; 4So; 3/2 | 2s22p2(3P)3s; 4P; 3/2 | 1.2 | 27* |
| 746.8 | 11.99 | 2s22p2(3P)3p; 4So; 3/2 | 2s22p2(3P)3s; 4P; 5/2 | 1.96 | 27* | |
| 821.6 | 11.85 | 2s22p2(3P)3p; 4Po; 5/2 | 2s22p2(3P)3s; 4P; 5/2 | 2.26 | 31* | |
| F | 685.6 | 14.50 | 2s22p4(3P)3p; 4Do; 7/2 | 2s22p4(3P)3s; 4P; 5/2 | 4.87 | 21 |
| 703.7 | 14.75 | 2s22p4(3P)3p; 2Po; 3/2 | 2s22p4(3P)3s; 2P; 3/2 | 4.23 | 17* | |
| Cl | 837.6 | 10.40 | 3s23p4(3P)4p; 4Do; 7/2 | 3s23p4(3P)4s; 4P; 5/2 | 2.8 | 36 |
| C | 247.9 | 7.68 | 2s22p3s; 1Po; 1 | 2s22p2; 1S; 0 | 2.8 | 36 |
| N2 | 268–545 | 11.05 | C3Пu | B3Пg | 2.44 | 40 |
| OH | 261–411 | 4.02 | A2Σ+ | Х2П | 0.125 | 720 |
| CO | 114–280 | 8.07 | A1П | Х1Σ+ | 9.61 | 15 |
| 412–668 | 10.78 | B1Σ+ | A1П | 4.21 | 30 | |
| CO+ | 180–315 | 5.69 | B2Σ | Х2Σ | 2 | 50 |
| CF | 197–220 | 6.1 | B2Δ | Х2П | 5.26 | 20 |
3.3. Локализация эмиссии неоднородных источников
Если плазма неоднородна, то даже для оптически тонкого слоя выражение (2) справедливо лишь в малой ее области. Реально происходит суммирование интенсивностей по лучу зрения. Для локализации интенсивностей в общем случае требуется решение томографической задачи с восстановлением профиля из наблюдений плазмы под разными ракурсами. Это математически сложная обратная задача [4]. На практике обычно ограничиваются случаями, когда плазма имеет ось симметрии. Экспериментально, без сложной математической обработки, восстановление возможно, если оптическая система формирования изображения на плоскость входной щели спектрометра допускает выделение и перемещение достаточно узких параллельных оси областей. Это не всегда реализуемо для протяженных объектов, и применяют наблюдения эмиссии по хордам поперек плазменного столба (рис. 4). Если ${{I}_{\nu }}$ – интенсивность по лучу зрения, а $I_{\nu }^{1}$ – интенсивность с единичной длины, то решение такой частной задачи восстановления $I_{\nu }^{1}$ по ${{I}_{\nu }}$ дается формулой Абеля
(10)
$\begin{gathered} I_{\nu }^{1}(x,y) = I_{\nu }^{1}(r) = - \frac{1}{\pi }\int\limits_r^R {\frac{{d{{I}_{\nu }}(x)}}{{dx}}\frac{{dx}}{{\sqrt {{{x}^{2}} - {{r}^{2}}} }} = } \\ \, = - \frac{1}{{\pi r}}\frac{d}{{dr}}\int\limits_r^R {{{I}_{\nu }}(x)\frac{{xdx}}{{\sqrt {{{x}^{2}} - {{r}^{2}}} }}} . \\ \end{gathered} $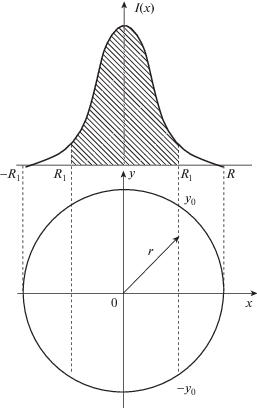
Для восстановленных $I_{\nu }^{1}$ формула (2) справедлива для области локализации. Если такие измерения и процедуры проведены для частиц А и Х, то и метод актинометрии может применяться с пространственным разрешением.
Формула (10) иллюстрирует общую для обратных задач чувствительность решения к точности интегральных измерений. В данном случае требуется дифференцирование ${{I}_{\nu }}(x)$ при наличии сингулярности на нижнем пределе. Практика показывает, что при типичных нормально распределенных погрешностях эксперимента (1–2)% погрешности такого восстановления составляют (10–15)%, и целесообразно тестирование полученных результатов на модельных задачах. Альтернативой является разработка методов трехмерной локализации экспериментальными средствами, основанными на других физических принципах измерений.
3.4. Поглощение в сплошном спектре
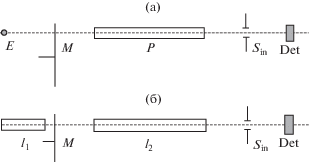
На рис. 5а показан случай, когда плазма Р просвечивается источником Е со сплошным спектром. Спектральный прибор Sin выделяет интервал частот $\Delta \nu $, включающий линию поглощения плазмы. В простейшем случае выделяется одна линия и Δν > Δνul, где Δνul – полуширина линии. Детектор Det имеет большой линейный диапазон, работает на частоте, определяемой модулятором М, и собственное излучение плазмы не регистрируется.
Рис. 5.
Измерения поглощения: а) – при использовании точечного источника сплошного спектра; б) – в варианте двух трубок.
Изменение интенсивности света от I0 до I после прохождения через плазму характеризуется полным поглощением на переходе u–l:
Безразмерная величина AL – функция поглощения. Для однородной плазмы
(12)
${{A}_{G}} = \int\limits_{\Delta \nu } {\left( {1 - \exp \left\{ { - {{\chi }_{{lu}}}(\nu )l} \right\}} \right)d\nu } .$Для доплеровского и лоренцевского уширений интеграл (12) выражается через табулированные функцию Ладенбурга–Леви S(χ0,luL) и функцию Ладенбурга–Райхе S′(χ0,luL) соответственно [4]. При смешанном уширении величины AG как функции оптической плотности в центре линии χ0,lul рассчитываются численно (кривые роста).
Из измеренного полного поглощения AG определяются либо интегральный коэффициент поглощения
либо связанный с ним коэффициент поглощения в центре линии χ0,lu. Из этих значений находятся концентрации частиц на нижнем, поглощающем, уровне l. При доплеровском или лоренцовском уширении(14)
${{\chi }_{{lu}}} = \frac{1}{2}{{\left( {\frac{\pi }{{\ln 2}}} \right)}^{{1/2}}}{{\chi }_{{0,lu}}}\Delta {{\nu }_{D}},$Если длина волны λ измеряется в нанометрах, Alu в с–1 , Nl в см –3, χ в см–1, то, в пренебрежении индуцированным излучением, при доплеровском уширении величина
(16)
${{\chi }_{{0,lu}}} = 1.23 \times {{10}^{{ - 33}}}\frac{{{{g}_{u}}}}{{{{g}_{l}}}}\frac{{{{A}_{{ul}}}}}{{\Delta {{\lambda }_{D}}}}\lambda _{{lu}}^{4}{{N}_{l}},$(17)
${{\chi }_{{0,lu}}} = 0.84 \times {{10}^{{ - 33}}}\frac{{{{g}_{u}}}}{{{{g}_{l}}}}\frac{{{{A}_{{ul}}}}}{{\Delta {{\lambda }_{D}}}}\lambda _{{lu}}^{4}{{N}_{l}}.$Здесь gu и gl – статистические веса уровней.
Говоря о концентрационной чувствительности этого метода, следует отметить, что:
– при заданных величинах χlu и χ0,lu она возрастает с длиной оптического пути l; интегрирование (12) показывает, что AG ~ ΔνD и AG ~ ΔνL, а измеряемая по уменьшению интенсивностей величина AL тем меньше, чем больше отношения Δν/ΔνD,lu и Δν/ΔνL,lu. Это соответствует уменьшению измеряемого относительного поглощения и, в конечном счете, ограничивает концентрационную чувствительность измерений на фоне сплошного спектра;
– измерения при малых Δν ≪ Δνul выходят за рамки сделанного выше предположения, но возможны. Для этого требуется высокое спектральное разрешение, сопровождаемое падением уровня детектируемого сигнала от просвечивающего плазму источника. По физическим соображениям ключевым здесь является то обстоятельство, что тепловые источники сплошного спектра обладают сравнительно невысокой яркостной температурой ТВ ≤ (2–2.5) × 103 К.
Для преодоления этих трудностей разработаны другие версии метода поглощения.
3.5. Поглощение излучения с линейчатым спектром
На рис. 5б источник Е со сплошным спектром заменен на источник с линейчатым спектром, линии которого перекрываются, хотя бы частично, с линиями поглощения в плазме. В этом случае функции спектрального прибора сводятся к выделению этих линий. Такой вариант называется методом линейчатого поглощения (или атомной абсорбции). Возможны разные варианты выбора и использования такого источника [4, 91]. Это может быть, например, такая же разрядная трубка, как и исследуемая Р. При пониженном давлении излучающие уровни u атомов возбуждаются электронным ударом, и даже при наличии, кроме ударов 2-го рода, тушения тяжелыми частицами “температура возбуждения” (см. Введение) для пары уровней u–l сопоставима с электронной – порядка нескольких эВ. Поэтому, помимо выигрыша в спектральном разрешении (и светосиле), в этом случае спектральная яркость просвечивающего излучения также будет намного выше, чем при использовании теплового источника сплошного спектра.
При принятом определении (11) функция поглощения AL находится из двух измерений I0 и I. В случае одинаковых трубок с учетом самопоглощения в протяженном источнике [4] ее связь с коэффициентами поглощения имеет вид
(18)
${{A}_{L}} = 2\left( {1 - \frac{{S{\text{*}}\left( {2{{\chi }_{{0,lu}}}l} \right)}}{{S{\text{*}}\left( {{{\chi }_{{0,lu}}}l} \right)}}} \right),$В табл. 3 приведены значения AL(χ0,luL) для трех случаев: AL – доплеровское уширение; $A_{L}^{'}$ – лоренцовское уширение; $A_{L}^{0}$ – монохроматический свет источника на частоте центра линии поглощения плазмы.
Таблица 3.
Функции поглощения при различных типах уширения линий
| χ0,lul | 0.1 | 0.2 | 0.4 | 0.6 | 0.8 | 1.0 | 2.0 | 4.0 |
|---|---|---|---|---|---|---|---|---|
| AL | 0.066 | 0.130 | 0.238 | 0.329 | 0.404 | 0.469 | 0.673 | 0.806 |
| $A_{L}^{'}$ | 0.049 | 0.090 | 0.163 | 0.225 | 0.278 | 0.319 | 0.445 | 0.527 |
| $A_{L}^{0}$ | 0.095 | 0.181 | 0.330 | 0.451 | 0.551 | 0.632 | 0.865 | 0.982 |
При фиксированных χ0,luL значания AL для всех случаев варьируются в пределах фактора 2, и использование источника с узкой линией к заметному увеличению чувствительности не приведет, если не касаться характеристик, важных для детектирования (интенсивность, шумы и пр.). Это характеризует удобство метода, поскольку обеспечить идентичность условий в просвечивающем источнике и исследуемой плазме часто затруднительно.
Последнее является одной из причин того, что метод широко используют для исследований широкого круга плазменных объектов, включая плазменные анализаторы. Разработано большое количество моделей коммерческих атомно-адсорбционных спектрометров для промышленности. В них, как правило, в качестве источника просвечивающего излучения используются разряды в полом катоде с излучением в видимой области спектра, а равновесная плазма (пламена, индуктивно-связанная плазма) служит атомизатором образцов. Для исследований неравновесной плазмы в последние годы разработаны и используются специальные виды разрядов с полыми микрокатодами. При малых размерах, позволяющих хорошо коллимировать излучение, они применимы в том числе в ВУФ-области спектра. Так, с их помощью определяются концентрации атомов Н по линии Lα (121.6 нм) [92], кислорода О (130.2 нм) [93], N (120.7 нм) [29] и др.
4. ЛАЗЕРНАЯ СПЕКТРОСКОПИЯ
Исследования плазмы, в т.ч. оптическими и спектральными методами, начали проводиться намного раньше появления лазеров. Лазеры сразу же были востребованы в этой области. По мере развития лазерной техники расширялось и поле лазерной спектроскопии плазмы. Условно методы лазерной спектроскопии плазмы можно разделить на три группы. Первая использует основные принципы методов классической спектроскопии, повышая пределы измерений. Вторая реализует измерения, не доступные классическим методам. Третья частично использует оба предыдущих признака и добавляет новые возможности косвенных измерений и детектирования оптических сигналов.
В рамках темы настоящего обзора мы остановимся на методах определения с помощью лазеров концентраций частиц, преимущественно в основных и метастабильных состояниях. Эти вопросы решаются, как и в классике, методами поглощения и, частично, рассеяния. Учитывая наличие большого числа обзорных работ в этой сфере (см. Введение), мы отметим только принципиальные возможности, их физическое обоснование, области применимости и ограничения, отсылая за деталями к литературе.
4.1. Трассовые измерения с полупроводниковыми лазерами
С 1970-х г.г. начались и продолжаются разработки доступных для пользователей полупроводниковых лазеров с перестраиваемой частотой в непрерывном и в импульсно-периодическом режимах. Лазеры на основе соединений А2В6 и А3В5 излучают от ближней УФ- до ближней ИК-области (0.4–2.5) мкм при комнатной температуре, на соединениях А4В6 (4–40) мкм – при охлаждении хладагентами или элементами Пельте [30]. С 1980-х годов в спектроскопии плазмы расширяется применение квантово-каскадных лазеров (3–30) мкм, работающих при комнатной температуре [31]. Эти диапазоны соответствуют частотам колебательно-вращательных переходов подавляющего большинства молекул. Типичная ширина линии излучения имеет порядок 10–4 см–1, перестройка частоты осуществляется током инжекции или температурой охлаждения. Перестроечная характеристика кусочно-непрерывная из-за переключения мод, но совместными изменениями температуры и тока все частоты из области перестройки становятся доступными. Миниатюрные лазеры являются практически точечными источниками. Расходимость излучения дифракционная, и при простейшей коллимации поперечные размеры пучка в объекте порядка 1 мм; возможна локализации измерений по координатам нормальным к оси распространения.
Трассовыми называются измерения, при которых длина оптического пути по оси распространения совпадает (с точностью до рефракции) с геометрическим размером объекта. Выше (разд. 3.4) показано, что сужение линии просвечивающего излучения не дает существенного выигрыша в концентрационной чувствительности. Заметное повышение чувствительности при применении лазеров связано с детектированием просвечивающего излучения [4].
Минимально фиксируемые изменения мощности за счет поглощения, ΔРmin, определяются квантовыми флуктуациями сигнала (дробовые шумы). При фотоэлектрической регистрации и пуассоновской статистике фотоэлектронов
(19)
$\Delta {{P}_{{\min }}} = \xi {\text{'}}\sqrt {\frac{{Ph{{\nu }_{{lu}}}}}{{\Delta t{{\eta }_{D}}}}} .$Здесь P – мощность лазера, Δt – время регистрации, ηD – квантовый выход фотодетектора, ξ' ~ 1. Пусть A – поперечное сечение пучка, I – плотность мощности (интенсивность), L – длина поглощающего объекта, σlu – сечение поглощения. Число частиц на уровне l при их концентрации на нижнем уровне l в зоне лазерного пучка равно N' = Nl AL. С учетом (19) для малых поглощений, в пренебрежении индуцированными переходами, и насыщающей плотности мощности Is минимально обнаружимое число молекул N' (максимальная чувствительность) будет
(20)
$N_{{\min }}^{'} = \sqrt {\frac{{Ph{{\nu }_{{lu}}}}}{{\Delta t{{\eta }_{D}}}}} \frac{{1 + I{\text{/}}{{I}_{s}}}}{{I{{\sigma }_{{lu}}}}}.$Наименьшее значение $N_{{\min }}^{'}$ достигается при I = Is. Величина насыщающей плотности мощности для двухуровневой системы выражается через время жизни τu верхнего уровня Is = hνlu/(σluτu), и для оценки чувствительности может быть использовано соотношение
(21)
$N_{{\min }}^{'} = 2\sqrt {\frac{{A{{\tau }_{u}}}}{{\Delta t{{\eta }_{D}}{{\sigma }_{{lu}}}}}} .$Чтобы перейти от числа частиц на основном поглощающем уровне к полному числу частиц, в (20), (21) следует использовать не $N_{{\min }}^{'}$, а Nmin= = $N_{{\min }}^{'}{{Q}_{{in}}}$, где Qin – внутренняя статистическая сумма. Так, если при определении концентрации атомов по резонансным переходам σlu ~ 10–12 см2, τu ~ 10–8 с, практически все атомы находятся в основном состоянии и Qin ~ 1, Δt ~ 1 с, то величина Nmin ~ 102. Для суммарной по уровням концентрации молекул чувствительность снижается до типичной величины Nmin ~ 108 из-за увеличения типичных τu ~ 10–4 с и Qin ~ 102 для молекул.
Достижение максимальной чувствительности при мощности просвечивающего излучения, равной насыщающей, приводит к нелинейной зависимости ΔР от Р и нарушению распределения частиц по уровням. Корректнее выбрать режим I ≪ Is [94], причем чувствительность ухудшается не слишком сильно: по формуле (20) для I = = 10‒2 × Is величина $N_{{\min }}^{'}$ возрастает в 5 раз. При этом абсолютные значения лазерной мощности остаются высокими, их мощность и спектральная яркость существенно превышает соответствующие параметры классических источников. Типичная интенсивность насыщения для резонансных переходов в атомах Is ~ 10 Вт · см–2. Достаточно, чтобы при сечении пучка 1 мм2 соответствующая мощность лазера была не более 0.1 Вт. Это многократно превышает типичные мощности классических источников после необходимой монохроматизации, порядка (10–6–10–4) Вт на интервал спектра, равный ширине линии поглощения.
Из-за большой (по сравнению с классическими источниками) плотности мощности лазерного излучения отклик детектора велик, и собственные тепловые шумы фотоприемника не оказывают критического влияния на измерения. Это особенно важно в ИК-области спектра. Для λ ~ 10 мкм, при современных детекторах с ηD ~ 0.4 и обнаружительной способностью D* ~ 1010 см · Гц1/2, тепловыми шумами в таких измерениях можно пренебречь при P > 1 мВт. Подробнее см. [94–96]. В работе [97] рассмотрены погрешности измерений концентраций, связанные с конечной точностью определения интенсивностей для шумов различного типа (помимо дробовых – джонсоновские, генерационно-рекомбинационные, темновые).
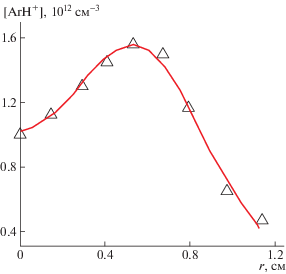
На рис. 6 приведен пример радиального распределения ионов ArH+ в положительном столбе тлеющего разряда в трубке в аргоне при давлении 6 Торр в трубке длиной 2 м и диаметром 25 мм при токе 1.25 А [4, 223]. Пучок диаметром 2 мм проходил параллельно оси разряда, регистрировались переходы v'' = 0, J'' = 3 → v' = 1, J' = 2 (2525.475 см–1) и v'' = 0, J'' = 3 → v' = 1, J' = 2 (2525.414 см–1). Ионы возникали из-за появления в разряде примесей водорода (вероятно, со стенок).
4.2. Схемы с увеличением длины оптического пути
В основной закон поглощения Бугера–Ламберта–Бэра (БЛБ, см. (12)) входит произведение χL. Для регистрации малых составляющих с малыми χ желательно использовать трассы с большими L. Разработан ряд схем для реализации этого подхода.
А. Для исследования компактных объектов применяется зеркальная оптика, обеспечивающая многократные прохождения света между ними при ограниченной апертуре. Наиболее известны схемы (ячейки) Уайта, Эрио, Чернина. Эти и иные схемы подробно описаны в обзоре [98]. При расстоянии между отражателями 50 см обеспечивается до 200 проходов [99]. Схемы используются как с классическими, так и с лазерными источниками. При исследованиях плазмы с широкополосными классическими источниками схемы Уайта, Эрио комбинируются со спектральными приборами высокого разрешения, например, фурье-спектрометрами. В такой комбинации в работе [100] исследовался состав газов, образующихся при взаимодействии струи плазмы, истекающей из капиллярного разряда в аргоне с воздухом переменной влажности, по поглощению в ИК-области спектра. Измерения в 8-проходной ячейке Уайта с длиной пути L = 19.2 м показали вариации NO2 в пределах (1–8) × 1014 см–3 и О3 – (0.5–3) × 1014 см–3. Локализация практически отсутствует, результаты измерений усреднены по объему, близкому к объему ячейки.
Б. Исторически следующим за ячейками стал метод внутрирезонаторной спектроскопии (ВРЛС). Изучаемый объект и активная лазерная среда помещены в общий оптический резонатор. К началу 1970-х годов частотно-перестраиваемые диодные лазеры еще не получили распространения, но стали широко доступными лазеры на красителях и лазеры на кристаллах и стеклах с широким спектральным контуром усиления. Развитие этого метода до настоящего времени идет, в основном, с использованием твердотельных, волоконных, полупроводниковых лазеров, оптических параметрических генераторов (OPО) и лазеров на красителях.
Качественно ВРЛС отличается от пассивных многопроходных кювет тем, что потери на зеркалах компенсируются усилением активной среды, а селективность поглощения объекта – наличием многих добротных резонаторных мод при широком контуре усиления. При этом эффективная длина оптического пути значительно выше, чем в ячейках и может достигать десятков километров при расстоянии между зеркалами порядка метра. Спектральное разрешение обеспечивается внешним спектральным прибором, а пространственное – областью перекрытия объемов объекта и каустики резонатора. Фундаментальное ограничение чувствительности связано с динамикой мод резонатора, зависит от типа используемого лазера, и, согласно оценкам работы [101], эффективный оптический путь может достигать для лазера Ti:sapphire значения 1.3 × 109 км, а для Ga-AlAs – 250 км. Реальные ограничения связаны с рядом факторов: спонтанным излучением лазерной среды, рассеянием Рэлея, нелинейными взаимодействиями третьего порядка и рядом других причин. Так, для Ti:.sapphire более реалистичны длины 70 000 км, для GaAlAs – 40 км, и эти значения экспериментально подтверждаются, причем в первом случае основным фактором выступает нелинейное взаимодействие, во втором – спонтанное излучение. Из исследованных наибольший путь достигается для лазеров Ti:sapphire, наименьший 1.5 м – для OPO, последнее значение связано с ограничением по длительности импульса генерации.
Метод, как аналитический, детально описан в оригинальных работах, обзорах и монографиях [101–105] и продолжает развиваться.
В качестве примеров применения к исследованию следовых частиц (вновь образованных и примесных) в плазме можно привести измерения концентраций синглетного метастабильного кислорода O2(a1Δg) на квадрупольном переходе a1Δg–b1Σ+g в области 1.9 мкм. В послесвечении ВЧ-разряда в кислороде при давлении 1.9 Торр концентрации варьировались в диапазоне (2–5) × 1014 см–3 [105]. Мониторинг метастабильных молекул азота в импульсном разряде в СО2 : N2 : He при давлении 240 Торр на переходах 1+ системы (575–610) нм показал концентрации [N2(А3Σ, v = 2–8)] ~ ~ (0.2–2) × 1014 см–3 [104]. Область локализации определяется каустикой резонатора.
В. Поглощение и затухание света в резонаторе. Эта техника, являясь развитием техники классических многопроходных ячеек, ориентирована исключительно на применение лазеров, активно развивается. Она может быть условно разделена на несколько групп
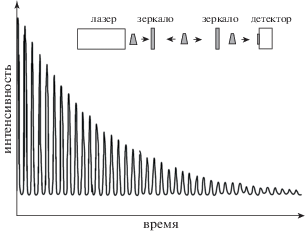
В первой многопроходные ячейки не разделяют пучки в пространстве, а конфигурированы так, чтобы пучок испытывал многократные отражения, распространяясь по оси в виде коротких разделенных во времени импульсов, интенсивность которых спадает во времени из-за диссипативных потерь при каждом новом отражении зеркалами. Схематически это обычно иллюстрируется в виде, аналогичном рис. 7 [4]. Спектральная ширина излучения в таких импульсах определяет разрешение и, как правило, превышает интервал c/2d между аксиальными модами ячейки длины d, что также способствует подавлению интерференционной структуры детектируемого сигнала. Эта техника, введенная в 1980-х годах [106] для измерения коэффициентов отражения зеркал, впоследствии стала широко применяться в лазерной спектроскопии и подробно описана, например, в монографии [107] (Cavity Ring Down Spectroscopy – CRDS).
При каждом круговом обходе кюветы часть энергии импульса теряется не только на пропускание зеркал, но и на поглощение в среде между зеркалами. Для пропускания эта доля равна (1 – R), а для поглощения A = 1–exp(−χct). Если зеркала имеют одинаковое отражение, то при A, (1 – R) ≪ 1 интенсивность импульса спадает во времени по закону
что и фиксируется как огибающая серии импульсов от детектора. Для пустой кюветы имеем А = 0, и характерное время затухания τ0= L/(c(1 – R)). Современные технологии позволяют изготовлять зеркала с коэффициентами отражения R > > 99.99%, поэтому затухание может быть достаточно “медленным”, удобным для анализа. Так, при длине кюветы 50 см и R = 99.99% затухание τ0 = 17 мкс, а эффективное число обходов кюветы светом будет ~5000. Величина поглощения А определяется из (22) путем сравнения времен затухания импульсов для пустой и заполненной кювет:Здесь время затухания
Из измерений τ определяются последовательно коэффициент поглощения χ и концентрации частиц на поглощающем уровне (16), (17). Чувствительность измерений близка к таковой в методе ВРЛС. При отражении зеркал R = 99.99% и длине ячейки 50 см эффективная длина оптического пути оказывается 10 км.
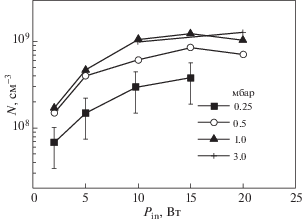
В качестве примера можно привести измерения данным методом концентраций атомов кремния [108]. Появление примесей этих атомов связывается с эрозией кварца в плазме. Использовался ВЧ барьерный разряд в аргоне с кварцевой пластиной в схеме CRDS с отражением зеркал ~99.7% на длине волны 252 нм. На рис. 8 приведены результаты измерений концентраций атомов Si в зависимости от мощности разряда при разных давлениях аргона. Пределы наблюдаемых концентраций составили (107–109) см–3. При перемещении разряда поперек оси ячейки достигалось пространственное разрешение ~(5–10) мм в пространстве над кварцевой пластиной до 100 мм.
К этой же группе следует отнести вариант, когда используется не импульсный, а непрерывный лазер и одновременно детектируется интегральный сигнал от лучей, прошедших через ячейку различное количество раз. Этот вариант имеет несколько используемых в литературе аббревиатур: CRDS, ICOS, CEAS. Он также широко используется в спектроскопии плазмы и подробно проанализирован в монографии [107].
Другие версии основаны на использовании резонансных свойств ячеек, в которых увеличение оптического пути достигается для собственных аксиальных продольных мод TEMm0 [107], а ширина лазерной линии меньше межмодового расстояния. Эти системы несомненно перспективны, но пока применяются мало, находятся в стадии исследований, имеются нерешенные вопросы согласования лазерных мод с модами внешнего резонатора и учета их неизбежных рассогласований в реальных системах. Одним из перспективных предложений является использование трехлучевой схемы (RICOS), в которой перед вводом в ячейку лазерный луч после отражения от входного зеркала отводится на отдельный детектор и является опорным [109, 110]. При рассогласовании частоты лазерного излучения с частотой аксиальной моды одновременно меняются пропускание и отражение ячейки, при этом интенсивности в отраженном и прошедшем ячейку лучами антикоррелированы. Это позволяет эффективно исключить влияние нестабильности резонатора на результаты измерений, что было успешно продемонстрировано при мониторинге метана в атмосфере [109, 110]. Для исследований плазмы такой подход пока не применялся.
К третьей группе версий следует отнести резонансные ячейки с неаксиальным вводом излучения, по аналогии с классическими интерферометрами [111]. Используемая в литературе аббревиатура – “off-axis ICOS”. В этом случае возбуждается набор поперечных мод TEMmn с m, n ≠ 0. Спектр возбуждаемых мод становится достаточно плотным, чтобы не терялись и резонансные свойства ячейки, и облегчалось согласование мод лазера и ячейки. Отклонение входящего пучка от оси не может быть велико, а апертура ячейки ас достаточна для выполнения условия малости дифракционных потерь ${{Z}_{F}} = a_{c}^{2}{\text{/}}\lambda L \gg 1$, где ZF – число Френеля, λ – длина волны света, L – расстояние между зеркалами. Данная группа по идеологии оказывается промежуточной между двумя предыдущими. Компромисс наиболее часто используется при измерениях концентраций частиц, начиная с работы [112], для газового анализа и применительно к исследованиям плазмы (например, обзоры [30, 31, 33]).
Эта группа методов развивается наиболее активно, дополняясь новыми приемами. Так, в работе [113] повышение чувствительности достигается за счет нестационарного токового нагрева активного элемента диодного лазера. В [109, 110, 114] развивается техника фазовых измерений при глубокой модуляции без потери разрешения. Проводятся исследования по измерению концентраций парамагнитных частиц (О2, ОН, NO, HO2…) по разности углов поворота s- и p-поляризаций излучения в резонансных ячейках при наложении магнитного поля [115–117]. По оценкам, сделанным в обзоре [33], такой метод может в перспективе обеспечить чувствительность поляризационных измерений в магнитном поле ~(10–1000) Гс, например, радикалов [НО2] ~ 107 см–3, тогда как проведенные измерения методами традиционной CRDS и ее версии “off-axis ICOS” показали чувствительность ~[НО2] ~ 1010 см–3 [117]. При этом трудно пока сказать, как это сказывается на свойствах плазмы.
Анализ проводимых исследований показывает, что в настоящее время большинство версий измерения концентраций частиц с использованием больших оптических путей в плазме проводятся с применением частотно-перестраиваемых полупроводниковых лазеров.
При измерениях концентраций частиц альтернативой методу измерения непосредственно поглощения света может быть регистрация изменения того или иного параметра объекта, вызванного поглощением. Это могут быть, например, нагрев объекта и связанные с ним рефракционные и акустические эффекты, дополнительные люминесценция, проводимость и др. Измеренные количественные характеристики этих проявлений могут быть связаны с величиной поглощения и концентрациями, что используется в газовом анализе [96]. В отличие от газов, для которых они не имеют собственного фона, плазма сама продуцирует эти же явления, и речь может идти о влиянии поглощения на них в той или иной мере. В отдельных случаях это удается успешно реализовать.
Г.1. Лазерно-индуцированная флуоресценция. Метод ЛИФ (LIF) имеет много общего с методом спонтанной эмиссии плазмы. Интенсивность люминесценции определяется выражением (2) с той разницей, что скорость kXu возбуждения свечения частицы X с уровня u определяется поглощением света. Если поглощение происходит из основного состояния g–u, дополнительная люминесценция на переходе u–l (для исключения помех от рассеяния l ≠ g) может быть отделена от иных многочисленных возможных каналов возбуждения этого же уровня в плазме. Отпадает, в частности, необходимость в знании свойств электронной компоненты, но остается необходимым учет тушения QXu.
В такой версии однофотонного возбуждения g–u–l метод LIF используется для определения концентраций в плазме ряда атомов (F (1–20) × × 1013 см–3 [16]), радикалов NO (2–40) × 1014 см–3 [118], СН (1–6) × 1012 см–3 [119], С2 (1–6) × 1010 cм–3 [120], CF (1–6) × 1012 cм–3 [16] и ряда других молекул, если доступны лазеры с величиной кванта излучения, соответствующей переходу g–u. Это простой и удобный метод, не требующий абсолютных измерений, поскольку может быть прокалиброван, например, по рассеянию Рэлея с той же линейной зависимостью интенсивности от интенсивности лазерного излучения, что и сигнал LIF. Для большинства частиц энергия между основным и возбуждаемым состояниями лежит в УФ- или ВУФ-областях, и эта простейшая версия не пригодна ввиду отсутствия излучения (и гармоник) частотно-перестраиваемых лазеров. Некоторый выход из положения может быть связан с использованием синхротронного излучения. В целом, по отношению к обсуждаемому методу, синхротронное излучение аналогично лазерному. Оно имеет малую спектральную ширину, может перестраиваться по частоте и коллимироваться. В работе [121] при исследовании высокояркостной галогенной лампы использовалось синхротронное излучение в области энергий фотонов (40–100) кэВ ((0.3–0.12) Å), монохроматичностью Δλ/λ ~ 10–3, расходимостью ~10–5 рад, сечением пучка 1 × 1 мм2. Локальность измерений была ~1 мм3. Спектры флуоресценции, возбуждаемой столь “жестким” излучением, соответствуют атомным переходам с участием внутренних электронных оболочек и в исследованном объекте содержали линии kα таких элементов, как I (наполнитель) и Cs, Dy (примеси из-за эрозии электродов). Калибровки по концентрациям не проводилось, хотя для атомных частиц это не составило бы больших проблем при использовании газов или равновесных атомизаторов при контроле иными методами. По-видимому, ограниченность исследований в этой области связана со сложностью сочетания дорогостоящего синхротрона с изучаемыми объектами. Однако с развитием техники рентгеновских источников такой подход имеет перспективу.
В иных случаях доступны версии двойного оптического резонанса (DOR LIF) или двухфотонного возбуждения (TALIF).
В первом из них для возбуждения используются два лазера и промежуточный уровень i возбуждения g–i–u–l. Для получения количественных результатов этот случай требует анализа процессов, заселяющих уровень i в плазме, и сложен технически, и, хотя он позволяет проводить локализацию измерений при пресечении сфокусированных лазерных пучков, практически в спектроскопии плазмы не используется.
Гораздо более распространено применение версии TALIF. В этом случае калибровка по рассеянию Рэлея также не пригодна ввиду разных зависимостей интенсивностей рассеяния и люминесценции от мощности лазера. В данной ситуации было в очередной раз предложено использовать опыт классической актинометрии и проводить измерения одновременно для исследуемых частиц и частиц с известной концентрацией, например, атомов инертных газов. Подбираются актинометрические пары, аналогично табл. 1, но, в отличие от классической актинометрии, где роль играет близость порогов электронного возбуждения, в TALIF выбор ограничивается более жестко, в основном, возможностями подбора лазеров. Наиболее часто в спектроскопии малых составляющих плазмы этот метод применяется для измерений концентраций атомов H, O и N. За счет нелинейности процесса при фокусировке пучков достигается трехмерная локализация с линейным размером <100 мкм. Эта техника развита начиная с работ [122–125], где можно найти примеры применений, а соответствующие комбинации пар показаны на диаграмме рис. 9.
Для практического удобства при работе с неравновесной плазмой, в работе [124] в равновесных условиях выполнены (с точностью ±5%) измерения отношения сечений соответствующих двухфотонных возбуждений:
(25)
$\frac{{\sigma _{u}^{{{\text{K}}r}}\left( {5p{\text{'}}\,{{{[3{\text{/}}2]}}_{2}}} \right)}}{{\sigma _{u}^{{\text{H}}}(3{}^{2}{{D}_{J}})}} = 0.62,$(26)
$\frac{{\sigma _{u}^{{{\text{K}}r}}\left( {5p{\text{'}}\,{{{[3{\text{/}}2]}}_{2}}} \right)}}{{\sigma _{u}^{{\text{N}}}(3{}^{2}{{D}_{J}})}} = 0.67,$(27)
$\frac{{\sigma _{u}^{{{\text{Xe}}}}\left( {5p{\text{'}}\,{{{[3{\text{/}}2]}}_{2}}} \right)}}{{\sigma _{u}^{{\text{O}}}(3p{}^{3}{{P}_{J}})}} = 0.36.$Г.2. Спектроскопия фотоотрыва. В плазме, содержащей частицы, способные захватывать электроны, образуются отрицательные ионы (ОИ). При облучении электроны могут быть освобождены. Сечения фотоотрыва σd для многих частиц приведены в книге [4]. В плазме появляются избыточные электроноы, и их число может быть измерено методом зондов. Это число дается соотношением:
(28)
$\Delta {{n}_{e}}(t) = N_{0}^{ - }\left[ {1 - \exp \left\{ { - \frac{{{{\sigma }_{a}}(\nu )}}{{h\nu }}\int {I(\nu )dt} } \right\}} \right],$Развитие обсуждаемых в настоящем обзоре высокочувствительных методов спектроскопии частиц малых концентраций создало альтернативу и этому известному методу. Фотоотрыв связан с поглощением света с характерными сечениями σd~ (10–18–10–16) см2 в области спектра от УФ до ближней ИК. Коэффициенты поглощения и соответствующие концентрации могут быть измерены описанными в предыдущих разделах методами. Так, в работе [129] измерялись концентрации ионов О– в планарном ВЧ-разряде в кислороде методом CRDS в диапазоне [O–] ~ (1010–1011) cм–3. При разряде в водороде в этой же системе и при тех же давлениях (0.075–0.4) Торр концентрации [H–] ~ (109–1010) см–3. При разряде в трубке в магнитном поле в водороде при давлении (0.3–1.8) × 10–2 Торр методом поглощения установлен диапазон концентраций [H–] ~ (2–15) × 109 см–3 [130].
Г.3. Оптико-гальванические измерения. Этот метод, как и метод фотоотрыва, связан с влиянием излучения на ионизационный баланс в плазме, однако механизм его более сложен. Оптогальванический эффект (ОГЭ) возникает из-за перераспределения заселенностей уровней атомов или молекул при поглощении света. В результате различных радиационных и (или) столкновительных и (или) коллективных процессов изменяются плотности заряженных частиц, их подвижности и энергии, что сказывается на ионизационном балансе и проводимости. Метод прост в техническом отношении. Спектры поглощения могут регистрироваться как изменение тока или напряжения на электродах либо зондовым методом. Возможна локализация измерений, как и в случае ЛИФ, при использовании двойного оптического резонанса или двухфотонного возбуждения.
Для аналитических плазменных анализаторов, где модель ЛТР справедлива, ОГЭ используется как количественный метод. С его помощью измеряются концентрации большинства металлов, имеющих линии поглощения в видимой и ближней УФ-областях. При этом его чувствительность в большинстве случаев превосходит методы поглощения, излучения, ЛИФ.
При работе в условиях неравновесной плазмы ОГЭ используется для установления структуры спектров слабых переходов. Проблема количественных калибровок пока не решена, поэтому ОГЭ может использоваться как чувствительный индикатор наличия тех или иных частиц в плазме. Подробный анализ метода проведен в обзоре [131], книгах [4, 132, 133], работы продолжаются (см., например, [134]).
5. МЕТОДЫ ИЗМЕРЕНИЙ МАЛЫХ КОНЦЕНТРАЦИЙ ЧАСТИЦ НА УСТАНОВКАХ, МОДЕЛИРУЮЩИХ УСЛОВИЯ, СХОДНЫЕ С ПЛАЗМОЙ ПРИСТЕНОЧНЫХ СЛОЕВ РЕАКТОРОВ
5.1. Проблема примесей
Начиная с 1950-х годов стали воплощаться в жизнь идеи и проекты создания термоядерных реакторов с магнитным удержанием плазмы при температурах, достаточных для протекания управляемой термоядерной реакции D + T → He (3.5 МэВ) + n (14.2 МэВ). К настоящему времени уже создано несколько поколений таких устройств разного масштаба и оснащенности, накоплен большой опыт, выражающийся, в том числе, в формулировке ряда проблем, которые необходимо решить при подготовке к созданию реакторов последующих поколений. В отличие от предыдущих, экспериментальный реактор ИТЭР [15, 135–147] планируется уже как прототип устройства промышленной энергетики, хотя, конечно, в процессе его создания еще должен быть решен ряд вопросов, ответы на которые не были даны в предшествующих экспериментах. Мы коснемся вопроса о чистоте плазмы.
Для протекания указанной реакции выделяемое в ней тепло должно быть достаточным для компенсации диссипативных потерь энергии реактора. Это требование выражается критерием Лоусона. Он удовлетворяется, если при температуре ионов Ti ~ 15 кэВ, концентрации электронов ne и времени удержания τE выполняется условие Ti neτE > 6 × 1020 кэВ · с ⋅ м–3. При τE = 1 с плотность должна быть ne > 1020 м–3 = 1014 см–3.
При столь высоких температурах в зоне горячей плазмы у всех не самых тяжелых атомов происходит полная ионизация, и, по условию детального равновесия, протекает интенсивная рекомбинация, сопровождаемая излучением. Рекомбинационное излучение оказывается одним из важных (если не самых важных, наряду с выносом энергии нейтронами) механизмов охлаждения загрязненной примесями плазмы. Мощность Рrad рекомбинационного излучения для ионов с зарядом Z выражается формулой [136–147]
(29)
${{P}_{{rad}}} = 1.78 \times {{10}^{{ - 32}}}{{n}_{e}}{{n}_{Z}}\frac{{{{Z}^{4}}}}{{\sqrt {{{T}_{e}}} \;({\text{кэВ}})}}{\text{, Вт}} \cdot {\text{с}}{{{\text{м}}}^{{ - {\text{3}}}}}.$Радиационная мощность резко зависит от Z, и для “рабочих” элементов H, D, T она значительно меньше, чем для более тяжелых элементов примесей. Если радиационная мощность составляет значительную долю от мощности в плазме, происходит охлаждение плазмы, а вместе с ним увеличивается Рrad. Процесс охлаждения развивается лавинообразно, плазма не может существовать в самоподдерживающемся режиме, и наступает т.н. радиационный коллапс. С учетом таких соображений и особенностей переноса частиц (аномальный и неоклассический) может быть рассчитана “летальная доза” примесей как доля от плотности электронов ξd = nz/ne. В табл. 4 приведены эти значения для условий работы реактора ИТЭР [15]. Там же выписаны значения реальных доз ξc, допускаемых разработчиками проекта.
Таблица 4.
“Летальная доза” примесей
| Элемент | C | O | Ne | Ar | Fe | Mo | W |
|---|---|---|---|---|---|---|---|
| Z | 6 | 8 | 10 | 18 | 20 | 42 | 74 |
| ξd | 1.65 | 0.52 | 0.21 | 0.04 | 4.7 × 10–3 | 6.9 × 10–4 | 7.1 × 10–5 |
| ξc | 0.05 | 0.05 | 0.02 | 0.005 | 5 × 10–4 | 5 × 10–5 | 5 × 10–6 |
Для ИТЭР величина ne ~ 1014 см–3 и определение содержания примесей на таком уровне требует взвешенного выбора метода измерений с учетом возможностей, представленных выше в разделах 2–4.
5.2. Предшествующие, текущие и требуемые исследования
Негативное влияние примесей отмечалось в процессе работы практически всех поколений конкретных установок с магнитным удержанием плазмы. Именно загрязнения плазмы традиционно становились причинами длительного простоя исследовательских установок. Анализ, предшествовавший формулировке требований проекта ИТЭР, показал, что “слабым местом” сложной конструкции из многочисленных модулей (~103 индивидуальных частей) является появление в плазме кислорода, носителем которого могут быть молекулы воды.
Вода является одновременно и охладителем стенок реактора, и его рабочим телом. В рабочем режиме температура охладителя ~450 К, давление парогазовой среды ~4 МПа (~40 атм), и проникновение молекул воды внутрь многоэлементной охлаждаемой конструкции вполне реально [15, 148]. Мониторинг этих молекул в пристеночной зоне стал задачей для многих групп. С учетом соображений разд. 5.1, размеров камеры ИТЭР (1400 м3), производительности откачки и дополнительной инжекции частиц взамен выбывших при неполном рециклинге (восстановление нейтралов при электрон-ионной рекомбинации на стенке), в проекте [15] задан максимально допустимый поток Q молекул воды в камеру. Он составил 10–7 Па ⋅ м3 с–1 , или 2.65 × 1013 с–1 по числу поступающих частиц (величине 1 Па ⋅ м3 с–1 = = 10‒2 мбар ⋅ м3 с–1 = 10 мбар ⋅ л ⋅ с–1 = 1 Вт соответствует поток 2.65 × 1020 с–1).
Это подразумевает необходимую чувствительность измерений концентраций. Планируемое время удержания плазмы 10 с при длительности разряда (400–3000) задает необходимое быстродействие. Важным условием является отыскание мест (локализация) проникновения частиц в камеру от первой стенки (680 м2, 480 модулей бланкета в ИТЭР [135–143]). В совокупности это делает проблему диагностики весьма сложной, не имеющей известных аналогов.
Вывод реактора на рабочий режим будет проводиться поэтапно в течение нескольких лет. После завершения монтажа должна проводиться диагностика вакуумной плотности конструкции. На следующем этапе будут проводиться обработка внутренней поверхности первой стенки разрядом, тестовые испытания с разрядами в водороде, затем в дейтерии и в их смесях и только затем в смеси D–T. Это связано с отладкой работы систем при экономии трития и ресурса радиационной стойкости самой установки. В соответствии с этим следует ориентироваться на три группы диагностических методов и средств. Первая используется для контроля реактора в “холодном” состоянии не только после завершения строительства, но и во время технологических “окон” при ремонте, профилактике, внесении конструктивных коррекций. При этом, однако, не будут замечены возможные дефекты, образующиеся при нагрузках, определяемых разрядом. На следующем этапе с разрядом в водороде и дейтерии вторая группа диагностик должна быть уже полноценной, с выявлением таких дефектов, и, хотя некоторые вариации условий допустимы, после этой стадии средства третьей группы для мониторинга рабочего режима должны быть обеспечены. Вполне вероятно, что методы второй и третьей групп будут близки, хотя выявленные возможные трудности должны быть учтены. Общим для всех групп является условие дистанционности измерений. Для второй и третьей групп это очевидно, для первой диктуется требованиями безопасности даже в перерывах между запусками реактора с разрядами в водороде и дейтерии также по требованиям безопасности.
С другой стороны, несмотря на возможность такого поэтапного ввода диагностик, специфика, усложняющая задачу в целом, связана с тем, что уже отработанные на действующих ректорах методы, хотя и требуют адаптации, но ориентированы на измерения в активной горячей области плазмы. Среди них можно выделить классический метод рассеяния Томсона, многократно апробированный на лабораторных плазменных установках и нескольких поколениях реакторов, но требующий применения современных мощных лазеров с короткими импульсами, а также методы активной спектроскопии. Первым методом исследуются, в основном, параметры электронной составляющей, но, поскольку в этой зоне модель ЛТР хорошо выполняется, из измерений могут быть извлечены и иные параметры. Второй основан на инжекции узкого диагностического пучка атомов водорода. При скоростях атомов, сопоставимых со скоростями движения электронов на атомных орбитах, обеспечивается проникновение пучка в центр плазмы и максимальное (резонансное) сечение перезарядки. Для ИТЭР обсуждается использование атомного пучка с энергией ~100 кэВ и мощностью 2.2 МВт. Спектры каскадных переходов с высоких уровней образующихся ионов могут использоваться для измерения концентраций частиц различного сорта, включая примеси. Оба метода количественные и способны обеспечить достаточно острую локальность. При этом, однако, область плазмы вблизи стенки, до границы транспортного барьера (Н-мода, на которую рассчитан ИТЭР) с малыми энергиями и концентрациями электронов остается вне поля зрения. Помимо технических вопросов, связанных с чувствительностью, попытки адаптировать эти методы к пристеночному слою, встречают принципиальные трудности, связанные с отсутствием равновесия, выделяя эту проблему в самостоятельную.
В упомянутой выше первой группе проводится ряд работ, связанных с моделированием газодинамики течей и исследованием камер на вакуумную плотность. Так, в [148, 149] проведены расчеты газодинамики течей газа через узкий канал в стенке. При температуре стенки 450 К, внешнем давлении 40 атм и длине канала 1 см расчетная скорость натекания молекул воды составляет при диаметре капилляра 1 мкм величину Q = 1.5 × × 10‒4 Па ⋅ м3 с–1, при 0.1 мкм – 10–8 Па ⋅ м3 с–1, при 0.03 мкм – 10–10 Па ⋅ м3 с–1, т.е. допустимое натекание возникает при наличии 10 одинаковых каналов диаметром 0.1 мкм. В работе [150] эта модель модифицирована на случай скрытых течей, когда молекулы проникают через узкое отверстие на внешней стороне стенки и затем мигрируют в камеру через систему каналов и трещин бланкета. При некоторых предположениях скорость поступления молекул в камеру может быть на 2–3 порядка выше, чем в случае узкого канала такого же диаметра, что и у отверстия в тонком слое элемента оболочки.
Эксперименты первой группы проводились в различных условиях, в той или иной мере моделирующих ИТЭР. В этих работах используются адаптированные средства традиционного газового анализа высокой чувствительности.
Ведется разработка многоцелевого носителя “робота–руки” [151–153], тестированного на токамаке Tore Supra. Это устройство учитывает особенности геометрии конкретной камеры и может размещать внутри нее различные приборы, например, течеискатели, масс-спектрометры, лазерные спектрометры и пр. Другая группа использует метод прямых измерений потоков воды с помощью лазерной абсорбционной спектроскопии в трассовой схеме [154]. Эксперименты проводятся на установке TITAN, созданной в 2011 г. В ней объем вакуумной камеры 18 м3, стенки можно нагревать до 250°С. В систему введен калиброванный натекатель, позволяющий создавать потоки He или H2O на уровне 10–5 Па ⋅ м3 с–1, однако реальные измерения проводились при больших потоках. Прямые измерения по поглощению света молекулами воды продемонстрировали чувствительность метода на уровне потоков 10–2 Па ⋅ м3 с–1, предельная экстраполяция прямых измерений дает результат 10–5 Па ⋅ м3 с–1. Планируемый уровень обнаруживаемых течей после предполагаемой модификации измерительной схемы с использованием внешнего резонатора (разд. 4) – около 10–6 Па ⋅ м3 с–1, что пока тоже недостаточно для ИТЭР.
В работе [155] предложена сенсорная система сигнализации появлении течи по изменению давления в системе охлаждения реактора. Подобная система также тестировалась ранее на установке Tore Supra [156]. Кроме определения конкретного контура охлаждения, из которого происходят утечки теплоносителя, в [156] предложена схема удаления попавших в камеру паров воды. Поскольку в конструкции предусмотрено 5 контуров охлаждения, на каждый из которых приходится несколько десятков элементов бланкета, более детальная локализация места течи воды в камеру проблематична.
В более ранней работе [157] предлагалось добавлять малые (на уровне 0.1%) маркеры в воду охлаждения. При помощи масс-спектрометра в канале откачки камеры реактора они будут обнаружены в случае образовании течи. Однако установление необходимой для регистрации концентрации маркера в воде занимает в лучшем случае несколько часов (для наиболее приемлемого маркера Хе), и проблема локализации та же, что и указанная выше.
Использование традиционных методов с гелиевыми течеискателями также встречает трудности, поскольку требует тщательной осушки контуров охлаждения.
Вторая группа работ практически полностью ориентирована на спектральные методы. Как на одно из немногих исключений укажем работы [158, 159], где используется эффект возникновения автоколебаний разряда при наличии молекул воды. Этот контактный метод основан на влиянии оксидной пленки на эмиссионную способность некоторых материалов катода, продемонстрированная чувствительность (10–4 Па ⋅ м3 с–1) пока недостаточна.
Из предшествовавших разделов 3 и 4 видно, что к настоящему времени разработано большое количество спектроскопических методов прямых измерений концентрации атомов и молекул с высокой чувствительностью в плазме при отсутствии в ней равновесия для многих объектов. Предметный анализ этих и ряда других методов показывает, однако, что в отношении применимости к токамакам типа ИТЭР большинство из них встречают серьезные проблемы. Главная – техническая сложность, радиационные нагрузки и ограниченная доступность к активной среде. При этих обстоятельствах предпочтительно выглядят наиболее простые в техническом отношении методы, использующие спектры спонтанного излучения плазмы. Парадоксально, но, как отмечалось в разд. 3, именно они требуют наиболее детального физического обоснования для получения количественных результатов при использовании их для исследований конкретных неравновесных плазменных объектов. Работы в этом направлении ведутся рядом групп.
Поскольку ИТЭР пока не работает, потом еще длительное время будет вводиться в рабочий режим, и натурные эксперименты пока невозможны, на стадии подготовки необходимо:
− использовать и/или специально разработать установки для экспериментального моделирования условий, по возможности близких к таковым в пристеночной плазме реактора;
− отработать на модельных установках приемлемые количественные спектральные методы определения концентраций молекул воды и их фрагментов, определить потоки их поступления в плазму, выяснить условия локализации измерений;
− поскольку полное воспроизведение условий для уникального строящегося реактора на модельных установках проблематично, необходимо создать расчетную модель превращений частиц в плазме модельных установок, протестировать ее для совокупности возможно большего числа различных частиц и условий экспериментов с целью масштабирования на условия плазмы реактора.
Еще на стадии разработки проекта [15] по заданиям ИТЭР были выполнены несколько экспериментальных исследований по обсуждаемой теме. Спектроскопически были измерены концентрации атомов кислорода по излучению тлеющего разряда в ИК-области (см. [150]), достигнуты предельные чувствительность 10–6 Па ⋅ м3 с–1 и пространственное разрешение несколько м2.
В работах [149, 150] проведены эксперименты с разрядами на установке “пробкотрон” ПР-2 при давлениях (10–2–10–4) Торр в инертных газах, водороде, дейтерии, воздухе и их смесях. Изучение спектров излучения показало, что, независимо от состава плазмообразующего газа, в области (230–350) нм доминируют полосы гидроксила ОН. Интенсивности этих полос сопоставлены с результатами параллельных масс-спектрометрических измерений концентраций молекул воды, и на основе экстраполяции сделан вывод, что в этих экспериментах полосы ОН могут быть заметны при давлениях паров воды до 2 × 10–4 Торр. Остальные оценки носили качественный характер. Важный вывод из результатов этих работ состоял в том, что полосы ОН, наблюдаемые, в том числе, в спектрах инертных газах без добавок воды, могут служить чувствительным индикатором влажности.
Предложение [148–150] было более подробно изучено в работе [160], в которой измерения интенсивностей свечения полос радикалов ОН в разряде с полым катодом (1 × 5 × 10 см3) проводилось с параллельными калибровками при помощи инертных газов известной влажности. Достигнутая чувствительность к потоку натекания воды составила 10–5 Па ⋅ м3 с–1.
Комплекс спектроскопических исследований проведен на стеллараторе Л-2М (большой радиус 1 м, малый 0.115 м) [161–163]. В них также проведены измерения по свечению ОН по методике работы [160], подтвердившие чувствительность 10–5 Па ⋅ м3 с–1 [162, 163]. Параллельно проверялась идея работ [153, 157] об использовании Хе в качестве маркера [161, 163], но в данном случае маркер детектировался по спектру излучения. В отличие от [153, 157], ксенон не растворялся в воде, а напускался непосредственно в камеру стелларатора с помощью специального натекателя с контролируемым потоком через малое отверстие. Хордовые измерения показали чувствительность к потоку ксенона QХе ~ 10–6 Па ⋅ м3 с–1, точность локализации течи ~5 мм. При концентрации Хе, растворенного в охлаждающей воде в количестве 1%, близком к возможному, чувствительность данной методики к потоку молекул воды могла бы составить ${{Q}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$ ~ 10–4 Па ⋅ м3 с–1. Для установок, охлаждаемых потоком воды с большим расходом, использование растворяемого благородного газа в качестве маркера, помимо недостаточной чувствительности к молекулам воды, затратно, требует специальной подготовки воды и необходимости технологически сложной обработки стенок вакуумной камеры после каждого аварийного случая или тестового запуска системы диагностики из-за взаимодействия маркера со стенкой.
Таким образом, после многих экспериментов с эмиссионными спектрами наибольшая чувствительность была продемонстрирована с помощью регистрации интенсивностей полос гидроксила ОН(Х2П-А2Σ), но прогресс остановился на уровне потоков Q ~ (10–5–10–6) Па ⋅ м3 с–1. И даже при этом оставался вопрос о том, насколько адекватно во всех предыдущих работах делалось соотнесение интенсивности этих полос с концентрацией “родительских” молекул Н2О.
5.3. Модельные установки
Большая часть исследований проводились на двух установках. Установка “Течь” (рис. 10, 11), моделирует свойства плазмы разряда на этапах первичной обработки бланкета первой стенки реактора (планируется, в том числе, тлеющий разряд) и в режимах с разрядами в водороде и дейтерии [47, 49, 55–58, 72, 84–86, 164–167]. Бланкетная конструкция первой стенки в местах соединения элементов структуры при горении в камере разряда потенциально наиболее подвержена появлению течей либо непосредственно через стыки, либо в результате миграции молекул по внутренним каналам конструкции. Моделью этих областей может быть разряд, поддерживаемый полым катодом. Вторая установка – с тлеющим разрядом в трубке (рис. 12, 13) – используется для моделирования условий в положительном столбе и для сравнения различных методов измерений [58, 82, 83, 168–172].
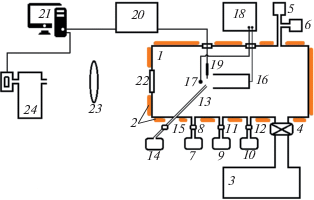


Установка “Течь”. Объем камеры 1 из нержавеющей стали составляет 22 л. Нагреватель 2 варьировал температуру стенок (295–450) К, остаточное давление 10–7 мбар обеспечивалось вакуумной станцией 3, соединяемой с камерой шибером 4. Датчики 5, 6 контролировали давление газов (10–9–1000) мбар, напускаемых из баллонов 7, 9, 10 и контейнера с водой 14 через натекатели 8, 11, 12, 15. Кроме паров воды использовались газы He, Xe, Ar, Н2, D2. Использовался разряд, поддерживаемый полым катодом 16 100 × 50 × 10 мм3 с открытой стороной 100 × 10 мм2, обращенной к аноду 17 в виде вольфрамовой п-роволоки диаметром 1.5 мм или вольфрамовой сетки (100 × 10 мм) на расстоянии 10–15 мм от края катода. Выходящее через окно 22 излучение разряда коллимировалось и фокусировалось оптической системой 23 на щель спектрографа 24 со сменными дифракционными решетками и фотоэлектрической регистрацией. Сигнал с ФЭУ поступал на компьютер 21.
Для измерений абсолютных интенсивностей излучения в спектральных линиях блок полого катода (16, 17) замещался калиброванной ленточной лампой. Для измерения параметров электронов использовалась схема одиночного зонда Ленгмюра 19 из вольфрамовой проволоки диаметром 0.04–0.1 мм, выступающей из керамического изолятора на 2–3 мм. Зонд мог устанавливаться в различных местах разряда. О методиках обработки зондовых характеристик, см. [167]. На рис. 11 приведен вид вакуумной камеры.
Установка с тлеющем разрядом (рис. 12, 13) предназначена для проведения сопоставительных эмиссионных и абсорбционных лазерных измерений с контролем напряженности аксиального электрического поля. Кварцевая трубка 1 использовалась как разрядная с электродами 2, 2', 3 и высоковольтным источником тока 4. Три электрода использовались для исключения приэлектродных падений напряжения при контроле аксиального электрического поля в положительном столбе; 5, 6 – каналы откачки и напуска газов; 7, 7 '– ввод и вывод воды в рубашку охлаждения трубки; 8 – диодный лазер, 9, 10 – зеркала с отраженим 0.99–0.9998 на длине волны 1392 нм; 11 – делительная пластина; 12 – эталон Фабри-Перо; 13, 16 – детекторы PDA10CS-EC, ThorLabs; 14 – линза; 15 – интерференционный фильтр для выделения участка сперктра при лазерных измерениях; 17 – плата PCI-6120, National Instruments, осуществлявшая вывод сигналов управления лазером 8 и ввод сигналов с детекторов 13, 16 в компьютер 18 со средой LabVIEW, в которой генерировались сигналы управления и обрабатывались сигналы с детекторов. В актинометрических измерениях зеркало 10 заменялось кварцевым окном 10', фильтр 15 устранялся, а на место детектора 16 помещался монохроматор 16' (МДР-204) с ФЭУ, сигнал с которого поступал на компью-тер 18. Фрагмент установки показан на рисунке 13.
5.4. Измерения абсолютных концентраций молекул воды по тонкой структуре эмиссионного электронного спектра гидроксила
Отмеченная выше проблема неоднозначности соотнесения интенсивностей свечения фрагментов молекул воды с концентрацией Н2О путем различных калибровок [160, 163] рассмотрена в работе [164] принципиально иным способом.
При экзотермической реакции в неравновесных условиях избыток энергии перераспределяется между различными степенями свободы продуктов. Такая ситуация изучалась в отношении возбуждения электронных излучающих состояний атомов и молекул в различных элементарных процессах [4]. Это, в частности, относится и к процессам возбуждения радикалов ОН [173–180]. Последнее иллюстрируется рис. 14 [164].
Рис. 14.
Два механизма возбуждения свечения гидроксила электронным ударом: I – прямое возбуждение гидроксила из основного состояния; II – диссоциативное возбуждение молекул воды.
Известны два основных механизма возбуждения состояния ОН(A2Σ), излучающего фиолетовые полосы.
I. Если в плазме каким-то образом образовался свободный гидроксил в основном состоянии ОН(Х2П), то он может возбуждаться электронами в состояние ОН(A2Σ):
(30)
$\begin{gathered} е + {\text{ОН(}}{{{\text{Х}}}^{2}}{\text{П}}) \to е + {\text{О}}{{{\text{Н}}}^{{\text{с}}}}{\text{(}}{{{\text{A}}}^{2}}\Sigma ) \to \\ \to е + {\text{ОН}}({{{\text{Х}}}^{2}}{\text{П}}) + h\nu . \\ \end{gathered} $В электронном спектре при таком процессе (30) преобладают структуры полос “холодной” группы ОНс(A2Σ) [173–176] с вращательной температурой, близкой к температуре газа, поскольку в основном состоянии вращения молекулы находятся в равновесии с кинетическим движением газа, а электрон при возбуждении электронного состояния молекулы не может передать ей значительный момент импульса. Поэтому по интенсивностям линий во вращательной структуре полосы, возбуждаемой в процессе I, можно определять температуру газа.
II. В процессе диссоциативного возбуждения молекул воды электронами молекула Н2О переходит из основного связанного состояния Х1А1 в отталкивательное В1А1 и распадается на атом водорода в основном состоянии и возбужденную молекулу ОН(A2Σ) с последующим излучением.
В основном состоянии молекула воды изогнута под углом 105° (рис. 14), и при переходе в линейное состояние В1А1 избыток энергии деформации переходит к тепловому, колебательному и вращательному движениям. Легкий водород принимает основную долю кинетической энергии. Более тяжелый радикал ОН(A2Σ), напротив, мало меняет скорость, но начинает быстро вращаться, формируя т.н. “горячие” вращательные ансамбли ОНh(A2Σ):
(31)
$\begin{gathered} е + {{{\text{Н}}}_{{\text{2}}}}{\text{О(}}{{{\text{Х}}}^{{\text{1}}}}{{{\text{А}}}_{{\text{1}}}}{\text{)}} \to е + {{{\text{Н}}}_{{\text{2}}}}{\text{О(}}{{{\text{В}}}^{{\text{1}}}}{{{\text{А}}}_{1}}{\text{)}} \to \\ \to е + {\text{О}}{{{\text{Н}}}^{{\text{h}}}}({{{\text{A}}}^{2}}\Sigma ) + H \to е + {\text{ОH(}}{{{\text{Х}}}^{{\text{2}}}}{\text{П)}} + H + h\nu . \\ \end{gathered} $В электронном спектре электронно-колебательных полос это проявляется в виде развитой вращательной структуры полос ОН(А–Х) – “горячие” группы.
На рис. 15 приведен фрагмент вращательной структуры спектра (0, 0) полосы перехода ОН(A2Σ–Х2П), полученный на установке “Течь” [164]. Разряд в смеси Н2О : Не = 0.075 : 0.925, давление 1 мбар, ток 0.23 А. Отмечены линии ветви Q1(K) по данным [4, 181] для спектров высокого разрешения, K – вращательное квантовое число полного момента без спина. На вставке в полулогарифмическом масштабе показана зависимость Z = IK(SKνK4)–1 от энергии вращательного терма F(K) = BK(K + 1) состояния ОН(A2Σ). Здесь IK – интенсивности линий, SK – факторы Хёнля– Лондона, νK – частоты переходов (для Q1 ветви значения K нижнего и верхнего уровня совпадают), В ~ 18 см–1 – вращательная постоянная.
Рис. 15.
Фрагмент спектра вращательной структуры (0, 0) полосы перехода ОН(A2Σ–Х2П). Указаны линии Q1(K) ветви. На вставке – зависимости интенсивностей линий от энергии вращательного терма [164].
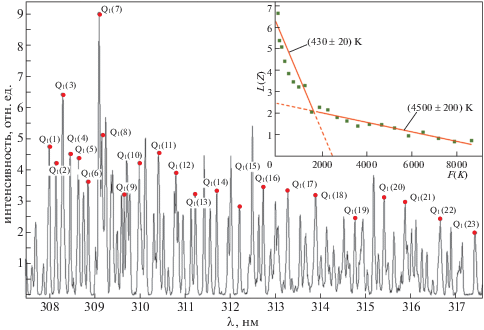
Видно, что молекулы ОН(A2Σ) разбиваются на две больцмановские группы. Вращательная температура “холодных” молекул, образующихся в процессе (30), соответствует кинетической температуре нейтрального газа плазмы Tс ~ 400 К. Это, как указывалось выше, связано с ограничениями на передачу момента импульса молекуле электроном. Уровни с K > 10 (310–320 нм) заселены практически только “горячими” молекулами c вращательными температурами Th ~ 4.5 × 103 К. В этом случае наличие парциальной температуры объясняется статистическим законом распада возбужденного комплекса Н2О(В1А1) [173–176]. В экспериментах [164] с разными влажными газами эти температуры менялись: Tс ~ (320–500) K, Th ~ ~ (4–7) × 103 К, но всегда резко различались между собой, что позволяло их надежное разделение. Из условия баланса для концентрации возбужденных молекул горячей группы $N_{{{\text{OH}}}}^{h}$ в стационарных условиях следует, что
(32)
${{N}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}} = \frac{1}{{{{\tau }_{{{{A}^{2}}\Sigma }}}}}\frac{{N_{{OH}}^{h}}}{{{{n}_{e}}\left\langle {{{v}_{e}}{{\sigma }_{{de}}}} \right\rangle }}.$Здесь ${{\tau }_{{{{A}^{2}}\Sigma }}}$ – время жизни состояния ОН(A2Σ), к которому принадлежат вращательно-горячие молекулы, ${{v}_{е}}$ – скорости электронов, σde – сечение диссоциативного возбуждения в процессе (31). Время жизни определяется радиационным распадом и тушением:
(33)
$\frac{1}{{{{\tau }_{{{{A}^{2}}\Sigma }}}}} = {{A}_{{{{A}^{2}}\Sigma }}} + \sum\limits_i {{{N}_{i}}\left\langle {{{v}_{i}}{{\sigma }_{{iq}}}} \right\rangle } ,$Таблица 5.
Константы скоростей тушения
| Частицы излучающие | λ, нм | AA,X, с–1 τA,X, нс | Тушащие частицы i$q_{{{\text{X,A}}}}^{i}$, 10–10 см3 ⋅ с–1 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| H2O | He | Ar | Xe | D2 | Н2 | О2 | |||
| OH A2Σ+ – X2П | 306–320 | 1.29×106 773 ± 47 [90] | T = 300 K [178] | ||||||
| 5 | ≤0.0001 | ≤0.0004 | 1.8 [179] | 0.7 | 0.92 | 1.0 | |||
| T = 450 Ka) | |||||||||
| 6.1 | ≤0.00012 | ≤0.00049 | 2.2 | 0.86 | 1.0 | 1.2 | |||
a) Зависимость констант скоростей тушения определяется зависимостью $q_{{{\text{X,A}}}}^{i}\sim {{T}^{0}}^{{.5}}$ [180]. Константы (300 К) тушения на молекулаx из разных экспериментальных работ разумно согласуются друг с другом. Для тушения на He и Ar число работ невелико и разброс констант составляет несколько порядков. Поскольку, однако, их величины не превышают 0.1 для Не и 0.01 для Ar, мы приводим для них данные из критического обзора [178].
Поскольку добавки инертных газов, водорода и дейтерия к основному инертному газу (в проводимых экспериментах обычно гелий) малы, ~(1–10)%, как и константы скоростей тушения с их участием, основную роль в тушении могут играть молекулы воды при ${{N}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$ > 1015 cм–3. Для рассматриваемых условий интерес представляют следы Н2О с ${{N}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$ < (1014–1013) cм–3, можно полагать (${{\tau }_{{{{A}^{2}}\Sigma }}}$)–1 ≈ ${{A}_{{{{A}^{2}}\Sigma }}}$, и формула (32) будет иметь вид
(34)
${{N}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}} = \frac{{{{A}_{{{{A}^{2}}\Sigma }}} \cdot N_{{{\text{OH}}}}^{h}}}{{{{n}_{e}}\left\langle {{{v}_{e}}{{\sigma }_{{de}}}} \right\rangle }} = \frac{{I_{{}}^{h}}}{{{{n}_{e}}\left\langle {{{v}_{e}}{{\sigma }_{{de}}}} \right\rangle }}\frac{1}{{h\nu }},$Таким образом, для измерений концентрации молекул воды в плазме требуется определить абсолютную интенсивность группы линий во вращательной структуре перехода ОН(A2Σ–Х2П), концентрацию электронов и их распределение по энергиям. Это стандартные методики [182, 183], и их выполнение предусмотрено на установке “Течь” [164, 167]. Результаты измерений продемонстрировали возможность измерения концентраций молекул воды на уровне 1012 см–3. При этом надежно контролируются изменения концентрации молекул 1013 см–3 с–1, что удовлетворяет ранее не достигавшимся экспериментально требованиям проекта ИТЭР по обнаружению потоков молекул H2O в камеру на уровне 10–7 Па ⋅ м3 с–1 (см. разд. 5.2).
5.5. Мультиспектральная актинометрия
Описанный в разд. 5.4 метод требует привлечения контактных зондовых методов и абсолютных измерений интенсивностей. Это легко выполнимо на модельных установках, но не всегда приемлемо для реальных объектов. Еще одна трудность в том, что соответствующий рабочий спектральный диапазон 306–320 нм. В установках с высоким радиационным фоном, что характерно для термоядерных реакторов, оптика подвержена наибольшей деградации именно в коротковолновой УФ-области. Сохраняя ценность данного метода как контрольного и калибровочного для других методов, можно рассмотреть приемы, минимизирующие эти трудности.
В частности, в этой связи в работах [49, 55] предложена новая версия классической актинометрии – мультиспектральная актинометрия (МА). Как отмечалось в разд. 3.2, актинометрический подход не требует знания концентрации электронов и сильно снижает требования к точности знания их энергетического спектра (-ФРЭЭ). Если, однако, измеряются малые концентрации частиц, остается проблема учета тушения излучающих состояний как детектируемой частицы, так и актинометра. Если в формуле (6) величина AXul ≤ QXu и AAul ≤ QAu, тушением излучающих состояний уже нельзя пренебречь.
Константы скоростей тушения многих состояний частиц известны из литературных баз данных, в первом приближении оценку влияния тушения можно провести исходя из коэффициентов Эйнштейна и химического состава плазмообразующей газовой смеси. Однако в реальной химически активной плазме заранее не известно, какие частицы следует учитывать при определении скорости тушения.
Для интерпретации результатов измерений интенсивностей IX,A в химически активной плазме предлагается процедура [49], которая включает ряд последовательных итераций, каждая из них использует два приближения.
В первом приближении для каждой актинометрической пары записывается система, состоящая из уравнений (6), в которых в членах QX,A присутствуют концентрации измеряемых частиц, известные концентрации актинометров и буферного газа:
(35)
${{N}_{{{{X}_{i}}}}} = {{N}_{{{{A}_{j}}}}}{{G}_{{{{X}_{i}}{{A}_{j}}}}}{{K}_{{{{X}_{i}}{{A}_{j}}}}}{{I}_{{{{X}_{i}}{{A}_{j}}}}}\frac{{{{Q}_{{{{X}_{i}}}}} + {{A}_{{{{X}_{i}}ul}}}}}{{{{Q}_{{{{A}_{i}}}}} + {{A}_{{{{A}_{j}}ul}}}}},$(36)
$\frac{{{{C}_{A}}}}{{{{C}_{X}}}}\frac{{{{\nu }_{A}}}}{{{{\nu }_{X}}}}\frac{{{{A}_{A}}}}{{{{A}_{X}}}} = {{G}_{{AX}}},\quad \frac{{{{k}_{A}}}}{{{{k}_{X}}}} = {{R}_{{AX}}},\quad \frac{{{{I}_{A}}}}{{{{I}_{X}}}} = {{I}_{{XA}}}.$Уравнения системы (35) нелинейные, поскольку скорости тушения Q зависят от концентраций частиц:
(37)
${{Q}_{{{{X}_{i}}}}} = {{q}_{{{{X}_{i}}}}}\sum\limits_{j,l} {({{N}_{{{{X}_{j}}}}} + {{N}_{{{{A}_{l}}}}}} ),$(38)
${{Q}_{{{{A}_{i}}}}} = {{q}_{{{{A}_{i}}}}}\sum\limits_{j,l} {({{N}_{{{{X}_{j}}}}} + {{N}_{{{{A}_{l}}}}})} .$В первой итерации определяются концентрации только тех частиц, помимо буферного газа и актинометров, которые составляют актинометрические пары. Точность любых измерений конечна. Если концентрация частиц одного сорта определяется независимо по нескольким (М) актинометрам, и результаты отличаются в пределах, допустимых точностью измерений интенсивностей и расхождения данных о сечениях в различных источниках без обоснованного предпочтения, берется среднее арифметическое
(39)
${{N}_{{{{X}_{i}}}}} = {{\left( {\sum\limits_j {{{N}_{{{{X}_{i}}{{A}_{j}}}}})} } \right)} \mathord{\left/ {\vphantom {{\left( {\sum\limits_j {{{N}_{{{{X}_{i}}{{A}_{j}}}}})} } \right)} M}} \right. \kern-0em} M}.$Второе приближение учитывает то, что в разряде, помимо исходных, образуются и другие частицы (например, вследствие диссоциации молекул и т.д.). Если одна или несколько “новых” частиц детектируются по спектру или концентрация именно этих частиц и измеряется, записывается брутто-химический баланс частиц [49] с сохранением числа атомов всех сортов. Это условие также включается в систему уравнений. При этом становится возможным определить концентрации компонент, измерения которых не проводились.
Концентрации всех частиц после второго приближения подставляются в члены QX,A, после чего система решается опять и находятся новые концентрации атомов и молекул. Итерации повторяются до сходимости в пределах необходимой точности определения концентрации NX. Такой метод был успешно применен в работе [49] для измерений концентраций H2O, O, H и ОН в смеси, изначально состоящей из Не и H2O, в разряде с полым катодом.
Частным случаем МА является H/D-метод для исследования плазмы, содержащей водород и дейтерий, что важно для термоядерных реакторов. В [184] при работе на установке “пробкотрон” ПР-2 было замечено, что если плазмообразующий газ содержит дейтерий, то соотношение интенсивностей линий Dα (656.09 нм) и Hα (656.27 нм) изменяется, и высказано предположение, что такое соотношение может использоваться в качестве индикатора влажности плазмы. Параллельные исследования на установке “Течь” [55] показали, при мультиспектральном подходе этот метод становится количественным. Пример записи спектра на установке “Течь” показан на рис. 16 [55].
Рис. 16.
Спектры дублета Dα–Hα. Начальное парциальное давление D2 равно ${{p}_{{{{{\text{D}}}_{2}}}}}$ = 0.05 мбар (p(He + Ar + Xe + $_{{{{{\text{D}}}_{2}}}}$) = 0.65 мбар). Кривая 1 – без добавок H2O; 2 – ${{p}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$ = 0.05 мбар; 3 –${{p}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$ = 0.1 мбар. Тонкая структура линий не разрешена.
Особенностью этой пары, если ее в рамках мультиспектрального подхода рассматривать как актинометрическую, является то, что для двух атомных изотопов сечения возбуждения соответственных состояний практически идентичны. Это означает, что при измерении концентраций параметры электронной компоненты исключаются полностью. При этом уравнение (6) дополнительно упрощается ввиду малости различия длин волн λH/λD ≈ СD/СH ≈ kD/kH ≈ 1, и, поскольку радиационные времена жизни излучающих состояний (табл. 2) 2D3/2 и 2P3/2,1/2 составляют (18–45) нс, при давлениях в несколько мбар и менее тушением можно пренебречь. Тогда (6) принимает вид
(40)
$\frac{{{{N}_{{\text{H}}}}}}{{{{N}_{{\text{D}}}}}} = \frac{{{{I}_{{\text{H}}}}}}{{{{I}_{{\text{D}}}}}}.$Метод МА расширяет возможности традиционной актинометрии. В частности:
− расширяется выбор актинометрических пар и числа частиц, концентрации которых могут быть измерены;
− в качестве актинометров могут быть использованы не только те частицы, концентрации которых по сравнению с плазмообразующим газом не меняются (в основном, инертные газы), но и вновь образованные, в том числе нестабильные, частицы. Они играют роль промежуточных актинометров, поскольку их концентрация определена с помощью основных актинометров;
− из-за расширения выбора актинометрических пар рабочая область спектра может варьироваться;
− из измерений одного актинометрического отношения может быть определена концентрация ряда других частиц.
Приведем несколько примеров.
Первый – в рассмотренном выше методе определения молекул Н2О по радикалам ОН с быстрым вращением требовалось проводить зондовые измерения параметров электронов. При введении малых добавок Хе можно использовать метод МА, пользуясь тем, что сечения возбуждения Хе (823 нм) и диссоциативного возбуждения Н2О имеют близкие пороги и подобны друг другу, определить концентрацию Н2О, не прибегая к зондовым измерениям. Даже при сильных вариациях ФРЭЭ связанная с этим погрешность не превышает 10%. При этом можно дополнительно определить концентрации О, Н, и полную концентрацию ОН в основном состоянии.
Другой пример. В работе [55] актинометрическая пара H-D использована для измерений потока ${{Q}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$ натекания молекул воды в условиях установки “Течь”. Для этого вначале производится калибровка при известных количествах $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ молекул воды в исходной смеси и находится коэффициент γ в соотношении
(41)
$N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0} = \frac{1}{\gamma }{{N}_{{\text{D}}}}\frac{{{{I}_{{\text{H}}}}}}{{{{I}_{{\text{D}}}}}},$Чувствительность к внешнему потоку молекул воды будет определяться как
(42)
$Q_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{}} = \frac{{\Delta \left( {N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}} \right)}}{{\Delta t}} = \frac{{\alpha \Delta \left( {N_{{\text{H}}}^{{}}{\text{/}}N_{{\text{D}}}^{{}}} \right)}}{{\Delta t}}.$Заметим также, что калибровка (41) универсальна и может быть сделана на лабораторной установке, поскольку отношение IH/ID от условий в плазме не зависит. Единственным условием является контроль атомов дейтерия в реальном объекте, но это делается простым способом традиционной актинометрии в удобной спектральной области. Весьма важно, что в данном случае метод МА позволяет значительно увеличить чувствительность при малых концентрациях дейтерия в модельной установке до ${{Q}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$ = 5 × 10–9 Па ⋅ м3 с–1 [55].
5.6. Локализация измерений в разряде, поддерживаемом полым катодом
На схеме установки “Течь” (рис. 10) показано, что ввод паров воды в плазму проводился из контейнера 14 через капилляр 13. Внутренний диаметр капилляра 1 мм. В экспериментах по влиянию паров воды при давлении 0.7 Торр при таком вводе проявлялась динамика установления однородного свечения разряда в гелии, поддерживаемого полым катодом [57, 160]. Мониторинг свечения разряда проводился с помощью скоростной камеры. Фотографировалась вся область разряда. При закрытом натекателе 15 слабое свечение гелия заполняло всю область напротив открытой стороны полости катода, практически повторяя ее форму. При быстром открытии натекателя в начальный момент в области инжекции на фоне свечения гелия возникало более яркое свечение из-за изменения локального состава плазмы (рис. 17). Интервал между снимками 3 мс. Из рис. 17б видно, что уже через (6–7) мс яркое свечение заполняет всю область разряда. Это согласуется с оценкой времени диффузии частиц. В условиях данного эксперимента стационарная трехмерная локализация оказывается невозможной из-за малых линейных размеров плазмы (10 × 2 × 1 см3), порядка длины диффузии. При этом, однако, 2D локализация существует в стационарном режиме, поскольку время жизни излучающих состояний частиц мало и свечение прекращается после ухода частицы из зоны возбуждения. В системах большего размера ситуация может быть иной и будет обсуждаться ниже.

5.7. Измерения концентраций изотопомеров воды в разрядной трубке
Проводились сопоставительные измерения методами диодной лазерной спектроскопии поглощения и эмиссионной актинометрии методами диодной лазерной спектроскопии (рис. 12, 13). Исходная газовая смесь – He : Xe (99 : 1) с добавками Н2О и D2 [82, 83].
Эмиссионные измерения проводились методом выделения “горячих” вращательных групп. В отсутствие дейтерия измерения концентрации молекул ОН в этих группах осуществлялись с использованием Хе как актинометра (линия 823 нм, табл. 2) без измерения параметров электронной компоненты. Концентрации О2 и Н2 для учета тушения находились из брутто-баланса $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ – ${{N}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$ = ${{N}_{{{{{\text{H}}}_{2}}}}}$ = = $2{{N}_{{{{{\text{O}}}_{2}}}}}$ , где $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ – начальная концентрация молекул воды в камере до зажигания разряда.
Спектр поглощения Н2О, записанный на лазерном спектрометре (ДЛС), показан на рис. 18.
Использовалась довольно большая относительная доля молекул воды H2O : He : Xe (148 : 99 : 1). При такой концентрации колебательная релаксация молекул происходит быстро, и внутренняя статистическая сумма, необходимая для расчета концентрации, может рассчитываться как равновесная, когда колебательные температуры всех мод Н2О совпадают с вращательной и газовой. Газовая температура измерялась по доплеровскому уширению в случае спектра лазерного поглощения и отождествлялась с температурой “холодной” группы ОН – при эмиссионных измерениях. Результаты совпадали.
И в отношении концентраций молекул H2O оба метода также дают согласующиеся в пределах погрешностей результаты (рис. 19).
При добавлении в исходный газ молекул дейтерия в спектре поглощения появляются линии молекул HDO (рис. 20) [168–172].
Рис. 20.
Экспериментальный спектр поглощения H2O и HDO в диапазоне перестройки лазера [168]. На врезке пример аппроксимации линии перехода 7181.31 см–1 HDO по Гауссу. 1 – измерения, 2 – аппроксимация.
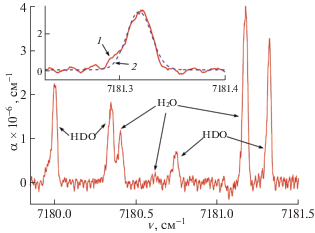
При обработке спектров поглощения использовались линии, для которых отношение интенсивностей на входе и выходе резонатора находилось в пределах 0.2 < I/I0 < 0.4, что обеспечивает лучшую точность измерений [97].
На рис. 21 показаны результаты измерений концентраций HDO в зависимости от времени горения разряда при токе 4 мА в гелии $N_{{{\text{He}}}}^{0}$ = 8.5 × × 1015 см–3 + $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 2.1 × 1014 см–3 и разных начальных концентрациях молекул дейтерия $N_{{{\text{D2}}}}^{0}$ [168, 169]. В отсутствие разряда молекулы HDO не регистрируются, но появляются сразу после включения разряда. Далее имеется слабая (но заметная на фоне погрешностей измерений) тенденция к снижению концентрации тяжелой воды в плазме. Поскольку этот процесс имеет характерное время десятки минут, он, по-видимому, связан с хемосорбцией.
Рис. 21.
Поведение молекул HDO после включения разряда при начальной концентрации молекул воды $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 8.4×1014 см–3 и разных начальных концентрациях молекул дейтерия $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$: 1 – 1.8 × 1015 см–3, 2 – 3.6 × 1015 см–3; 3 – 5.3 × 1015 см–3 [168].
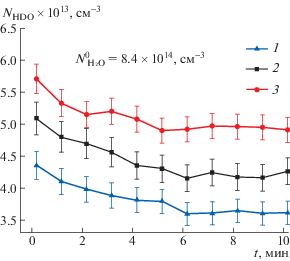
Аналогичные измерения концентраций молекул воды [168] показывают их быстрый распад уже на начальной стадии (время измерений ~30 с) на (20–30)% и дальнейшую стабилизацию с трендом к медленному уменьшению концентраций (это близко к тому, что наблюдалось в ранних работах по диссоциации СО2, см. разд. 2). При сопоставлении этих результатов можно ввести величины отношений k = ${{N}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$/NHDO в разряде. Оно оказалось практически постоянным k = 3 ± 0.5 и хорошо поддерживающимся во времени. Результаты приведены на рис. 22. Можно сделать предположение о существовании механизма, ограничивающего долю молекул Н2О на стенке, способных к преодолению барьера десорбции при взаимодействии с молекулами D2 [170].
Рис. 22.
Поведение отношения измеренных в разряде концентраций молекул H2O и HDO при разных начальных концентрациях N0 молекул H2O и D2 [168]: 1 – $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 2.4 × 1014 см–3, $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$ = 1.8 × 1015 см–3; 2 ‒ $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 1.7 × 1014 см–3, $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$ = 1.8 × 1015 см–3; 3 ‒ $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 2.0 × 1014 см–3, $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$ = 1.8 × 1015 см–3; 4 ‒ $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 2.1 × 1014 см–3, $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$ = 3.6 × 1015 см–3; 5 ‒ $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 2.7 × 1016 см–3, $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$ = 3.6 × 1015 см–3; 6 ‒ $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 1.8 × 1014 см–3, $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$ = 5.3 × 1015 см–3; 7 ‒ $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 2.1 × 1014 см–3, $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$ = 5.3 × 1015 см–3.
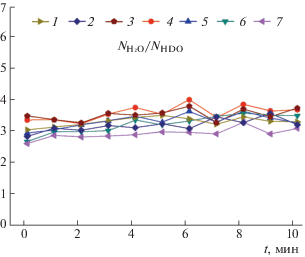
6. МОДЕЛИРОВАНИЕ ПРОЦЕССОВ В ПЛАЗМЕ ТЛЕЮЩЕГО РАЗРЯДА С ПРИМЕСЯМИ ВОДЫ И ДЕЙТЕРИЯ. СОПОСТАВЛЕНИЕ С ЭКСПЕРИМЕНТОМ
Превращения молекул и состав плазмы в положительном столбе тлеющего разряда в гелии с добавками H2O и D2 анализировались с помощью однородной кинетической модели [172]. Решались балансные уравнения для нейтральных и заряженных частиц совместно с уравнением Больцмана для функции распределения электронов по энергиям (ФРЭЭ) в двухчленном приближении.
Оценка длины релаксации энергии электрона λu для условий эксперимента дает значение λu = = DT/Ve ∼ 0.3–0.8 см, где DT – коэффициент поперечной диффузии электронов, а Ve – их скорость дрейфа. Значение λu меньше радиуса разрядной трубки, равного 1 см, что оправдывает использование локального приближения для ФРЭЭ.
В модели учитываются процессы в объеме и на поверхности с участием атомов, молекул и радикалов He, H2O, O2, H2, O, H, OH, H2O2, HO2, D2O, HDO, D2, D, OD, DO2, D2O, HD, D2O2, HDO2, положительных ионов He+, H2O+, ${\text{O}}_{2}^{ + }$, ${\text{H}}_{2}^{ + }$, D2O+, HDO+, ${\text{D}}_{2}^{ + }$, отрицательных ионов O–, H–, D–, OH–, D– и электронов. В модель включено уравнение для внешней электрической цепи, состоящей из балластного сопротивления и источника напряжения,
где Udisch – напряжение между электродами трубки, U0 – напряжение на источнике питания, Rb – балластное сопротивление; ток разряда Idisch = = eneVeS, ne – концентрация электронов, Ve – их скорость дрейфа, S – площадь сечения трубки. Высоковольтный источник напряжения работал в режиме стабилизации тока, поэтому его эффективное внутреннее сопротивление зависело от условий разряда и заданного тока. В уравнении (43) величина Rb включает в себя обе составляющие, Udisch измерялось независимо.Реакции взаимодействия электронов с атомом гелия и молекулами приведены в табл. 6 (процессы R1–R37).
Таблица 6.
Реакции взаимодействия электронов с атомом гелия и молекулами
| № | Реакция | № | Реакция |
|---|---|---|---|
| R1 | D2 + e < = > D2(Rot) + e | R19 | H2O + e = > OH– + H |
| R2 | D2 + e < = > D2(v) + e | R20 | H2O + e = > O– + H2 |
| R3 | D2 + e < = > D2(Tr) + e | R21 | H2O + e = > H– + OH |
| R4 | D2 + e < = > D2(Singl) + e | ||
| R5 | D2 + e = > ${\text{D}}_{2}^{ + }$ + e + e | R22 | D2O + e = > OD + D + e |
| R6 | D2 + e = > D– + D | R23 | D2O + e = > D2O+ + e + e |
| R24 | D2O + e = > OD– + D | ||
| R7 | He + e = > He+ + e + e | R25 | D2O + e = > O– + D2 |
| R26 | D2O + e = > D– + OD | ||
| R8 | H2 + e < = > H2(Tr) + e | ||
| R9 | H2 + e < = > H2(Singl) + e | R27 | HDO + e = > OD + H + e |
| R10 | H2 + e = > ${\text{H}}_{2}^{ + }$ + e + e | R28 | HDO + e = > OH + D + e |
| R11 | H2 + e = > H– + H | R29 | HDO + e = > HDO+ + e + e |
| R30 | HDO + e = > OD– + H | ||
| R12 | HD + e < = > HD(Tr) + e | R31 | HDO + e = > OH– + D |
| R13 | HD + e < = > HD(Singl) + e | R32 | HDO + e = > O– + HD |
| R14 | HD + e = > HD+ + e + e | R33 | HDO + e = > D– + OH |
| R15 | HD + e = > H– + D | R34 | HDO + e = > H– + OD |
| R16 | HD + e = > H + D– | ||
| R35 | O2 + e = > O + O + e | ||
| R17 | H2O + e = > OH + H + e | R36 | O2 + e = > ${\text{O}}_{2}^{ + }$ + e + e |
| R18 | H2O + e = > H2O+ + e + e | R37 | O2 + e = > O– + O |
В табл. 7 приведены процессы (R38–R57) с участием отрицательных ионов и ион-ионной рекомбинации.
Таблица 7.
Реакции отлипания электронов от отрицательных ионов, перезарядки отрицательных ионов и ион-ионной рекомбинации
| № | Реакция | Величина константы скорости реакции, см3 с–1 | № | Реакция | Величина константы скорости реакции, см3 с–1 |
|---|---|---|---|---|---|
| R38 | O– + H2 = > H2O + e | 6 × 10–10 [194] | R49 | H– + H2O < = > OH– + H2 | 3.8 × 10–10 [194] |
| R39 | O– + H = > OH + e | 5 × 10–10 [195] | R50 | D– + D2O < = > OD– + D2 | 3.8 × 10–10 |
| R40 | H– + H = > H2 + e | 1.3 × 10–10 [194] | R51 | H– + D2O < = > OH– + D2 | 3.8 × 10–10 |
| R41 | H– + O = > OH + e | 10–9 [195] | R52 | D– + H2O < = > OD– + H2 | 3.8 × 10–10 |
| R42 | O– + D2 = > D2O + e | 6 × 10–10 | R53 | D– + HDO < = > OH– + D2 | 3.8 × 10–10 |
| R43 | O– + HD = > HDO + e | 6 × 10–10 | R54 | H– + HDO < = > OD– + D2 | 3.8 × 10–10 |
| R44 | O– + D = > OD + e | 5 × 10–10 | R55 | D– + HDO < = > OD– + HD | 3.8 × 10–10 |
| R45 | D– + D = > D2 + e | 1.3 × 10–10 | R56 | H– + HDO < = > OH− + HD | 3.8 × 10–10 |
| R46 | H– + D = > HD + e | 1.3 × 10–10 | R57 | P+ + M– = > продукты, где P = He, D2,H2, HD, H2O, D2O, HDO, O2; M = O, H, D, OH, OD | 4 × 10–7 [196] |
| R47 | D– + H = > HD + e | 1.3 × 10–10 | |||
| R48 | D– + O = > OD + e | 10–9 |
Процессы (R58–R71) распада отталкивательных электронных синглетных и триплетных состояний изотопомеров водорода и процессы гетерогенной рекомбинации приведены в табл. 8.
Таблица 8.
Каналы распада электронных состояний H2, D2 и НD, и гетерогенные реакции
| № | Реакция | Вероятность реакции | № | Реакция | Частота, с–1 |
|---|---|---|---|---|---|
| R58 | D2(Tr) = > D + D | 1 | R67 | H + Hw = > H2 | 2 |
| R59 | D2(Singl) = > D + D | 0.1 | R68 | O + Ow = > O2 | 600 |
| R60 | D2(Singl) = > ${\text{D}}_{2}^{*}$ | 0.9 | R69 | OH + OH = > H2O + O | 600 |
| R61 | H2(Tr) = > H + H | 1 | R70 | D + Dw = > D2 | 12 |
| R62 | H2(Singl) = > H + H | 0.1 | R71 | H + ODw = > HDO | 50 |
| R63 | H2(Singl) = > ${\text{H}}_{2}^{*}$ | 0.9 | |||
| R64 | HD(Tr) = > H + H | 1 | |||
| R65 | HD(Singl) = > H + D | 0.1 | |||
| R66 | HD(Singl) = > HD* | 0.9 |
Наборы сечений взаимодействия электронов с атомами гелия и молекулами дейтерия брались из работ [185] и [186] соответственно. Расчеты скорости дрейфа электронов, коэффициентов продольной и поперечной диффузии, скорости ионизации величины с набором этих сечений дают результаты, хорошо согласующиеся с имеющимися в литературе экспериментальными данными.
В условиях проводимых экспериментов измеренные установившиеся значения приведенного электрического поля E/N ~ (55–90) Тд. С учетом этого в кинетическую модель были включены процессы возбуждения вращательных и колебательных уровней молекул дейтерия (процессы R1 и R2, в табл. 6), триплетных R3 (b3Σ$_{u}^{ + }$, c3Пu, a3Σ$_{g}^{ + }$, …) и синглетных R4 (B1Σ$_{u}^{ + }$, E1Σ$_{g}^{ + }$, C1Пu, …) электронных состояний молекул дейтерия. Учитывалось диссоциативное прилипание (R6) ионизация атомов гелия (R7) и молекул дейтерия (R5). Учет возбуждения электронных уровней атомов гелия дает вклад в баланс энергии при E/N = 70 Тд около 5%, поэтому данные процессы в расчетах не учитывались. Для молекул H2 и HD, в силу их заметно меньшей концентрации (см. ниже), учитывались только процессы возбуждения триплетных и синглетных электронных состояний, а также диссоциативное прилипание электронов к молекулам в основных колебательных состояниях и ионизация (R8–R16). Как следует из анализа базы данных [187], транспортные сечения рассеяния электронов на молекулах D2, H2 и НD отличаются незначительно. Слабо отличаются и сечения возбуждения электронных уровней и сечения ионизации. По данным работы [188], зависимость формы и величины сечения диссоциации от типа изотопа мала. Cечения возбуждения электронных уровней, ионизации и диссоциативного прилипания для молекул H2, НD и D2 в расчетах полагалось равными. При этом сечения диссоциативного прилипания к молекулам H2, НD и D2 считались равными сечению диссоциативного прилипания к молекуле H2.
Учитывалось, что диссоциативное прилипание к молекуле HD может происходить по двум каналам с равной вероятностью,
и их сечения σ1,2(u) равны 0.5 от сечения диссоциативного прилипания к молекуле D2. Нижнее состояние b3Σ$_{u}^{ + }$ отталкивательное, и его возбуждение приводит к диссоциации молекулы. Это сечение вносит основной вклад в сумму сечений триплетных состояний. Поэтому считалось, что возбуждение триплетных электронных состояний изотопомеров H2 приводит к диссоциации молекул (R58, R61, R64). Исходя из рекомендованных в обзоре [189] данных для интегральных сечений диссоциации молекул H2 и недавних расчетов, проведенных в работе [190], можно заключить, что при возбуждении синглетных электронных состояний диссоциирует около 10% возбуждаемых молекул. Причем в модели считалось, что диссоциация молекул происходит мгновенно. Вероятности каналов диссоциации электронных состояний и гетерогенные реакции приведены в табл. 8.Для молекул H2O сечения диссоциации, ионизации и диссоциативного прилипания брались из работы [191].
Для изотопомеров H2O учитывались сечения их диссоциации (R17, R22, R27, R28), ионизации (R18, R23, R29) и диссоциативного прилипания (R19–R21, R24–R26, R30–R34). Как и для HD, в случае появления двух каналов диссоциации или диссоциативного прилипания для НDO вероятности этих каналов считались равновероятными, а сами величины сечений брались по аналогии с H2O (R27–R28, R30–R31, R32–R34) [191].
Для молекул кислорода учитывались их диссоциация [192, 193], ионизация [193] и диссоциативное прилипание [193]. Учитывалось, что для молекулы O2 существует два канала диссоциации электронным ударом с порогом 5.6 эВ и 8.4 эВ [192]. В первом канале образуются два атома O в основном состоянии, во втором один из атомов находится в электронно-возбужденном состоянии O(1D). В кинетической схеме, как и в наших предыдущих работах [49, 56, 57, 72, 82, 83, 171, 172], величина константы диссоциации бралась равной сумме констант двух каналов.
В табл. 7 приведены процессы отлипания электронов от отрицательных ионов, перезарядки отрицательных ионов и ион-ионной рекомбинации. Если рядом с величиной константы скорости отсутствует ссылка, то величина константы взята равной константе скорости аналогичного процесса [172].
Считалось, что синглетные и триплетные электронные состояния H2, D2 и НD мгновенно диссоциируют на атомы. Вероятности каналов диссоциации электронных состояний и гетерогенные реакции приведены в табл. 8.
Оценки показали, что из-за низкого давления газовой смеси основную роль играют процессы гибели заряженных частиц на стенках разрядной трубки. Поэтому в модель, наряду с объемной гибелью, включены процессы амбиполярной диффузии заряженных частиц на стенки разрядной трубки. В плазмообразующим газе присутствуют молекулы, к которым происходит диссоциативное прилипание электронов (изотопомеры молекул воды и молекулярного водорода). Расчеты зарядового состава плазмы показали, что для рассматриваемых условий концентрация отрицательных ионов значительно превышает концентрацию электронов. В этом случае большая концентрация отрицательных ионов влияет на скорость диффузии электронов к стенке разрядной трубки. Поэтому для учета гибели электронов, отрицательных и положительных ионов на стенках разрядной трубки использовалось приближение, предложенное в работе [197]. Уравнение для концентрации электронов имеет вид
(46)
$\frac{{d{{n}_{e}}}}{{dt}} = {{\nu }_{{ion}}}{{n}_{e}} + \sum\limits_k {{{\nu }_{{det,k}}}} {{n}_{{n,k}}} - {{\nu }_{{att}}}{{n}_{e}} - {{\nu }_{D}}{{n}_{e}},$Константы скоростей процессов с участием тяжелых частиц: атомов, молекул и радикалов, – и их температурные зависимости в газовой фазе, взятые из работы [56], приведены в табл. 9.
Таблица 9.
Объемные реакции с участием нейтральных частиц
| № | Реакция | Величина константы скорости реакции k(T), см3 с–1, см6 с–1 | k(T) при T = 430 K, см3 с–1, см6 с–1 | Ссылка |
|---|---|---|---|---|
| R72 | OH + H + He < = > H2O + He | 4.3 × 10–25Т–2.6 | 6.1 × 10–32 | 198 |
| R73 | OH + OH < = > O + H2O | 5.56 × 10–20Т2.42e970/T | 1.3 × 10–12 | 199 |
| R74 | H + HO2 < = > OH + OH | 2.8 × 10–10e–440/T | 1.0 × 10–10 | 200 |
| R75 | H + HO2 < = > H2 + O2 | 1.75 × 10–10e–1030/T | 1.6 × 10–11 | 199 |
| R76 | OH + O < = > H + O2 | 2.0 × 10–10Т–0.352e123/T | 3.1 × 10–11 | 199 |
| R77 | OH + H < = > O + H2 | 8.0 × 10–21Т2.8e–1951/T | 2.0 × 10–15 | 201 |
| R78 | OH + HO2 < = > H2O + O2 | 4.8 × 10–11e250/T | 8.6 × 10–11 | 199 |
| R79 | OH + H2 < = > H2O + H | 3.6 × 10–16Т1.52e–1740/T | 6.3 × 10–14 | 199 |
| R80 | HO2 + O < = > OH + O2 | 2.7 × 10–11e224/T | 4.5 × 10–11 | 199 |
| R81 | H + H2O2 < = > HO2 + H2 | 2.8 × 10–12e–1888/T | 3.5 × 10–14 | 199 |
| R82 | H + H2O2 < = > OH + H2O | 1.7 × 10–11e–1798/T | 2.6 × 10–13 | 199 |
| R83 | H + HO2 < = > O + H2O | 5.0 × 10–11e–866/T | 6.7 × 10–12 | 200 |
| R84 | H + H + M < = > H2 + M | 1.76 × 10–30Т–1 | 4.1 × 10–33 | 202 |
| R85 | OH + O + M < = > HO2 + M | 4.0 × 10–32 | 4.0 × 10–32 | 203 |
| R86 | OH + OH + M < = > H2O2 + M | 8.0 × 10–31Т–0.76 | 8.0 × 10–33 | 200 |
| R87 | O + H2O2 < = > HO2 + OH | 4.65 × 10–11e–3222/T | 2.6 × 10–14 | 202 |
| R88 | O + H + M < = > OH + M | 1.3 × 10–29·Т–1 | 3.0 × 10–32 | 201 |
| R89 | H + O2 + M < = > HO2 + M | 5.5 × 10–30·Т–0.8 | 4.3 × 10–32 | 202 |
| R90 | O + O + M < = > O2 + M | 2.76 × 10–31Т–1 | 6.4 × 10–34 | 202 |
| R91 | OD + D + He < = > D2O + He | 4.3 × 10–25Т–2.6 а) | 6.1 × 10–32 а) | 198 |
| R92 | OD + H + He < = > HDO + He | 4.3 × 10–25Т–2.6 а) | 6.1 × 10–32 а) | 198 |
| R93 | OH + D + He < = > HDO + He | 4.3 × 10–25Т–2.6 а) | 6.1 × 10–32 а) | 198 |
| R94 | OD + OD < = > O + D2O | 2.5 × 10–13e170/T | 3.7 × 10–13 | 204 |
| R95 | OD + OH < = > O + HDO | 1.5 × 10–12 | 1.5 × 10–12 | 204 |
| R96 | D + DO2 < = > OD + OD | 2.8 × 10–10e–440/Tа) | 1.0 × 10–10 а) | 200 |
| R97 | D + HO2 < = > OD + OH | 2.8 × 10–10e–440/Tа) | 1.0 × 10–10 а) | 200 |
| R98 | H + DO2 < = > OD + OH | 2.8 × 10–10e–440/Tа) | 1.0 × 10–10 а) | 200 |
| R99 | D + DO2 < = > D2 + O2 | 1.75 × 10–10e–1030/Tа) | 1.6 × 10–11 а) | 199 |
| R100 | H + DO2 < = > HD + O2 | 1.75 × 10–10e–1030/Tа) | 1.6 × 10–11 а) | 199 |
| R101 | D + HO2 < = > HD + O2 | 1.75 × 10–10e–1030/Tа) | 1.6 × 10–11 а) | 199 |
| R102 | OD + O < = > D + O2 | 2.0 × 10–10Т–0.352e123/Tа) | 3.1 × 10–11 а) | 199 |
| R103 | OD + D < = > O + D2 | 8.0 × 10–21Т2.8e–1951/Tа) | 2.0 × 10–15 а) | 201 |
| R104 | OH + D < = > O + HD | 8.0 × 10–21Т2.8e–1951/Tа) | 2.0 × 10–15 а) | 201 |
| R105 | OD + H < = > O + HD | 8.0 × 10–21Т2.8e–1951/Tа) | 2.0 × 10–15 а) | 201 |
| R106 | D + H2 < = > HD + H | 4.0 × 10–18Т2.29e–2627/T | 9.5 × 10–15 | 205 |
| R107 | H + D2 < = > HD + D | 3.8 × 10–21Т3.21e–2850/T | 1.4 × 10–15 | 206 |
| R108 | OD + DO2 < = > D2O + O2 | 3.8 × 10–11 | 3.8 × 10–11 | 207 |
| R109 | OH + DO2 < = > HDO + O2 | 4.8 × 10–11e250/Tа) | 8.6 × 10–11 а) | 199 |
| R110 | OD + HO2 < = > HDO + O2 | 4.8 × 10–11e250/Tа) | 8.6 × 10–11 а) | 199 |
| R111 | OD + D2 < = > D2O + D | 7.93 × 10–18Т1.98e–1821/T | 1.9 × 10–14 | 208 |
| R112 | OH + D2 < = > HDO + D | 4.36 × 10–15Т1.18e–2333/T | 2.9 × 10–14 | 209 |
| R113 | OH + HD < = > HDO + H | 7.87 × 10–12e–2807/T | 1.2 × 10–12 | 210 |
| R114 | OH + HD < = > H2O + D | 4.7 × 10–12e–2259/T | 2.5 × 10–14 | 210 |
| R115 | OD + H2 < = > HDO + H | 7.4 × 10–15 | 7.4 × 10–15 | 211 |
| R116 | OH + D2 < = > D2O + H | 7.93 × 10–18Т1.98e–1821/Tа) | 1.9 × 10–14 а) | 209 |
| R117 | OD + H2 < = > H2O + D | 7.4 × 10–15 а) | 7.4 × 10–15 а) | 211 |
| R118 | OD + HD < = > D2O + H | 7.4 × 10–15 | 7.4 × 10–15 | 211 |
| R119 | OD + HD < = > HDO + D | 1.8 × 10–15 | 1.8 × 10–15 | 211 |
| R120 | DO2 + O < = > OD + O2 | 2.7 × 10–11e224/Tа) | 4.5 × 10–11 а) | 199 |
| R121 | D + D2O2 < = > DO2 + D2 | 2.8 × 10–12e–1888/Tа) | 3.5 × 10–14 а) | 199 |
| R122 | D + H2O2 < = > DO2 + H2 | 5.1 × 10–12e–2115/T | 3.7 × 10–14 | 212 |
| R123 | D + H2O2 < = > HO2 + HD | 5.1 × 10–12e–2115/T | 3.7 × 10–14 | 212 |
| R124 | H + D2O2 < = > DO2 + HD | 5.1 × 10–12e–2115/T | 3.7 × 10–14 | 212 |
| R125 | H + D2O2 < = > HO2 + D2 | 5.1 × 10–12e–2115/T | 3.7 × 10–14 | 213 |
| R126 | H + HDO2 < = > DO2 + H2 | 5.1 × 10–12e–2115/T | 3.7 × 10–14 | 213 |
| R127 | D + HDO2 < = > HO2 + D2 | 5.1 × 10–12e–2115/T | 3.7 × 10–14 | 212 |
| R128 | D + HDO2 < = > DO2 + HD | 5.1 × 10–12e–2115/T | 3.7 × 10–14 | 212 |
| R129 | H + HDO2 < = > HO2 + HD | 5.1 × 10–12e–2115/T | 3.7 × 10–14 | 212 |
| R130 | D + D2O2 < = > OD + D2O | 2.45 × 10–8e–7502/T | 6.5 × 10–16 | 213 |
| R131 | D + H2O2 < = > OD + H2O | 1.16 × 10–11e–2110/T | 8.6 × 10–14 | 212 |
| R132 | D + H2O2 < = > OH + HDO | 1.16 × 10–11e–2110/T | 8.6 × 10–14 | 212 |
| R133 | H + D2O2 < = > OH + D2O | 1.16 × 10–11e–2110/Tа) | 8.6 × 10–14 а) | 212 |
| R134 | H + D2O2 < = > OD + HDO | 1.16 × 10–11e–2110/Tа) | 8.6 × 10–14 а) | 212 |
| R135 | D + HDO2 < = > OD + HDO | 1.16 × 10–11e–2110/Tа) | 8.6 × 10–14 а) | 212 |
| R136 | D + HDO2 < = > OH + D2O | 1.16 × 10–11e–2110/Tа) | 8.6 × 10–14 а) | 212 |
| R137 | H + HDO2 < = > OD + H2O | 1.16 × 10–11e–2110/Tа) | 8.6 × 10–14 а) | 212 |
| R138 | H + HDO2 < = > OH + HDO | 1.16 × 10–11e–2110/Tа) | 8.6 × 10–14 а) | 212 |
| R139 | D + DO2 < = > O + D2O | 5.0 × 10–11e–866/Tа) | 6.7 × 10–12 а) | 200 |
| R140 | D + HO2 < = > O + HDO | 5.0 × 10–11e–866/Tа) | 6.7 × 10–12 а) | 200 |
| R141 | H + DO2 < = > O + HDO | 5.0 × 10–11e–866/Tа) | 6.7 × 10–12 а) | 200 |
| R142 | D + D + M < = > D2 + M | 1.49 × 10–32 | 1.49 × 10–32 | 214 |
| R143 | H + D + M < = > HD + M | 1.49 × 10–32 а) | 1.49 × 10–32 а) | 214 |
| R144 | OD + O + M < = > DO2 + M | 4.0 × 10–32 а) | 4.0 × 10–32 а) | 203 |
| R145 | OD + OD + M < = > D2O2 + M | 8.0 × 10–31Т–0.76 а) | 8.0 × 10–33 а) | 199 |
| R146 | OD + OH + M < = > HDO2 + M | 8.0 × 10–31Т–0.76 а) | 8.0 × 10–33 а) | 199 |
| R147 | O + D2O2 < = > DO2 + OD | 4.65 × 10–11e–3222/Tа) | 2.6 × 10–14 а) | 202 |
| R148 | O + HDO2 < = > HO2 + OD | 4.65 × 10–11e–3222/Tа) | 2.6 × 10–14 а) | 202 |
| R149 | O + HDO2 < = > DO2 + OH | 4.65 × 10–11e–3222/Tа) | 2.6 × 10–14 а) | 202 |
| R150 | O + D + M < = > OD + M | 1.3 × 10–29Т–1 а) | 3.0 × 10–32 а) | 202 |
| R151 | D + O2 + M < = > DO2 + M | 5.5 × 10–30Т–0.8 а) | 4.3 × 10–32 а) | 202 |
Для молекул D2, HD, D2O и HDO константы скорости диссоциации брались такими же, как и для молекул H2 и H2O соответственно. Для химических реакций в объеме с участием компонент, содержащих атомы D, константы скорости реакций брались из базы данных NIST [215]. При отсутствии в базе данных таких процессов их константы скорости брались такими же, как и для аналогичных реакций, но с компонентами, содержащими атомы H. Константы скоростей обратных процессов рассчитывались из принципа детального равновесия с использованием термодинамических данных [216].
В рассматриваемых условиях важными являются процессы гетерогенной рекомбинации частиц на поверхностях. Эти процессы учтены в модели реакциями R67 – R71. Расчеты выполнены в приближении постоянной температуры газа в объеме разряда (320 К). Для решения описанной системы уравнений использовался программный пакет Chemical WorkBench (CWB 4.1.18411, www.kintech.ru) [217]. Система уравнений решалась методом установления до достижения стационарных значений концентраций компонент плазмы.
Описанная выше кинетическая модель использовалась для расчета характеристик тлеющего разряда и концентраций компонент плазмы в трубке. Совместное решение кинетических уравнений для компонент плазмы с уравнением Больцмана для ФРЭЭ и уравнения для внешней цепи позволяет определить величину электрического поля и концентрацию электронов в положительном столбе плазмы. В эксперименте высоковольтный источник работал в режиме стабилизации тока, и подстройка внутреннего сопротивления в уравнении (43) включена в сопротивление Rb. Оно подбирались из условия равенства установившегося расчетного значения величины тока разряда измеренной величине.
Расчеты выполнены для концентрации атомов He равной 8.5 × 1015 см–3, начальные концентрации молекул H2O варьировались в диапазоне от 4.4 × 1014 см–3 до 9.6 × 1014 см–3, а молекул D2 – (1.8–5.3) × 1015 см–3 [172]. Начальная концентрация остальных молекул атомов и радикалов, включенных в модель, задавалась равной нулю. Начальная концентрация электронов задавалась раной 107 см–3. Температура газа считалась постоянной и равной измеренной в эксперименте T = 320 K. Внутренний диаметр трубки 2 см. Расчетная динамика поведения приведенного значения электрического поля E/N и тока разряда показаны на рис. 23. В начальный момент времени концентрация электронов мала и к разрядной трубке приложено напряжение U0. Происходит рост концентрации электронов, тока разряда и падения напряжения на балластном сопротивлении. В результате на временах около 1 мкс устанавливается стационарное значение тока разряда и напряжения на разрядной трубке. Маркеры соответствуют измеренным установившимся току разряда и E/N. Экспериментальное значение величины приведенного электрического поля находилось с учетом катодного падения напряжения, величина которого определялась по измерению напряжения на различных парах электродов с разрядами разной длины. Вначале происходит быстрое установление тока разряда и E/N на временах около 1 мкс, концентрация молекул воды на этих временах равна начальной. Далее происходит диссоциация молекул воды, eе концентрация падает в несколько раз и выходит на стационарное значение на временах порядка (50–100) с. При этом приведенное значение E/N падает всего на 4 Тд. Это связано с тем, что электроны гибнут, в основном, за счет их амбиполярной диффузии на стенки разрядной трубки. Окончательное установившееся расчетное значение приведенного электрического поля составляет 69 Тд, а среднее значение энергии электронов – 7.43 эВ.
Рис. 23.
Зависимость приведенного электрического поля и тока разряда в трубке от времени. Сплошные линии результаты моделирования. Маркеры результаты изменений в эксперименте. NHe = 8.5 × 1015 см–3, $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 8.8 × 1014 см–3, $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$ = 3.6 × 1015 см–3.
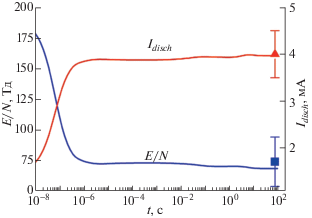
На рис. 24 представлена эволюция концентраций электронов, положительных и отрицательных ионов во времени. Концентрация электронов, как и тока разряда, устанавливается за время ~1 мкс. Установление концентрации ионов происходит в два этапа: вначале за времена около 1 мс, потом около 10 с. Это связано с тем, что вначале ионный состав устанавливается, когда молекулы H2O еще слабо диссоциированы, а на следующем этапе заканчивается формирование стационарного состава практически для всех нейтральных частиц (см. рис. 25). Радикалы OH и OD обладают энергией сродства к электрону 4.77 эВ. Для сравнения, энергия сродства к электрону для атомов H и О равна 0.754 эВ и 1.47 эВ, соответственно. Концентрация отрицательных ионов O–, H– и D– примерно в 250 раз ниже, чем концентрация ионов OH– и OD–. Концентрации ионов OH– и OD– примерно на порядок выше концентрации электронов. На рис. 26 из числа представленных на рис. 25 зависимостей выделены три, соответствующие поведению изотопомеров воды. Там же показаны данные измерений для молекул H2O, HDO. Видно, что подбором констант гетерогенных реакций удается получить хорошее согласие теории и эксперимента для этих молекул в стационарной стадии формирования химического состава плазмы.
Рис. 24.
Расчетная зависимость концентрации электронов, положительных и отрицательных ионов от времени в разряде в трубке. NHe = 8.5 × 1015 см–3, $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 8.8 × 1014 см–3, $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$ = 3.6 × 1015 см–3.
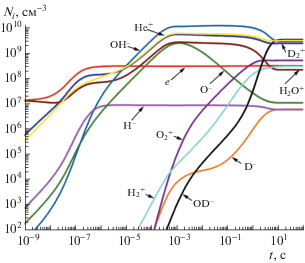
Рис. 25.
Расчетная зависимость концентрации нейтральных частиц от времени в разряде в трубке. NHe = = 8.5 × 1015 см–3, $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 8.8 × 1014 см–3, $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$ = 3.6 × × 1015 см–3.
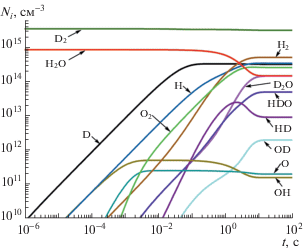
Рис. 26.
Расчетная зависимость концентраций H2O, HDO и D2O от времени в разряде в трубке. Маркеры – эксперимент. NHe = 8.5 × 1015 см–3, $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ = 8.8 × × 1014 см–3, $N_{{{{{\text{D}}}_{{\text{2}}}}}}^{0}$ = 3.6 × 1015 см–3.
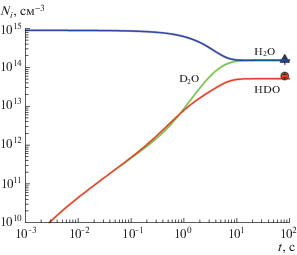
При моделировании плазмохимических процессов в смесях гелия и паров воды в разряде в полом катоде использовалась более простая кинетическая модель [49]. Учитывались следующие компоненты: He, H2O, O2, H2, H2O2, HO2, OH, H и O. Были включены процессы диссоциации молекул H2O, O2 и H2 электронами плазмы, объемные химические реакции, гетерогенная рекомбинация атомов O и H, учитывались радикалы ОН на поверхности стенок камеры. Скорости диссоциации электронами плазмы молекул H2O, O2 и H2 рассчитаны путем усреднения с максвелловской ФРЭЭ с электронной температурой 9 эВ и концентрацией электронов 109 см–3 по результатам зондовых измерений. Температура газа равна 430 К, что соответствует экспериментальным измерениям, давление 0.87 Торр, исходная газовая смесь H2O : He = 1 : 3. Предполагалось, что небольшие количества инертных актинометров, присутствующие в газовой смеси, не влияют на химические превращения.
Результаты моделирования показывают, что в квазистационарном состоянии главные каналы разрушения молекул H2O, O2 и H2 связаны с диссоциацией электронами, а их образование происходит путем рекомбинации на поверхности. Вероятности рекомбинации атомов О и Н и радикала ОН на поверхности зависят от ее материала и состояния. Они известны с большей погрешностью, чем константы химических реакций в объеме. Поэтому вероятности гетерогенной рекомбинации были выбраны из условия равенства концентрации молекул H2O, радикалов ОН и атомов О и Н измеренным значениям.
В эксперименте измерялись концентрации H2O, OH, O и H. Расчеты показали, что концентрация молекул HO2 и H2O2 значительно меньше, чем концентрации H2O, OН, О и H. Поэтому концентрации молекул H2 и O2 определялись из условия сохранения частиц в пренебрежении концентрациями HO2 и H2O2, и брутто-химический баланс частиц можно записать в виде
(47)
$\begin{gathered} {\text{А}}(\Delta {{{\text{H}}}_{{\text{2}}}}{\text{O}}) = {\text{В(ОН)}} + {\text{С}}({\text{Н}}) + \\ + \;{\text{D(О)}} + {\text{Е(}}{{{\text{Н}}}_{2}}{\text{)}} + {\text{F(}}{{{\text{O}}}_{2}}{\text{)}}, \\ \end{gathered} $На рис. 27 представлены результаты расчетов (сплошные линии) и результаты измерений (маркеры). Видно, что модель хорошо описывает экспериментальные данные, за исключением поведения атомов кислорода. Возможно, это связано с наличием молекул воды на поверхности стенок камеры, что проявляется через реакцию О + H2O (стенка) → Н2 + О2 (газ). Подробно объяснение такого расхождения приводится в [49, 50].
Рис. 27.
Поведение измеренных концентраций молекул и атомов во времени в разряде с полым катодом. Маркеры – экспериментальные данные, сплошные линии – результаты моделирования.
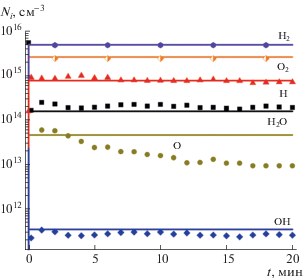
На рис. 28 представлены результаты моделирования для первоначальной фазы горения разряда. Согласно расчетам, характерное время установления химического равновесия составляет около 1 с. Рисунок 28 иллюстрирует сильную диссоциацию молекул воды до 97%. Такое сильное разложение молекул воды должно учитываться при диагностике утечек в электровакуумных приборах по спектрам плазмы.
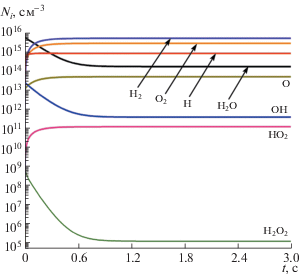
7. ОБ ОЖИДАЕМОЙ ЧУВСТВИТЕЛЬНОСТИ АКТИНОМЕТРИЧЕСКИХ ИЗМЕРЕНИЙ И ЛОКАЛИЗАЦИИ ИСТОЧНИКОВ ПРИМЕСЕЙ МОЛЕКУЛ ВОДЫ В УСЛОВИЯХ ИТЭР
Основываясь на анализе выполненных предварительных и модельных исследований по поиску течей воды и определению их потоков, описанных в разделах 5 и 6, применительно к крупному реактору разумным компромиссом выглядят методы, основанные на собственной эмиссии плазмы. В последние годы их возможности как количественных методов анализа в неравновесной плазме расширились за счет более глубокой методической и физической проработки и нацеленности именно на такие задачи. Для сложной системы невозмущающий характер и возможность удаленных измерений также принципиально важны. Предпочтительным представляется метод оптической актинометрии в ее новой мультиспектральной версии МА с включением пар промежуточных актинометров, например, атомов водорода и дейтерия с линиями Hα–Dα в удобной для экспериментов области 656 нм. С помощью введения дополнительного актинометра Хе контролируются концентрации атомов О, H и D, также, как и плотность “горячей” группы гидроксила, а через нее – концентрация молекул воды и их поток в плазму извне. Независимость I(Hα)/I(Dα) = = NH/ND от параметров электронной компоненты позволяет производить необходимые проверки и калибровки на модельных установках. Еще одним немаловажным фактором является отработанная и протестированная с помощью МА модель формирования химического нейтрального и ионного состава плазмы на основе газов, содержащих примеси воды и дейтерия (разд. 6). Прямой мониторинг реактора пока невозможен, и для оценки возможностей контроля примесей исходя из результатов предварительных экспериментов требуются либо полные расчеты, либо модели для экстраполяции.
7.1. Экстраполяция результатов модельных экспериментов, фактор неоднородности
Поскольку кинетическая модель хорошо описывает результаты экспериментов на двух различных модельных установках, можно с ее помощью предсказать результаты по концентрациям молекул в расширенном диапазоне условий. На рис. 29 приведены результаты расчетов концентраций молекул Н2О и атомов О, Н в плазме разряда и величины [H2O]0 в исходной смеси Не(0.5 Торр) + Н2О с добавками дейтерия [57]. Как и в разд. 6, экспериментальные результаты в области их измерений хорошо согласуются с расчетными результатами. Измерения для группы частиц проведены по одной паре промежуточных актинометров H–D. Из сравнения зависимостей 4, 4 ' и 7, 7 ' видно, в частности, что уменьшение концентрации D (что достигается уменьшением добавок D2) увеличивает чувствительность измерений концентраций. Это благоприятный фактор для диагностики реакторов типа ИТЭР, в некоторых режимах работы которых ожидаются малые концентрации дейтерия, [D2] ~ (1013–1011) см–3.
Рис. 29.
Концентрации N атомов О, Н, молекул Н2О в плазме и концентрация Н2О в исходной смеси. Маркеры – эксперимент, линии – расчет. 1, 1 ' – [H2O]0, 2, 2 ' – H2O, 3, 3 ' – O, 4, 4 ' – H при [D2]0 = 6.75 × × 1014 cм–3; 5, 5 '- [H2O]0, 6, 6 ' – H2O, 7, 7 ' – H при [D2]0 = 1.35 × 1015cм–3 [57].
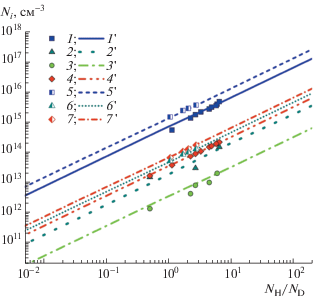
Измерения на модельных установках и моделирование проводились для условий однородной плазмы, тогда как в реакторе для плазмы вблизи стенки характерны градиенты параметров. Для учета влияния этого обстоятельства на измерения и моделирование мы использовали результаты расчетов, проводившихся в ряде работ. В табл. 10 приведены работы [218], полученные методом Монте-Карло в версии EIRENE, адаптированной для токамаков.
Таблица 10.
Распределения концентраций нейтральных частиц N, концентрации ne и температуры Те в направлении Х, нормальном к стенке ИТЭР
| X, см | 0 | 2 | 4 | 6 | 8 | 10 |
|---|---|---|---|---|---|---|
| N, отн. ед. | 1 | 1 | 1 | 0.4 | 0.15 | <0.1 |
| ND2, отн. ед. | 1 | 0.8 | 0.6 | 0.1 | – | – |
| ne, см–3 | 1011 | 5 × 1011 | 1012 | 4 × 1012 | 8 × 1012 | 1.5 × 1013 |
| Te, эВ | 4 | 6 | 12 | 20 | 40 | 70 |
Как отмечалось, нейтральные частицы в реакторе присутствуют только вблизи стенки, не далее 10 см, после чего плазма становится полностью ионизованной, а концентрация и температура электронов растут на этой дистанции от 0 до 1013 см–3 и от единиц до нескольких десятков эВ. Оценка температуры нейтралов дает при этом рост (430–1200) К.
7.2. Локализация источников проникновения молекул воды
Область, в которой источник примеси может быть локализован, определяется оптической схемой, регистрирующей спонтанное излучение, и процессами в плазме.
Оптическая система в диагностическом комплексе ИТЭР включает панорамный спектрометр для работы в видимой и ближней ИК-областях спектра (VisIR [221, 222]) с обратной линейной дисперсией не менее 1 нм/мм. Это достаточное разрешение для обсуждаемых методов. Панорама охватывает около 80% площади внутренней стенки при пространственном разрешении изображения R3 ~ 10 × 10 × 10 см3 = 1 л. Для установления места и потока течи необходимо, чтобы молекулы и их усредненные концентрации в плазме были измерены в пределах этого объема.
В модельных экспериментах для определения чувствительности к концентрации молекул воды в исходную смесь вводились контролируемые [H2O]0. В случае реальной течи и ограниченной области локализации следует связать плотность молекул с их потоком, Q = Δ(PV)/Δt. В отсутствие разряда, но при нагретой стенке молекулы, проникающие через стенку, не испытывают превращений, и их число в зоне наблюдения равно
(50)
${{N}_{0}} = {{\left[ {{{{\text{Н}}}_{{\text{2}}}}{\text{О}}} \right]}^{0}} = (dN{\text{/}}dt){{\tau }_{D}},$Таблица 11.
Соотношение величин, характеризующих натекание молекул в область наблюдения
| Q, Па м3 с–1 | 10–7 | 10–9 | 10–11 | 10–13 |
|---|---|---|---|---|
| Ω, с–1 | 6 × 1016 | 6 × 1014 | 6 × 1012 | 6 × 1010 |
| N0, см–3 | 3.6 × 1012 | 3.6 × 1010 | 3.6 × 108 | 3.6 × 106 |
При включении разряда химические процессы будут иметь разные локальные скорости. Мы провели расчеты динамики превращений молекул для различных комбинаций концентраций и температур электронов. Пример показан на рис. 30. Видно, что стационарные концентрации всех частиц устанавливаются за время ~2 × 10–2 c, меньшее, чем время ухода частиц из области локализации за счет диффузии. При больших концентрациях и температурах электронов это время еще уменьшается.
Рис. 30.
Динамика установления химического состава плазмы в пристеночной области. Плазмообразующий газ He (2.5 × 1016 cм–3) + D2 (1012 cм–3) + H2O (5 × × 1012 cм–3). Te = 10 эВ, ne = 1011 cм–3. 1 – H2O, 2 – D2, 3 – O, 4 – H2, 5 – D, 6 – O2, 7 – H, 8 – HD, 9 – OD, 10 – OH, 11 – D2O, 12 – HDO, 13 – HO2, 14 – H2O2.
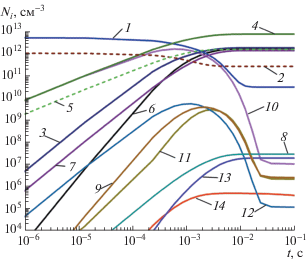
При усреднении результатов актинометрических измерений по области локализации использовались два способа. В первом из них использовались значения ne, Te (табл. 10) и температуры нейтрального газа T в центральной области (Х = = 4 см). При другой процедуре проводились вычисления концентраций для различных Х и соответствующих им данных из табл. 10 с последующим усреднением по X. Результаты совпадали с точностью не хуже 20%.
На рис. 31 показаны значения стационарных концентраций молекул воды в плазме и в отсутствие разряда, соответствующие различным отношениям плотностей [H] и [D] (равным отношениям интенсивностей в актинометрической паре H-D) при добавке ND2 = 1012 cм–3. Приведены примеры для X = 2 см и Х = 7 см. Видно, что поступающие в камеру частицы в этих областях глубоко диссоциированы. Изменение отношения H/D от 10–2 до 102 соответствует изменению концентрации $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{}}$ от 107 cм–3 до 1011 cм–3 в случае a) и от 105 cм–3 до 109 cм–3 в случае б). При тех же H/D, концентрации N для случаев а) и б) отличаются на 2 порядка величины. Напротив, при равных H/D концентрации $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ практически совпадают в пределах погрешностей измерений и данных табл. 10. При этом в той же области изменений H/D значения $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ варьируются в обоих случаях от 5 × 109 cм–3 до 5 × 1013 cм–3. Сильное различие случаев а) и б) в отношении $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{{}}$ и практическая независимость $N_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}^{0}$ при тех же H/D объясняются естественным образом. Изменения N связаны с изменениями в цикле плазмохимических процессов при различных параметрах электронов, тогда как N0 определяется только потоком Q молекул извне. Такой результат весьма важен, поскольку именно определение мощности и локализации течи в проекте ИТЭР выдвигаются на первый план.
Рис. 31.
Расчетные концентрации молекул Н2О в плазме (N) и в газе до зажигания разряда (N0), проникающих в камеру. а) – X = 2 см, Те = 8 эВ, ne = 6 × × 1011 см–3, T = 600 K; б) – X = 7 см, Te = 30 эВ, ne = = 1013 см–3 , T = 1000 К.
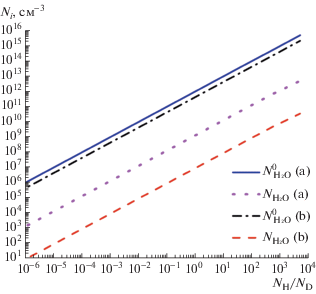
Как видно из рис. 31 и табл. 10, при измерениях Н-D пары при мультиспектральной актинометрии возможны локальные измерения потоков течей в диапазоне Q = (10–13–10–9) Па ⋅ м3 с–1. Диапазон может уточняться за счет вариации добавок D2 к инертному газу.
Исходя из требований проекта ИТЭР о максимально допустимом потоке молекул воды в камеру реактора Q < 10–7 Па ⋅ м3 с–1 , можно говорить о возможности нахождения локализации одновременно до 100 течей и определения потоков, если каждая течь дает поток Q < 10–9 Па ⋅ м3 с–1.
8. ЗАКЛЮЧЕНИЕ
В обзоре рассмотрено развитие и современные возможности методов измерения малых концентраций частиц в низкотемпературной плазме. Наибольший научный и практический интерес представляют объекты с неравновесной плазмой, и это критически сказывается на физическом обосновании и применимости методов их диагностики. Признак малости концентраций в разных ситуациях может свидетельствовать об их разной роли в плазменных процессах.
Не снижается ценность аналитических методов масс-спектрометрии. Трудность их применения связана с необходимостью отбора проб, и соответствие количества частиц в пробе их количеству в плазме нуждается в обосновании. Эта проблема также постепенно решается, но, как правило, под контролем методов оптической спектроскопии.
Часто речь идет о частицах, концентрации которых малы из-за высокой реакционной способности и, как следствие, их ключевой роли в формировании состава плазмы, наработке целевых продуктов в объеме плазмы и активном взаимодействии плазмы с поверхностями. Это, как правило, радикалы в виде атомов, двух- и многоатомных молекул, включая щелочные металлы, ОН, СН, СN, СН2О2 и др. Эти частицы важны, в частности, для селективной химии углеводородов. Для применений в микроэлектронике интерес представляют галогены и галогениды. Развивается направление с использованием плазменных источников атмосферного давления, генерирующих струи с активными атомами и молекулами. Это интересно для многих приложений, на данном этапе наиболее востребовано для биологических и медицинских применений. Важную роль в физике и химии плазмы играют электронно-возбужденные метастабильные частицы, аккумулирующие энергию плазмы и расходующие ее на инициирование химических реакций, возбуждение лазерных уровней и пр. Для многих из таких активных частиц существуют разрешенные в дипольном приближении переходы с участием наиболее заселенного основного электронного состояния. Они лежат в области спектра от ближнего УФ- до среднего ИК-диапазонов, достаточно удобных при использовании методов поглощения. С развитием лазеров с перестраиваемой частотой они постепенно заменяют средства классической абсорбционной спектроскопии и позволяют использовать как однофотонные, так и многофотонные процессы, прямую и косвенную регистрацию слабого поглощения (лазерная флуоресценция, гальванические явления, …). Каждый успех в развитии этих направлений сопровождается многочисленными публикациями, обзорами, монографиями.
Вместе с тем, во многих работах, если не в большинстве, лазерные методы и методы отбора проб дополняются классическими абсорбционными и эмиссионными методами. Они более просты в техническом отношении и для ряда измерений в неравновесной плазме получили надежные физические обоснования. Это относится, например, к измерению газовых, вращательных и колебательных температур, необходимых для расчета внутренней статистической суммы при лазерном измерении поглощения.
Наконец, лазерные методы или методы отбора проб часто трудно применимы для диагностики реальных объектов из-за недопустимости даже малых возмущений, внесения недопустимых дополнений к сложной конструкции, ограниченной доступности по причинам удаленности, безопасности и др.
Известно, конечно, что в ряде случаев наличие малых фракций частиц не несет положительных функций и, напротив, мешает достижению желаемого результата в плазменных технологиях; их чаще классифицируют как примеси, и они также нуждаются в мониторинге.
В данном обзоре мы, наряду с анализом общей ситуации с развитием высокочувствительной спектроскопии частиц в плазме, рассматриваем ситуацию, в которой на выбор средств измерений, помимо перечисленных трудностей, накладывается требование пространственной локализации источников примесей. Наряду с общим интересом к созданию таких методов, ближайший прикладной интерес связан с предстоящим запуском термоядерного реактора ИТЭР, для которого необходимо обеспечение защиты от паров воды в рабочей камере, играющей роль и охладителя, и рабочего тела. Опыт работы с реакторами предыдущих поколений позволил сформулировать уровень допустимых ограничений на примеси. В течение ряда лет ряд групп, включая нашу, работали в этом направлении. Оптимальным на сегодня решением является эмиссионный метод в новом варианте мультиспектральной актинометрии. При этом подходе используются не только классические актинометры инертных атомов, но и промежуточные нестабильные частицы. Это позволило повысить чувствительность измерений и расширить спектр детектируемых частиц, являющихся продуктами превращений молекул воды. Техника отрабатывалась на модельных установках со взаимным контролем, включая лазерный. Важным результатом явилось создание кинетической модели реакций в плазме, адекватно описывающей модельные эксперименты.
Наряду с высокой чувствительностью метода, принцип локализации реализуется за счет того, что уже вблизи первой стенки продукты химических реакций достигают стационарных концентраций за время, меньшее времени диффузионного ухода частиц из зоны измерений, определяемой пространственным разрешением оптической системы.
Это позволило сделать положительный прогноз возможностей мониторинга примесей на вводящемся в строй реакторе.
Необходимо сказать, что полученные результаты отвечают на вопрос о контроле примесей на этапе тестирования реактора с разрядами в газах, содержащими дейтерий. Разработка диагностик в режиме протекания термоядерных реакций, несмотря на созданную основу, потребует дополнительных исследований.
Исследование выполнено при финансовой поддержке РФФИ в рамках научного проекта № 19-12-50237.
Funding: The reported study was funded by RFBR, project number 19-12-50237.
Список литературы
Очкин В.Н., Фортов В.Е. Энциклопедия низкотемпературной плазмы, Серия Б: Справочные приложения, базы и банки данных, т. III-2. Термодинамические, оптические и транспортные свойства низкотемпературной плазмы. Часть I, Оптические свойства низкотемпературной плазмы. Предисловие / Ред. Ф.Е. Фортов, отв. ред. В.Н. Очкин. М.: Янус-К, 2008. С. 1–4.
Оптическая пирометрия плазмы / Под ред. Н.Н. Со-болева. М.: ИЛ, 1960.
Лохте-Хольтгревен В. Методы исследования плазмы. Спектроскопия, лазеры, зонды / Перевод с англ. под ред. С.Ю. Лукьянова. М.: Мир, 1971.
Очкин В.Н. Спектроскопия низкотемпературной плазмы. М.: Физматлит, 2010. [Ochkin V.N. Spectroscopy of Low-Temperature Plasma. New York: W-ILEY-VCH, 2009] https://doi.org/10.1002/9783527627509
Биберман Л.М., Воробьев В.С., Якубов И.Т. Кинетика неравновесной низкотемпературной плазмы. М.: Наука, 1982.
Смирнов Б.М. Введение в физику плазмы. М.: Наука, 1982.
Русанов В.Д., Фридман А.А. Физика химически активной плазмы. М.: Наука, 1984.
Grim H.R. Principles of plasma spectroscopy. N.Y.: Cambridge University Press, 1997. https://doi.org/10.1017/CBO9780511524578
Словецкий Д.И. Механизмы химических реакций в неравновесной плазме. М.: Наука, 1980.
Лебедев Ю.А., Шахатов В.А. Кинетические модели в оптической диагностике неравновесной плазмы газовых разрядов / Энциклопедия низкотемпературной плазмы, Серия Б: Справочные приложения, базы и банки данных, т. III-2. Термодинамические, оптические и транспортные свойства низкотемпературной плазмы. Часть I, Оптические свойства низкотемпературной плазмы / Ред. Ф.Е. Фортов, отв. ред. В.Н. Очкин. М.: Янус-К, 2008. С. 69–162.
Witteman W.J. The CO2 laser. Berlin: Springer-Verlag, 1987. https://doi.org/10.1007/978-3-540-47744-0
Трубачеев Э.А. Изучение физико-химических свойств плазмы СО лазера. В сб. Газовые лазеры и их применения / Под. ред. Н.Г. Басова. М.: Наука, 1977.
Трубачеев Э.А., Очкин В.Н., Азатян В.В. // Краткие сообщения по физике. 1973. № 3. С. 3.
Volchenok V.I., Egorov N.P., Komarov V.N., Kupriya-nov S.E., Ochkin V.N., Sobolev N.N., Trubacheev É.A. // Sov. J. Quantum Electronics. 1976. V. 6. P. 1173. https://doi.org/10.1070/QE1976v006n10ABEH011912
ITER, Final Design Report, 2001. G31 DDD 14 01_07-19 W0.1. Section 3.1. Vacuum pumping and fuelling systems.
Susuki C., Sasaki K., Kadota K. // Japan. J. Appl. Phys. 1998. V. 37. P. 5763. https://doi.org/10.1143/JJAP.37.5763
Lopaev D.V., Volynets A.V., Zyryanov S.M., Zoto-vich A.I., Rakhimov A.T. // J. Phys. D: Appl. Phys. 2017. V. 50. 075202. https://doi.org/10.1088/1361-6463/50/7/075202
Proshina O.V., Rakhimova T.V., Zotovich A.I., Lopa-ev D.V., Zyryanov S.M., Rakhimov A.T. // Plasma Sources Sci. Technol. 2017. V. 26. 075005. https://doi.org/10.1088/1361-6595/aa72c9
Hori M., Goto T. // Applied Surface Sci. 2002. V. 192. P. 135. https://doi.org/10.1016/S0169-4332(02)00024-7
Ibbotson D.E., Flamm D.L., Donnelly V.M. // J. Appl. Phys. 1983. V. 54. P. 5974. https://doi.org/10.1063/1.331775
Penetrante B.M., Hsiao M.C., Bardsley J.N., Merritt B.T., Vogtlin G.E., Kuthi A., Burkhart C.P., Baylessz J.R. // Plasma Sources Sci. Technol. 1997. V. 6. P. 251. https://doi.org/10.1088/0963-0252/6/3/002
Takeuchi W., Sasaki H., Kato S., Takashima S., Hiramatsu M., Hori M. // J. Appl. Phys. 2009. V. 105. 113305. https://doi.org/10.1063/1.3091279
Benedikt J., Wisse M., Woen R.V., Engeln R., San-den M.C.M. // J. Appl. Phys. 2003. V. 94. P. 6932. https://doi.org/10.1063/1.1622116
Ionin A.A., Frolov M.P., Ochkin V.N., Podmar’kov Y.P., Savinov S.Y., Seleznev L.V., Sinitsyn D.V., Tere-khov Y.V., Vagin N.P., Yuryshev N.N., Kochetov I.V., Napartovich A.P., Rulev O.A. // Proc. 6th AIAA Plasmadynamics and Lasers Conf., Toronto, Canada, 2005.
Lua X., Naidis G.V., Laroussi M., Reuter S., Graves D.B., Ostrikov K. // Physics Reports. 2016. V. 630. P. 1. https://doi.org/10.1016/j.physrep.2016.03.003
Zhang K., Perussello C.A., Milosavljević V., Cullen P.J., Da-Wen Sun, Tiwari B.K. // Critical Reviews in Food Science and Nutrition. 2019. V. 59. P. 812. https://doi.org/10.1080/10408398.2018.1564731
Iseki S., Hashizume S., Takeda J.F., Ishikawa K., Ohta T., Ito M., Hori M. // Applied Physics Express. 2011. V. 4. 116201. https://doi.org/10.1143/APEX.4.116201
Gessel A.F.H, Grootel S.C., Bruggeman P.J. // Plasma Sources Sci. Technol. 2013. V. 22. 055010. https://doi.org/10.1088/0963-0252/22/5/055010
Tada S., Takashima S., Ito M., Hori M., Goto T., Sakamoto Y. // J. Appl. Phys. 2000. V. 88. P. 1756. https://doi.org/10.1063/1.1305559
Ropcke J., Lombardi G., Rousseau A., Davies P.B. // Plasma Sources Sci. Technol. 2006. V. 15. P. S148 https://doi.org/10.1088/0963-0252/15/4/S02
Ropcke J., Davies P.B., Lang N., Rousseau A., Welzel S. // J. Phys.D: Appl. Phys. 2012. V. 45. 423001. https://doi.org/10.1088/0022-3727/45/42/423001
Reuter S., Sousa J.S., Stancu G.D., Helden J.-P.H. // Plasma Sources Sci. Technol. 2015. V. 24. 054001. https://doi.org/10.1088/0963-0252/24/5/054001
Peverall R., Ritchie G.A.D. // Plasma Sources Sci. Technol. 2019. V. 28. 073002. https://doi.org/10.1088/1361-6595/ab2956
Macken J.A., Matovich E., Brandewie R.A. // Bull. American Phys. Soc. 1967. V. 12. P.669.
Lotkova E.N., Makarov V.I., Polack L.S., Sobolev N.N. // High Energy Chemistry. 1968. V. 2–3. P. 278.
Witteman W.J., Werner H.W. // Physics Lett. A. 1968. V. 26. P. 454. https://doi.org/10.1016/0375-9601(68)90784-6
Волченок В.И., Егоров Н.Н., Комаров В.Н., Куприянов С.Е., Очкин В.Н. // Краткие сообщения по физике. 1976. № 11. С. 21.
Гасилевич Е.С., Иванов В.А., Лоткова Э.Н., Очкин В.Н., Соболев Н.Н., Ярославский Н.Г. // ЖТФ. 1969. Т. 39. № 6. С. 126.
Lotkova E.N., Ochkin V.N., Sobolev N.N. // IEEE Journal of Quantum Electronics. 1971. V. 7. P. 396. https://doi.org/10.1109/JQE.1971.1076813
Schneider S., Dünnbier M., Hübner S., Reuter S., Benedikt J. // J. Phys. D: Appl. Phys. 2014. V. 47. 505203. https://doi.org/10.1088/0022-3727/47/50/505203
Agarwal S., Hoex B., Sanden M.C.M., Maroudas D., Aydil E.S. // Appl. Phys. Lett. 2003. V. 83. P. 4918. https://doi.org/10.1063/1.1630843
Волченок В.И., Егоров Н.Н., Комаров В.Н., Куприянов С.Е., Очкин В.Н., Соболев Н.Н. // Доклады АН СССР. 1977. Т. 232. № 5. С. 1052.
Mrázková M., Vašina P., Kudrle V., Tálský A., Pintassilgo C.D., Guerra V. // J. Phys. D: Appl. Phys. 2009. V. 42. 075202. https://doi.org/10.1088/0022-3727/42/7/075202
Wu H., Sun P., Feng H., Zhou H., Wang R., Liang Y., Lu J., Zhu W., Zhang J., Fang J. // Plasma Processes and Polymers. 2012. V. 9. P. 417. https://doi.org/10.1002/ppap.201100065
Малышев В.И. Введение в экспериментальную спектроскопию. М.: Наука, 1979.
Ochkin V.N., Savinov S.Yu., Sobolev N.N. Electron-excited molecules in nonequilibrium plasma / Ed. by N.N. Sobolev N.Y.: Nova Science Publishers, 1989.
Bernatskiy A.V., Ochkin V.N., Bafoev R.N. // Bull. Lebedev Physics Institute. 2016. V. 43. P. 195. https://doi.org/10.3103/S1068335616060038
Иванов В.В., Клоповский К.С., Лопаев Д.В., Рахи-мов А.Т., Рахимова Т.В. // Физика плазмы. 2000. Т. 26. С. 1038. [Ivanov V.V., Klopovskii K.S., Lopa-ev D.V., Rakhimov A.T., Rakhimova T.V. // Plasma Phys. Rep. 2000. V. 26. P. 972. https://doi.org/10.1134/1.1323562]
Bernatskiy A.V., Ochkin V.N., Kochetov I.V. // J. Phys. D: Appl. Phys. 2016. V. 49. 395204. https://doi.org/10.1088/0022-3727/49/39/395204
Lopaev D.V., Malykhin E.M., Zyryanov S.M. // J. Phys. D: Appl. Phys. 2011. V. 44. 015201. https://doi.org/10.1088/0022-3727/44/1/015201
Granier A., Chéreau D., Henda K., Safari R., Leprin-ce P. // J. Appl. Phys. 1994. V. 75. P. 104. https://doi.org/10.1063/1.355897
Volynets A.V., Lopaev D.V., Zyryanov S.M., Bogdano-va M.A., Rakhimov A.T. // J. Phys. D: Appl. Phys. 2019. V. 52. 395203. https://doi.org/10.1088/1361-6463/ab28dc
d’Agostino R., Cramarossa F., De Benedictis S., Ferra-ro G. // J. Appl. Phys. 1981. V. 52. P. 1259. https://doi.org/10.1063/1.329748
Walkup R.E., Saenger K.L., Selwyn G.S. // J. Chemical Phys. 1986. V. 84. P. 2668. https://doi.org/10.1063/1.450339
Bernatskiy A.V., Ochkin V.N. // Plasma Sources Sci. Technol. 2017. V. 26. 015002. https://doi.org/10.1088/0963-0252/26/1/015002
Bernatskiy A.V., Kochetov I.V., Ochkin V.N. // Phys. Plasmas. 2018. V. 25. P. 083517. https://doi.org/10.1063/1.5042839
Bernatskiy A.V., Kochetov I.V., Ochkin V.N. // Plasma Sources Sci. Technol. 2019. V. 28. 105002. https://doi.org/10.1088/1361-6595/ab4301
Бернацкий А.В. Спектроскопические методы детектирования примесей молекул воды и их производных в плазме инертных газов электровакуумных установок: диссертация на соискание учёной степени кандидата физико-математических наук. М.: Физический институт им. П.Н. Лебедева РАН, 2017.
Henriques J., Villeger S., Levaton J., Nagai J., Santa-na S., Amorim J., Ricard A. // Surface and Coatings Technology. 2005. V. 200. P. 814. https://doi.org/10.1016/j.surfcoat.2005.01.051
Saloum S., Naddaf M., Alkhaled B. // Vacuum. 2008. V. 82. P. 742. https://doi.org/10.1016/j.vacuum.2007.10.009
Saloum S., Naddaf M. // Vacuum. 2007. V. 82. P. 66. https://doi.org/10.1016/j.vacuum.2007.04.031
Saloum S., Naddaf M., Alkhaled B. // Vacuum. 2010. V. 85. P. 439. https://doi.org/10.1016/j.vacuum.2010.08.007
Ricard A., Henriques J., Cousty S., Villeger S., Amo-rim J. // Plasma Processes and Polymers. 2007. V. 4. S965. https://doi.org/10.1002/ppap.200732308
Naz M.Y., Shukrullah S., Khan Y., Ghaffar A., Reh-man N.U., Ullah S. // High Energy Chemistry. 2015. V. 49. P. 449. https://doi.org/10.1134/S0018143915040116
Booth J.P., Joubert O., Pelletier J., Sadeghi N. // J. A-ppl. Phys. 1991. V. 69. P. 618. https://doi.org/10.1063/1.347395
Karakas E., Donnelly V.M., Economou D.J. // Appl. Phys. Lett. 2013. V. 102. 034107. https://doi.org/10.1063/1.4789435
Karakas E., Donnelly V.M., Economou D.J. // J. Appl. Phys. 2013. V. 113. 213301. https://doi.org/10.1063/1.4807298
Zhu Y., Lepikhin N.D., Orel I.S., Salmon A., Kloch-ko A.V., Starikovskaia S.M. // Plasma Sources Sci. Technol. 2018. V. 27. 075020. https://doi.org/10.1088/1361-6595/aac95f
Иванов В.В., Клоповский К.С., Лопаев Д.В., Рахи-мов А.Т., Рахимова Т.В. // Физика плазмы. 2000. Т. 26. С. 1046. [Ivanov V.V., Klopovskii K.S., Lopa-ev D.V., Rakhimov A.T., Rakhimova T.V. // Plasma Phys. Rep. 2000. V. 26. P. 980. https://doi.org/10.1134/1.1323563]
Kułakowska-Pawlak B., Jamro’z P. // Plasma Chemistry and Plasma Processing. 2010. V. 30. P. 641. https://doi.org/10.1007/s11090-010-9238-3
Mavadat M., Turgeon S., Ricard A. Laroche G. // J. Phys. D: Appl. Phys. 2012. V. 45. 315201. https://doi.org/10.1088/0022-3727/45/31/315201
Бернацкий А.В., Очкин В.Н., Кочетов И.В. // Письма в ЖТФ. 2017. Т. 43. С. 71. [Bernatskiy A.V., Och-kin V.N., Kochetov I.V. // Technical Physics Lett. 2017. V. 43. P. 902. https://doi.org/10.1134/S1063785017100030]
Filippov A.V., Mankelevich Y.A., Pal A.F., Rakhi-mov A.T., Serov A.O., Suetin N.V. // Proc. SPIE – The International Society for Optical Engineering. 2002. V. 4460. P. 285. https://doi.org/10.1117/12.459406
Huai Y., Li S.-Z., Li H., Wu K., Zhang J., Wang S., Wang Y.-X. // IEEE Trans. Plasma Sci. 2010. V. 42. P. 1648. https://doi.org/10.1109/TPS.2014.2320263
Coburn J.W., Chen M. // J. Appl. Phys. 1980. V. 51. P. 3134. https://doi.org/10.1063/1.328060
Абачев М.К., Газаров К.В., Гальперин В.А., Жогун В.Н., Пашков В.Ю., Орликовский А.А., Тябликов А.В. // Микроэлектроника. 1997. Т. 26. С. 225. [Aba-chev M.K., Gazarov K.V., Galperin V.A., Zhogun V.N., Pashkov V.Y., Ponomareva N.A., Orlikovskii A.A., Tyab-likov A.V. // Russian Microelectronics. 1997. V. 26. P. 192]
Shogun V., Tyablikov A., Abachev M., Pashkov V., Scharff W., Wallendorf T. // Surface and Coatings Technology. 1998. V. 98. P. 1406. https://doi.org/10.1016/S0257-8972(97)00259-4
d’Agostino R., Cramarossa F., Colaprico V., d’Ettole R. // J. Appl. Phys. 1983. V. 54. P. 1284. https://doi.org/10.1063/1.332193
Petitjean L., Ricard A. // J. Phys. D: Appl. Phys. 1984. V. 17. P. 919. https://doi.org/10.1088/0022-3727/17/5/008
d’Agostino R., Cramarossa F., Benedictis S., Fracassi F. // Plasma Chemistry and Plasma Processing. 1984. V. 4. P. 163. https://doi.org/10.1007/BF00566839
Gottscho R.A., Donnelly V.M. // J. Appl. Phys. 1984. V. 56. P. 245. https://doi.org/10.1063/1.333954
Bernatskiy A.V., Lagunov V.V., Ochkin V.N., Tskhai S.N. // Laser Physics Lett. 2016. V. 13. 075702. https://doi.org/10.1088/1612-2011/13/7/075702
Bernatskiy A.V., Ochkin V.N., Kochetov I.V. // Bull. Lebedev Physics Institute. 2017. V. 44. P. 147. https://doi.org/10.3103/S1068335617050062
Бернацкий А.В., Очкин В.Н., Бафоев Р.Н., Антипенков А.Б. // Физика плазмы. 2016. Т. 42. С. 949. https://doi.org/10.7868/S0367292116100012 [Bernatskiy A.V., Ochkin V.N., Ba-foev R.N., Antipenkov A.B. // Plasma Phys. Rep. 2016. V. 42. P. 990. https://doi.org/10.1134/S1063780X16100019]
Bernatskiy A.V., Ochkin V.N., Bafoev R.N. // J. Physics: Conf. Series. 2016. V. 747. 012013. https://doi.org/10.1088/1742-6596/747/1/012013
Bernatskiy A.V., Ochkin V.N. // Bull. Lebedev Physics Institute. 2015. V. 42. P. 273. https://doi.org/10.3103/S1068335615090055
Kiss L.D.B., Nicolai J.P., Conner W.T., Sawin H.H. // J. Appl. Phys. 1992. V. 71. P. 3186. https://doi.org/10.1063/1.350961
Karakas E., Kaler S., Lou Q., Donnelly V.M., Econo-mou D.J. // J. Phys. D: Appl. Phys. 2014. V. 47. 085203. https://doi.org/10.1088/0022-3727/47/8/085203
NIST Atomic Spectra Database Lines Data https://physics.nist.gov/PhysRefData/ASD/lines_form.html
Кузнецова Л.А., Кузьменко Н.Е., Кузяков Ю.Я., Пластинин Ю.А. Вероятности переходов двухатомных молекул / Под ред. Р. В. Хохлова. М.: Наука, 1980.
Фриш С.Э. Определение концентраций нормальных и возбужденных атомов и сил осцилляторов методами испускания и поглощения света. В кн. Спектроскопия газоразрядной плазмы / Ред. С.Э. Фриш. Ленинград: Наука, 1970. С. 7 – 62.
Takashima S., Hori M., Goto T., Koto A., Ito M., Yeda K. // Appl. Phys. Lett. 1999. V. 75. P. 3929. https://doi.org/10.1063/1.125497
Takeda K., Kato M., Jia F., Ishikawa K., Kano H., Sekine M. Hori M. // J. Phys. D: Appl. Phys. 2013. V. 46. 464006. https://doi.org/10.1088/0022-3727/46/46/464006
Копылов С.М., Лысой Б.Г., Серегин С.Л., Чередниченко О.Б. Перестраиваемые лазеры на красителях и их применения. М.: Радио и связь, 1991.
Демтредер В. Лазерная спектроскопия. Основные принципы и техника эксперимента. М.: Наука, 1985.
Лазерная аналитическая спектроскопия / Под ред. В.С. Летохова. М.: Наука, 1986.
Очкин В.Н. // Физика плазмы. 2015. Т. 41. С. 381. [Ochkin V.N. // Plasma Phys. Rep. 2015. V. 41. P. 350. https://doi.org/10.1134/S1063780X15040042]
Chernin S.M. // J. Modern Optics. 2001. V. 44. P. 619. https://doi.org/10.1080/09500340108230936
Nadezhdinskii A.I., Berezin A.G., Chernin S.M., Er-shov O., Kutnyak V. // Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy. 1999. V. 55. P. 2083. https://doi.org/10.1016/S1386-1425(99)00080-3
Schmidt-Bleker A., Winter J., Iseni S., Dunnbier M., Weltmann K.-D., Reuter S. // J. Phys. D: Appl. Phys. 2014. V. 47. 145201. https://doi.org/10.1088/0022-3727/47/14/145201
Baev V.M., Latz T., Toshek P.E. // Appl. Phys. B. 1999. V. 69. P. 171. https://doi.org/10.1007/s003400050793
Пахомычева Л.А., Свириденков Э.А., Сучков А.Ф., Титова Л.В., Чурилов С.С. // Письма ЖЭТФ. 1970. Т. 12. С. 60.
Лукьяненко С.Ф., Макогон М.М., Синица С.Н. Внутрирезонаторная лазерная спеткроскопия. Новосибирск: Наука, 1995.
Podmar'kov Yu.P., Raspopov N.A., Frolov M.P. // Proc. SPIE. 2002. V. 4766. P. 188. https://doi.org/10.1117/12.475325
Pazyuk V.S., Podmar’kov Yu.P., Raspopov N.A., Fro-lov M.P. // Quantum Electronics. 2001. V. 31. P. 363. https://doi.org/10.1070/QE2001v031n04ABEH001952
O’Keefe A., Deacon D.A.G. // Rev. Scientific Instrum. 1988. V. 59. P. 2544. https://doi.org/10.1063/1.1139895
Berden G.,Engeln R. Cavity Ring-Down Spectroscopy: Techniques and Applications. Wiley, 2009. https://doi.org/10.1002/9781444308259
Schwabedissen A., Brockhaus A., Georg A., Engemann J. // J. Phys. D: Appl. Phys. 2001. V. 34. P. 1116. https://doi.org/10.1088/0022-3727/34/7/314
Nikolaev I.V., Ochkin V.N., Spiridonov M.V., Tskhai S.N. // Laser Physics. 2011. V. 21. P. 2088. https://doi.org/10.1134/S1054660X11220014
Nikolaev I.V., Ochkin V.N., Tskhai S.N. // Laser Physics Lett. 2013. V. 10. 115701. https://doi.org/10.1088/1612-2011/10/11/115701
Herriott D.R., Kogelnik H., Kompfner H.R. // Optics. 1964. V. 3. P. 523. https://doi.org/10.1364/AO.3.000523
O’Keefe A., James J. // Chemical Physics Lett. 1999. V. 307. P. 343. https://doi.org/10.1016/S0009-2614(99)00547-3
Andreev S.N., Nikolaev I.V., Ochkin V.N., Savinov S.Yu., Spiridonov M.V., Tskhai S.N. // Quantum Electronics. 2007. V. 37. P. 399. https://doi.org/10.1070/QE2007v037n04ABEH013311
Nikolaev I.V., Ochkin V.N., Peters G.S., Spiridonov M.V., Tskhai S.N. // Laser Physics. 2013. V. 23. 035701. https://doi.org/10.1088/1054-660X/23/3/035701
Brumfield B., Sun W.T., Wang Y., Ju Y.G., Wysocki G. // Optics Lett. 2014. V. 39. P. 1783. https://doi.org/10.1364/OL.39.001783
Engeln R., Berden G., VandenBerg E., Meijer G. // J. Chemical Physics. 1997. V. 107. P. 4458. https://doi.org/10.1063/1.474808
Gianella M., Pinto T.H.P., Wu X., Ritchie G.A.D. // J. Chemical Physics. 2017. V. 147. P. 054201. https://doi.org/10.1063/1.4985900
Welzel S., Guaitella O., Lazzaroni C., Pintassilgo C.D., Rousseau A., Ropcke J. // J. Plasma Sources Sci. and Technol. 2011. V. 20. 015020. https://doi.org/10.1088/0963-0252/20/1/015020
Juchmann W., Luque J., Jeffries J.B. // Appl. Optics. 2000. V. 39. P. 3704. https://doi.org/10.1364/AO.39.003704
Luque J., Juchmann W., Jeffries J.B. // J. Appl. Phys. 1997. V. 82. P. 2072. https://doi.org/10.1063/1.366017
Curry J.J., Adler H.G., Shastri S.D. Using synchrotron radiation as a laser: X-ray induced fluorescence in high pressure lighting plasmas / Proc. X Int. Conf. on laser aided plasma diagnostics. Fukuoka, Japan, 2001. P. 362.
Katsch H.M., Tewes A., Quandt E., Goehlich A., Kawetzki T., Döbele H.F. // J. Appl. Phys. 2000. V. 88. P. 6232. https://doi.org/10.1063/1.1315332
Gottsho R.A., Davis G.P., Burton R.H. // J. Vacuum Sci. & Technology A. 1983. V. 1. P. 622. https://doi.org/10.1116/1.572192
Francis A., Gans T. Niemi, K. Czarnetzki U., Schulz-von der Gathen V., Döbele H.F // Proc. SPIE. 2002. V. 4460. P. 122. https://doi.org/10.1117/12.459391
Goechlich A., Kavetski T., Doebele H.F. // J. Chemical Physics. 1998. V. 108. P. 9362. https://doi.org/10.1063/1.476388
Gottsho R.A., Gaebe C.E. // IEEE Trans. Plasma Sci. 1986. V. PS-14. P. 92.
Pealat M., Taran J.P., Bacal M., Hillion F. // J. Chemical Physics. 1985. V. 82. P. 4943. https://doi.org/10.1063/1.448668
Grundberg K.E., Hebner G.A., Verduen G.T. // Appl. Physics Lett. 1984. V. 44. P. 299. https://doi.org/10.1063/1.94731
Grangeon F., Monard C., Dorier J.-L., Howling A.A., Hollenstein Ch., Romanini D., Sadeghi N. // Plasma Sources Sci. Technol. 1999. V. 8. P. 448. https://doi.org/10.1088/0963-0252/8/3/315
Quandt E., Kraemer I., Dobele H.F. // Europhysics Lett. 1999. V. 45. P. 32. https://doi.org/10.1209/epl/i1999-00127-x
Очкин В.Н., Преображенский Н.Г., Соболев Н.Н., Шапарев Н.Я. // УФН. 1986. Т. 148. С. 473. [Och-kin V.N., Preobrazhenskii N.G., Sobolev N.N., Shapa-rev N.Ya. // Sov. Phys. – Uspekhi. 1986. V. 29. P. 260. https://doi.org/10.1070/PU1986v029n03ABEH003187]
Очкин В.Н., Преображенский Н.Г., Шапарев Н.Я. Оптогальванический эффект в ионизованом газе. М.: Наука, 1991.
Ochkin V.N., Preobrazhensky N.G., Shaparev N.Y. Optogalvanic effect in ionized gas. London: Gordon and Breach Science Publishers, 1998.
Zhechev D., Bundaleska N., Costello G.T. // J. Phys. D: Appl. Phys. 2005. V. 38. P. 2237. https://doi.org/10.1088/0022-3727/38/13/023
ITER Physics Basis Editors, ITER Physics Expert Group Chairs and Co-Chairs and ITER Joint Central Team and Physics Integration Unit. Chapter 1: Overview and summary // Nucl. Fusion. 1999. V. 39. P. 2137. https://doi.org/10.1088/0029-5515/39/12/301
ITER Physics Expert Group on Confinement and Transport, ITER Physics Expert Group on Confinement Modelling and Database and ITER Physics Basis Editors. Chapter 2: Plasma confinement and transport // Nucl. Fusion. 1999. V. 39. P. 2175. https://doi.org/10.1088/0029-5515/39/12/302
ITER Physics Expert Group on Disruptions, Plasma Control, and MHD and ITER Physics Basis Editors. Chapter 3: MHD stability, operational limits and disruptions // Nucl. Fusion. 1999. V. 39. P. 2251. https://doi.org/10.1088/0029-5515/39/12/303
ITER Physics Expert Group on Divertor, ITER Physics Expert Group on Divertor Modelling and Database and ITER Physics Basis Editors. Chapter 4: Power and particle control // Nucl. Fusion. 1999. V. 39. P. 2391. https://doi.org/10.1088/0029-5515/39/12/304
ITER Physics Expert Group on Energetic Particles, Heating and Current Drive and ITER Physics Basis Editors. Chapter 5: Physics of energetic ions // Nucl. Fusion. 1999. V. 39. P. 2471. https://doi.org/10.1088/0029-5515/39/12/305
ITER Physics Expert Group on Energetic Particles, Heating and Current Drive and ITER Physics Basis Editors. Chapter 6: Plasma auxiliary heating and current drive // Nucl. Fusion. 1999. V. 39. P. 2495. https://doi.org/10.1088/0029-5515/39/12/306
ITER Physics Expert Group on Diagnostics and ITER Physics Basis Editors. Chapter 7: Measurement of plasma parameters // Nucl. Fusion. 1999. V. 39. P. 2541. https://doi.org/10.1088/0029-5515/39/12/307
ITER Physics Expert Group on Disruptions, Plasma Control, and MHD, ITER Physics Expert Group on Energetic Particles, Heating and Current Drive, ITER Physics Expert Group on Diagnostics and ITER Physics Basis Editors. Chapter 8: Plasma operation and control // Nucl. Fusion. 1999. V. 39. P. 2577. https://doi.org/10.1088/0029-5515/39/12/308
ITER Physics Basis Editors, ITER Physics Expert Group Chairs and Co-Chairs and ITER Joint Central Team and Physics Integration Unit. Chapter 9: Opportunities for reactor scale experimental physics // Nucl. Fusion. 1999. V. 39. P. 2627. https://doi.org/10.1088/0029-5515/39/12/309
Burkart W. // Nucl. Fusion. 2005. V. 45. https://doi.org/10.1088/0029-5515/45/10A/E01
International Fusion Research Council (IFRC) // Nucl. Fusion. 2005. V. 45. P. A1. https://doi.org/10.1088/0029-5515/45/10A/001
Ikeda K. // Nucl. Fusion. 2007. V. 47. P. E01. https://doi.org/10.1088/0029-5515/47/6/E01
Воронов Г.С. Энциклопедия низкотемпературной плазмы, Серия Б: Справочные приложения, базы и банки данных, т. III-2. Термодинамические, оптические и транспортные свойства низкотемпературной плазмы. Часть I, Оптические свойства низкотемпературной плазмы. Предисловие / Ред. Ф.Е. Фортов, отв. ред. В.Н. Очкин. М: Янус-К, 2008. С. 286–323.
Визгалов И.В., Курнаев В.А., Колесников В.Н., Мозгрин Д.В., Смирнов В.М., Трифонов Н.Н. // ВАНТ. Серия Термоядерный синтез. 2002. Т. 25. Вып. 3–4. С. 125.
Антипенков А.Б., Афонин О.Н., Визгалов И.В., Курнаев В.А., Колесников В.Н. // ВАНТ. Серия Термоядерный синтез. 2006. Т. 29. Вып. 3. С. 46.
Антипенков А.Б., Афонин О.Н., Визгалов И.В., Колесников В.Н., Курнаев В.А. // ВАНТ. Серия Термоядерный синтез. 2007. Т. 30. Вып. 4. С. 44.
Gargiuloa L., Bayetti P., Bruno V., Hatchressian J.-C., Hernandez C., Houry M., Keller D., Martins J.-P., Measson Y., Perrot Y., Samaille F. // Fusion Engineering and Design. 2009. V. 84. P. 220. https://doi.org/10.1016/j.fusengdes.2008.11.043
Houry M., Bayetti P., Keller D., Gargiulo L., Bruno V., Hatchressian J.C., Hernandez C., Martins J.P., Measson Y., Perrot Y., Russotto F.X. // Fusion Engineering and Design. 2010. V. 85. P. 1947. https://doi.org/10.1016/j.fusengdes.2010.06.030
Houry M., Gargiulo L., Balorin C., Bruno V., Keller D., Roche H., Kammerer N., Measson Y., Carrel F., Scho-epff V. // Fusion Engineering and Design. 2011. V. 86. P. 1868. https://doi.org/10.1016/j.fusengdes.2010.11.004
Durocher Au., Bruno A., Chantant M., Gargiulo L., Gherman T., Hatchressian J.C., Houry M., Le R., Mou-yon D. // Fusion Engineering and Design. 2013. V. 88. P. 1390. https://doi.org/10.1016/j.fusengdes.2013.02.078
Worth L.B.C., Pearce R.J.H., Wikus P., Chantant M. // Fusion Engineering and Design. 2013. V. 88. P. 2115. https://doi.org/10.1016/j.fusengdes.2013.01.064
Martinez A., Samaille F., Chantant M., Hatchres-sian J.C. // Fusion Engineering and Design. 2007. V. 82. P. 1989. https://doi.org/10.1016/j.fusengdes.2007.07.038
Caldwell-Nichols C.J., Glugla M., Day C., Haas H., Murdoch D.K., Wykes M.E.P. // Proc. Symposium on Fusion Engineering (21st IEEE/NPS Symposium on Fusion Engineering SOFE 05). 2006. 4018953. https://doi.org/10.1109/FUSION.2005.252919
Sorokin I., Vizgalov I., Gutorov K., Podolyako F. // Physics Procedia. 2015. V. 71. P. 116. https://doi.org/10.1016/j.phpro.2015.08.327
Сорокин И.А., Визгалов И.В., Гуторов К.М., Подоляко Ф.С. // Ядерная физика и инжиниринг. 2015. Т. 6. С. 73. https://doi.org/10.1134/S2079562915010157
Антипенков А.Б., Афонин О.Н., Очкин В.Н., Савинов С.Ю., Цхай С.Н. // Физика плазмы. 2012. Т. 38. С. 221. [Antipenkov A.B., Afonin O.N., Ochkin V.N., Savinov S.Yu., Tskhai S.N. // Plasma Phys. Rep. 2012. V. 38. P. 197. https://doi.org/10.1134/S1063780X12020018]
Воронов Г.С., Батанов Г.М., Бережецкий М.С., Бондарь Ю.И., Борзосеков В.Д., Вафин И.Ю., Васильков Д.Г., Гребенщиков С.Е., Гришина И.А., Колик Л.В., Кончеков Е.М., Ларионова Н.Ф., Лету-нов А.А., Логвиненко В.П., Малахов Д.В., Мещеряков А.И., Плешков Е.И., Петров А.Е., Сарксян К.А., Сатунин С.Н., Скворцова Н.Н., Степахин В.Д., Харчев Н.К., Хольнов Ю.В., Федянин О.И., Цыганков В.А., Курнаев В.А., Визгалов И.В., Урусов В.А., Антипенков A., Пирс Р., Ворх Л. // Физика плазмы. 2012. Т. 38. С. 769. [Voronov G.S., Batanov G.M., Berezhetsky M.S., Bondar’ Yu.F., Borzosekov V.D., Vaphin I.Yu., Vasilkov D.G., Grebenshchikov S.E., Grishina I.A., Kolik L.V., Konchekov E.M., Larionova N.F., Letunov A.A., Logvinenko V.P., Malakhov D.V., Meshcheryakov A.I., Pleshkov E.I., Petrov A.E., Sark-syan K.A., Satunin S.N., Skvortsova N.N., Stepa-khin V.D., Kharchev N.K., Khol’nov Yu.V., Fedyanin O.I., Tsygankov V.A., Kurnaev V.A., Vizgalov I.V., Urusov V.A., Antipenkov A., Pearce R., Worth L. // Plasma Phys. Rep. 2012. V. 38. P. 708. https://doi.org/10.1134/S1063780X12090073]
Воронов Г.С., Бережецкий М.С., Бондарь Ю.И., Вафин И.Ю., Васильков Д.Г., Воронова Е.В., Гребенщиков С.Е., Гришина И.А., Ларионова Н.Ф., Летунов А.А., Логвиненко В.П., Мещеряков А.И., Плешков Е.И., Хольнов Ю.В., Федянин О.И., Цыганков В.А., Щепетов С.В., Курнаев В.А., Визгалов И.В., Уру-сов В.А., Сорокин И.А., Подоляко Ф.С., Антипен-ков A., Пирс Р., Ворх Л. // Физика плазмы. 2013. Т. 39. С. 313. [Voronov G.S., Berezhetskii M.S., Bon-dar’ Yu.F., Vafin I.Yu., Vasil’kov D.G., Voronova E.V., Grebenshchikov S.E., Grishina I.A., Larionova N.F., Letunov A.A., Logvinenko V.P., Meshcheryakov A.I., Pleshkov E.I., Khol’nov Yu.V., Fedyanin O.I., Tsygan-kov V.A., Shchepetov S.V., Kurnaev V.A., Vizgalov I.V., Urusov V.A., Sorokin I.A., Podolyako F.S., Antipenkov A., Pearce R., Worth L. // Plasma Phys. Rep. 2013. V. 39. P. 277. https://doi.org/10.1134/S1063780X13040090]
Kurnaev V., Afonin O., Antipenkov A., Koborov N., Mu-khammedzyanov T., Ochkin V., Pearce R., Pleshkov E., Podolyako F., Sorokin I., Urusov V., Vizgalov I., Voro-nov G., Vukolov K., Worh L., L-2M team // Fusion Engineering and Design. 2013. V. 88. P. 1414. https://doi.org/10.1016/j.fusengdes.2012.12.022
Бернацкий А.В., Очкин В.Н., Афонин О.Н., Антипенков А.Б. // Физика плазмы. 2015. Т. 41. С. 767. [Bernatskiy A.V., Ochkin V.N., Afonin O.N., Antipen-kov A.B. // Plasma Phys. Rep. 2015. V. 41. P. 705. https://doi.org/10.1134/S1063780X15090032] https://doi.org/10.7868/S0367292115090036
Антипенков А.Б., Афонин О.Н., Бернацкий А.В., Очкин В.Н. // Ядерная физика и инжиниринг. 2014. Т. 5. С. 644. https://doi.org/10.1134/S2079562914070021
Andreev S.N., Bernatskiy A.V., Ochkin V.N. // J. Physics: Conference Series. 2019. V. 1370. 012011. https://doi.org/10.1088/1742-6596/1370/1/012011
Andreev S.N., Bernatskiy A.V., Ochkin V.N. // Vacuum. 2020. – в пeчaти
Bernatskiy A.V., Lagunov V.V., Ochkin V.N. // Quantum Electronics. 2019. V. 49. P. 157. https://doi.org/10.1070/QEL16819
Bernatskiy A.V., Lagunov V.V., Ochkin V.N. // Physics of Atomic Nuclei. 2019. V. 82. No. 10, P. 1382. https://doi.org/10.1134/S1063778819100065
Bernatskiy A.V., Lagunov V.V., Ochkin V.N. // Physics of Wave Phenomena. 2019. V. 27. P. 165. https://doi.org/10.3103/S1541308X19030014
Bernatskiy A.V., Kochetov I.V., Lagunov V.V., Och-kin V.N. // J. Physics: Conference Series. 2019. V. 1370. P. 012012. https://doi.org/10.1088/1742-6596/1370/1/012012
Bernatskiy A.V., Kochetov I.V., Lagunov V.V., Och-kin V.N. // Phys. Plasmas. 2019. V. 26. 083511. https://doi.org/10.1063/1.5118830
Очкин В.Н., Савинов С.Ю., Соболев Н.Н. // ЖТФ. 1977. Т. 47. С. 1168.
Ochkin V.N., Savinov S.Yu., Sobolev N.N. // J. Applied Spectroscopy. 1977. V. 26. P. 647. https://doi.org/10.1007/BF00611848
Очкин В.Н., Савинов С.Ю., Соболев Н.Н. // Краткие сообщения по физике. 1980. № 2. С. 9.
Очкин В.Н., Савинов С.Ю., Соболев Н.Н. Механизмы формирования распределений электронно-возбужденных молекул по колебательно-вращательным уровням в газовом разряде. Электронно-возбужденные молекулы в неравновесной плазме. Труды Физического института им. П.Н. Лебедева АН СССР. Том 157. М.: Наука, 1985. С. 6.
Кузнецова Л.А., Кузьменко Н.Е., Кузяков Ю.Я., Пластинин Ю.А. Вероятности оптических переходов двухатомных молекул / Под ред. Р.В. Хохлова. М.: Наука, 1980.
Schofield K. // J. Physical and Chemical Reference Data. 1979. V. 8. P. 723. https://doi.org/10.1063/1.555606
Wysong I.J., Jeffries J.B., Crosley D.R. // J. Chemical Physics. 1990. V. 92. P. 5218. https://doi.org/10.1063/1.458558
Tamura M., Berg P.A., Harrington J.E., Luque J., Jeffries J.B., Smith G.P., Crosley D.R. // Combustion and Flame. 1998. V. 114. P. 502. https://doi.org/10.1016/S0010-2180(97)00324-6
Dieke G.H., Crosswhite H.M. // J. Quantitative Spectroscopy and Radiative Transfer. 1962. V. 2. P. 97. https://doi.org/10.1016/0022-4073(62)90061-4
Демидов В.И., Колоколов Н.Б., Кудрявцев А.А. Зондовые методы исследования низкотемпературной плазмы. М.: Энергоатомиздат, 1996.
Godyak V.A., Alexandrovich B.M., Kolobov V.I. // Phys. Plasmas. 2019. V. 26. P. 033504. https://doi.org/10.1063/1.5088706
Vizgalov I.V., Sorokin I.A., Kurnaev V.A. // J. Physics: Conference Series. 2016. V. 747. 012020. https://doi.org/10.1088/1742-6596/747/1/012020
Дятко Н.А., Кочетов И.В., Напартович А.П., Та-ран М.Д. // ТВТ. 1984. Т. 22. С. 1048. [Dyatko N.A., Kochetov I.V., Napartovich A.P., Taran M.D. // High Temperature. 1984. V. 22. P. 795]
Biagi (transcription of data from SF Biagi’s Fortran code, Magboltz) www.lxcat.net/Biagi
www.lxcat.net
Scarlett L.H., Tapley J.K., Fursa D.V., Zammit M.C., Savage J.S., Bray I. // Phys. Rev. A. 2017. V. 96. 062708. https://doi.org/10.1103/PhysRevA.96.062708
Yoon J.-S., Song M.-Y., Han J.-M., Hwang S.H., Chang W.-S., Lee B., Itikawa Y. // J. Physical and Chemical Reference Data. 2008. V. 37. P. 913. https://doi.org/10.1063/1.2838023
Scarlett L.H., Tapley J.K., Fursa D.V., Zammit M.C., Savage J.S., Bray I. // The European Physical Journal D. 2008. V. 72. P. 34. https://doi.org/10.1140/epjd/e2017-80649-8
Itikawa Y., Mason N. // J. Physical and Chemical Reference Data. 2005. V. 34. P. 1. https://doi.org/10.1063/1.1799251
Ionin A.A., Kochetov I.V., Napartovich A.P., Yury-shev N.N. // J. Phys. D: Appl. Phys. 2007. V. 40. P. R25. https://doi.org/10.1088/0022-3727/40/2/R01
Eliasson B., Kogelschatz U. // Basic data for modelling of electrical discharge in gases: oxygen Report KLR-86-11C (Report Brown Boveri Forschungszentrum CH-5405 Baden). 1986.
McEwan M.J., Phillips L.F. Chemistry of the atmosphere. London: Edward Arnold, 1975.
Millar T.J., Farquhar P.R.A., Willacy K. // Astronomy and Astrophysics Supplement Series. 1997. V. 121. P. 139. https://doi.org/10.1051/aas:1997118
Sutherland C.D., Zinn J. Chemistry computations for irradiative hot air. Los Alamos Scientific Laboratory Inform Report LA-6055-MS. Los Alamos, NM., 1975.
Adamson S., Astapenko V., Chernysheva I., Chorkov V., Deminsky M., Demchenko G., Demura A., Demyanov A., Dyatko N., Eletzkii A., Knizhnik A., Kochetov I., Napartovich A., Rykova E., Sukhanov L., Umanskii S., Vetchinkin A., Zaitsevskii A., Potapki B. // J. Phys. D: Applied Phys. 2007. V. 40. P. 3857. https://doi.org/10.1088/0022-3727/40/13/S06
Zellner R., Erler K. Field D. // Symposium (International) on Combustion. 1977. V. 16. P. 939. https://doi.org/10.1016/S0082-0784(77)80386-X
Baulch D.L., Bowman C.T., Cobos C.J., Cox R.A., Just T., Kerr J.A., Pilling M.J., Stocker D., Troe J., Tsang W., Walker R.W., Warnatz J. // J. Physical and Chemical Reference Data. 2005. V. 34. P. 757. https://doi.org/10.1063/1.1748524
Baulch D.L., Cobos C.J., Cox R.A., Esser C., Frank P., Just T., Kerr J.A., Pilling M.J., Troe J., Walker R.W., Warnatz J. // J. Physical and Chemical Reference Data. 1992. V. 21. P. 411. https://doi.org/10.1063/1.555908
Tsang W., Hampson R.F. // J. Physical and Chemical Reference Data. 1986. V. 15. P. 1087. https://doi.org/10.1063/1.555759
Warnatz J. Combustion Chemistry. New York: Springer, 1984.
Baulch D.L., Drysdale D.D., Horne D.G., Lloyd A.C. Evaluated Kinetic Data for High Temperature Reactions. Vol 2. Cleveland: CRC Press, 1973.
Bedjanian Y., Le. Bras G., Poulet G. // J. Physical Chemistry A. 1999. V. 103. P. 7017. https://doi.org/10.1021/jp991146r
Michael J.V., Fisher J.R. // J. Physical Chemistry. 1990. 94. P. 3318. https://doi.org/10.1021/j100371a022
Pratt G., Rogers D. // J. Chemical Soc., Faraday Transactions 1: Physical Chemistry in Condensed Phases. 1976. V. 72. P. 1589. https://doi.org/10.1039/F19767201589
Bedjanian Y., Riffault V., Le Bras G., Poulet G. // J. Physical Chemistry A. 2001. V. 105. P.6154. https://doi.org/10.1021/jp010369q
Fisher J.R., Michael J.V. // J. Physical Chemistry. 1990. V. 94. P. 2465. https://doi.org/10.1021/j100369a048
Ravishankara A.R., Nicovich J.M., Thompson R.L., Tul-ly F.P. // J. Physical Chemistry. 1981. V. 85. P. 2498. https://doi.org/10.1021/j150617a018
Zhang D.H., Zhang J.Z.H., Zhang Y., Wang D., Zhang Q. // J. Chemical Physics. 1995. V. 102. P. 7400. https://doi.org/10.1063/1.469052
Talukdar R.K, Gierczak T., Goldfarb L., Rudich Y., Rao B.S.M., Ravishankara A.R. // J. Physical Chemistry. 1996. V. 100. P. 3037. https://doi.org/10.1021/jp9518724
Albers E.A., Hoyermann K., Wagner H.G., Wolfrum J. // Symposium (International) on Combustion. 1971. V. 13. P. 81. https://doi.org/10.1016/S0082-0784(71)80012-7
Baldwin R.R., Rossiter B.N., Walker R.W. // Transactions of the Faraday Society. 1970. V. 66. P. 2004. https://doi.org/10.1039/TF9706602004
Bennett J.E., Blackmore D.R. // J. Chemical Physics. 1970. V. 53. P. 4400. https://doi.org/10.1063/1.1673957
NIST (http://kinetics.nist.gov/kinetics)
www.kintech.ru
Deminsky M., Chorkov V., Belov G., Cheshigin I., Knizhnik A., Shulakova E., Shulakov M., Iskandarova I., Alexandrov V., Petrusev A., Kirillov I., Strelkova M., Uman-ski S., Potapkin B. // Computational Materials Science. 2003. V. 28. P. 169. https://doi.org/10.1016/S0927-0256(03)00105-8
Lisitsa V.S., Kadomtsev M.B., Kotov V., Neverov V.S., Shurygin V.A. // Atoms. 2014. V. 2. P. 195. https://doi.org/10.3390/atoms2020195
Физические величины. Справочник / Под ред. И.С. Григорьева и Е.З. Мейлихова. М.: Энергоатомиздат, 1991.
Полак Л.С., Словецкий Д.И., Соколов А.С., Федосеева Т.В. Исследование механизма диссоциации СО2 в тлеющем разряде. В кн. Экспериментальное и теоретическое исследование неравновесных физико-химических процессов. М.: Наука, 1974. С. 188–226.
Reicle R. System design description (DDD) 55.G1 Vis/IR equatorial port wide-angle viewing system ITER Report. DCHGAP IO/DG/DIP/CHD/DLAG ITER. 2015
Palermo I., Mota F., Rios L., Catalán J.P., Alonso J., Ibarra A. // Fusion Engineering and Design. 2015. V. 100. P. 629. https://doi.org/10.1016/j.fusengdes.2015.08.014
Pan F., Oka T. // Phys. Rev. A. 1987. V. 36. P. 2297. https://doi.org/10.1103/PhysRevA.36.2297
Дополнительные материалы отсутствуют.