

Физиология растений, 2021, T. 68, № 4, стр. 439-448
Особенности применения O2-электродов Кларка в биологических исследованиях
В. Б. Бородин *
Институт фундаментальных проблем биологии Российской академии наук, Федеральный исследовательский центр “Пущинский научный центр биологических исследований Российской академии наук”
Пущино, Россия
* E-mail: borodin_ibbp@mail.ru
Поступила в редакцию 05.05.2020
После доработки 09.07.2020
Принята к публикации 10.07.2020
Аннотация
Статья посвящена особенностям проведения кислородных (O2) измерений в биологических средах (культивационные среды, реакционные среды, среды для изолированных клеточных и субклеточных структур) и иных сложносоставных средах амперометрическим методом с помощью электродов Кларка. А именно, рассмотрен вопрос о влиянии качественного и количественного состава жидких сред на показания этих O2-датчиков. На примерах сахарозы, сорбитола, NaCl и фосфатного буфера показано, что, при условии постоянства содержания растворенного O2, увеличение содержания сопутствующих кислороду веществ в исследуемой среде приводит к усилению сигнала у находящегося в этой среде электрода Кларка. На основе полученных и литературных данных произведена оценка степени влияния качественного и количественного состава сред на показания электродов Кларка. В свою очередь, само это влияние объяснено тем, что электроды Кларка в действительности измеряют активность растворенного O2, которая обычно возрастает при увеличении содержания сопутствующих кислороду веществ. Поскольку электроды Кларка измеряют активность, а не истинное содержание O2 в среде, и поскольку соотношение между активностью и истинным содержанием O2 часто неизвестно, в статье рассмотрен также вопрос о калибровке электродов Кларка. Отмечено, что использованные в настоящей работе способы добавления известного количества молекул O2 в жидкие среды (посредством реакции Хилла, каталазной реакции или впрыскивания насыщенной чистым O2 микропорции воды) для демонстрации влияния состава среды на показания электрода Кларка могут быть применены для калибровки электродов Кларка в единицах истинного содержания O2 и, таким образом, могут быть использованы для учета влияния сред на показания этих O2-датчиков. Высказано мнение, что, благодаря почти безграничным возможностям применения и своим другим достоинствам, калибровка, основанная на добавке в среду с электродом Кларка O2-насыщенной микропорции воды или, при необходимости, O2-насыщенной микропорции другой жидкости, является во всех смыслах самым выгодным способом калибровки. Можно ожидать, что приобщение данного способа калибровки к другим, уже известным способам калибровки и введение его в исследовательский обиход позволит во многих затруднительных случаях сделать измерения электродами Кларка более точными.
ВВЕДЕНИЕ
Кислородные измерения (O2-измерения) являются важной и часто необходимой частью большого числа биологических исследований. Чаще всего при проведении биологических исследований O2-измерения производят амперометрическим методом с помощью электродов Кларка (электрохимические O2-измерительные устройства, у которых анод и катод защищены от окружающей среды O2-проницаемой мембраной) [1–6]. Особенность O2-измерений в биологических исследованиях состоит в том, что их обычно производят в богатых по составу биологических средах, оказывающих по этой причине заметное влияние на активность растворенных в них молекул O2. Между тем известно, что электроды Кларка и родственные им гальванические O2-датчики измеряют как раз активность, а не истинное содержание растворенных в жидких средах молекул O2 [2–4, 6–10]. Последнее было показано на примере среды для изолированных хлоропластов, а также на примерах растворов, содержащих NaCl, моносахариды, дисахариды, длинно-цепочечные спирты, пирофосфатный буфер [2–4, 6, 7, 10, 11]. Если отмеченную выше особенность электродов Кларка не учитывать и представлять результаты O2-измерений этими устройствами в количественном виде, то представленные результаты будут тем ошибочнее, чем больше отличаются друг от друга значения видимого содержания O2 (активности O2) и истинного содержания O2 в среде при проведении O2-измерений электродом Кларка.
Поскольку электроды Кларка получили очень широкое распространение в качестве инструмента для O2-измерений [12, 13], отрицательные последствия от ошибок при их использовании могут быть значительными. К ошибкам, совершаемым при использовании электродов Кларка, можно отнести также вольное или невольное пренебрежение отмеченным выше влиянием качественного и количественного состава жидких сред на показания этих O2-измерительных устройств. Указанная методическая ошибка совершается по трем причинам: (1) из-за незнания вопроса о влиянии сред на показания электродов Кларка вообще, (2) из-за незнания того, каким образом можно учесть влияние некоторых сред на показания электрода Кларка при знании вопроса в целом, (3) из-за невозможности учесть влияние некоторых сред на показания электрода Кларка с нужной точностью в принципе. Вопросу влияния состава жидких сред на показания электродов Кларка и, таким образом, вопросу повышения точности проводимых электродами Кларка измерений через устранение причин их неправильного использования и посвящена настоящая статья.
МАТЕРИАЛЫ И МЕТОДЫ
В работе был использован самодельный электрод Кларка классического типа с платиновым катодом (рабочий электрод; диаметр 4.0 мм) и серебряным анодом (вспомогательный Ag/AgCl электрод). Корпус электрода Кларка представлял собой полый эбонитовый цилиндр (внутренний диаметр 6.0 мм, внешний диаметр 9.0 мм). Внутренняя часть корпуса электрода Кларка с находящимися в ней рабочим электродом, вспомогательными электродом и электролитом (0.1 М KCl) была отделена от окружающей среды тефлоновой мембраной (толщина 15 мкм, рабочая площадь мембраны 28.3 мм2). Плато вольт-амперной кривой у электрода Кларка при его нахождении в воде, насыщенной атмосферным воздухом, начиналось и заканчивалось при -500 мВ и -800 мВ, соответственно. При рабочем напряжении, соответствующем диффузионному току и 25оС, сигнал погруженного в воду электрода Кларка линейно зависел от содержания O2 в пределах 0–100% насыщения воды кислородом. Для проведения O2-измерений электрод Кларка подсоединяли к полярографу LP7 (“Laboratorni Pristroje”, Чехия), позволяющего переводить датчик в рабочее состояние и записывать производимый им сигнал.
O2-измерения проводили в стеклянной цилиндрической термостатируемой O2-измерительной ячейке (объем 3.8 мл). Электрод Кларка был вставлен в ячейку с боковой стороны. Заполнение и опорожнение ячейки производили через отверстие в ее верхней части. Для перевода ячейки в рабочее положение это отверстие закрывали стеклянным запорным приспособлением, имеющим канал (диаметр 0.8 мм, длина 20.0 мм) для ввода добавок внутрь запертой O2-измерительной ячейки. Содержимое O2-измерительной ячейки перемешивали с помощью магнитной мешалки, на которой ячейка располагалась.
Изменение содержания O2 в среде с помощью реакции Хилла. Реакцией Хилла называется перенос электронов от воды к акцептору электронов в освещенных изолированных хлоропластах, который сопровождается разложением воды и образованием из нее молекул O2. Одним из часто используемых акцепторов электронов в реакции Хилла является феррицианид калия (ФЦ). Реакция Хилла с ФЦ была использована для точного изменения содержания O2 в среде в связи с ее протеканием до полного исчерпания ФЦ и строгим соответствием количества образованного O2 количеству восстановленного хлоропластами ФЦ (реакция 1).
ФЦ-восстанавливающие хлоропласты для проведения реакции Хилла извлекали из листьев гороха и хранили при плотности 1.8–2.5 мг Хл/мл [14]. Чтобы при проведении O2-измерений весь добавленный к хлоропластам ФЦ расходовался только на реакцию Хилла и не тратился, в том числе и на окисление оставшихся в препарате хлоропластов эндогенных растительных восстановителей, хлоропласты при извлечении тщательно промывали. ФЦ-зависимое выделение O2 хлоропластами измеряли при атмосферном содержании растворенного O2, 25оС и при плотности хлоропластов 30–45 мкг Хл/мл. Реакцию Хилла в изолированных хлоропластах производили в реакционной среде, содержащей сахарозу (0–1.2 M) или сорбитол (0–1.2 M), NaCl (50.0 мМ), NaH2PO4 (6.0 мМ), MgCl2 (2.0 мМ), трис-HCl (10.0 мМ), АДФ (1.0 мМ) при рН 7.9 (рис. 1а) или рН 8.0 (рис. 2). При проведении опытов в реакционной среде изменяли содержание только того вещества, влияние которого на показания электрода Кларка изучали. Содержание других компонентов среды при этом оставалось неизменным. В O2-измерительную ячейку вносили 3.75 мл реакционной среды и 50 мкл хлоропластов, после чего ячейку закрывали и начинали запись показаний электрода Кларка. Вначале записывали изменение содержания O2 в суспензии хлоропластов в темноте. Затем в закрытую ячейку к хлоропластам добавляли 30 мкл водного раствора ФЦ (50 мМ) и записывали увеличение содержания O2 (ΔO2) в суспензии на свету вплоть до исчерпания всего добавленного ФЦ (рис. 2). Для уверенности, что при проведении O2-измерений добавленный в суспензию хлоропластов ФЦ не расходуется на окисление возможно не до конца удаленных промывкой хлоропластов эндогенных растительных восстановителей, на одной и той же суспензии хлоропластов делали 2–3 измерения ΔO2. При определении ΔO2 ФЦ добавляли в закрытую ячейку микрошприцом через предназначенный для этого канал (см. выше).
Рис. 1.
Зависимость сигнала электрода Кларка, производимого добавкой одного и того же количества молекул O2 в среду в O2-измерительную ячейку, от содержания сахарозы и сорбитола в этой среде. (а) Испытательная среда – суспензия изолированных хлоропластов гороха с разным содержанием сахарозы и сорбитола. Нормированную добавку O2 в среду (добавляли 97.9 нмоль O2/мл среды) делали при помощи реакции Хилла в хлоропластах с одним и тем же количеством ФЦ (391.7 нмоль Фц/мл среды). Представлены средние значения результатов 9 опытов с сахарозой и 4 опытов с сорбитолом с доверительным интервалом 95%. (б) Испытательная среда – раствор каталазы и 50 мМ фосфатного буфера с разным содержанием сахарозы и сорбитола. Нормированную добавку O2 в среду (в зависимости от опыта добавляли 194.3–388.2 нмоль O2/мл среды) делали при помощи каталазной реакции с одним и тем же количеством H2O2 (в зависимости от опыта 388.5–776.3 нмоль H2O2/мл среды). Представлены средние значения результатов 4 опытов с сахарозой и 3 опытов с сорбитолом с доверительным интервалом 95%. (в) Испытательная среда – вода с разным содержанием сахарозы. Нормированную добавку O2 в среду (32.6 нмоль O2/мл среды) делали посредством впрыскивания в нее одного и того же количества O2-насыщенной воды (26.3 мкл/мл среды). Представлены средние значения результатов 2 опытов, в каждом из которых измерения сделаны в 4–7 кратной повторности. Во всех случаях ответ электрода Кларка на добавку O2 при нулевом содержании сахарозы и сорбитола принят за 100%.
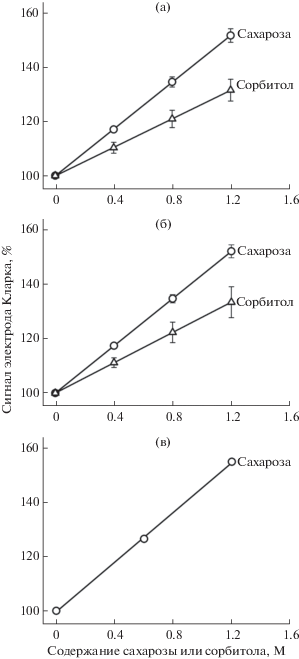
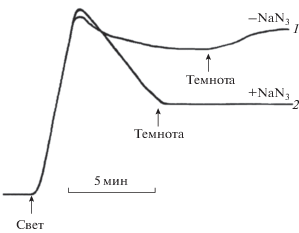
Рис. 2.
Приборные записи изменения содержания O2 в суспензии изолированных хлоропластов гороха до и после добавления к ним ФЦ. Записи (типичный результат) произведены при 0.4 М содержании сахарозы в суспензии хлоропластов плотностью 14.4 мкг Хл/мл в отсутствии и в присутствии NaN3 (0.5 мМ). ФЦ добавлен к хлоропластам в конечной концентрации 259.07 нмоль/мл непосредственно перед их освещением белым светом мощностью 250 Вт/м2. Примечание к рисунку: демонстрируемый кривыми 1 и 2 переход хлоропластов на свету от выделения O2 к поглощению O2 обусловлен исчерпанием добавленного к хлоропластам ФЦ и переключением хлоропластов с нециклического транспорта электронов (от H2O к ФЦ) на псевдоциклический транспорт электронов (от H2O к O2 с образованием H2O2), называемый также реакцией Мейлера. Сильное поглощение O2 хлоропластами на свету в присутствии ингибитора каталазы азида натрия, NaN3, (кривая 2) обусловлено тем, что образованная H2O2 без существенных изменений накапливается в хлоропластной среде. Более слабое поглощение O2 хлоропластами на свету в отсутствие NaN3 (кривая 1) является следствием газообмена O2 в суспензии хлоропластов в двух противоположных направлениях: поглощение O2 в реакции Мейлера с образованием H2O2 и выделение O2 в каталазной реакции из образованной в реакции Мейлера H2O2 (реакция 2). Следующее за выключением света довольно быстрое обратное повышение содержания O2 в среде в отсутствие NaN3 до соответствующего концу реакции Хилла уровня (кривая 1) также обусловлено каталазным разложением накопившейся в суспензии хлоропластов за счет реакции Мейлера H2O2 на O2 и H2O.
Обычно, благодаря хорошей промывке хлоропластов при их извлечении из листьев, величины ΔO2 после первой и второй добавок ФЦ были одинаковыми. В этом случае, обе величины ΔO2 использовали для вычисления среднего значения ΔO2. В тех случаях, когда величина ΔO2 после первой добавки ФЦ из-за постороннего расходования ФЦ была постоянно меньше, чем величина ΔO2 после второй и третьей добавок ФЦ, значение первого ΔO2 при вычислении среднего значения ΔO2 не учитывали. При проведении измерений исходили из допущения, что поглощение O2 хлоропластами и электродом Кларка в темноте до начала реакции Хилла продолжается с той же скоростью на свету в ходе реакции Хилла. Поэтому, при вычислении окончательного ΔO2 к показанному прибором ΔO2 прибавляли количество O2, суммарно поглощенное хлоропластами и электродом Кларка за время измерения ΔO2.
Для освещения хлоропластов использовали диапроектор с лампой накаливания мощностью 400 Вт. Данные, представленные на рис. 1а, получены на красном свету интенсивностью 21 Вт/м2 или 45 Вт/м2. Данные, представленные на рис. 2, получены на белом свету интенсивностью 250 Вт/м2. Интенсивность и качество падающего на хлоропласты света изменяли при помощи стеклянных нейтральных и красного светофильтров. Интенсивности падающего на хлоропласты света измеряли при помощи термостолбика Козырева.
Изменение содержания O2 в среде с помощью каталазной реакции. Каталазная реакция – реакция разложения перекиси водорода (H2O2) на O2 и H2O ферментом каталазой (H2O2 : H2O2-оксидоредуктаза, КФ 1.11.1.6). Каталазная реакция была использована для точного изменения содержания O2 в водной среде в связи с тем, что каталаза осуществляет реакцию разложения H2O2 на O2 и H2O с высокой скоростью, до полного исчерпания H2O2 и в строгом соответствии количеств добавленных к каталазе молекул H2O2 и количеств образовавшихся из H2O2 молекул O2 (реакция 2).
Каталазную реакцию осуществляли в водной среде, содержащей сахарозу (0–1.2 M) или сорбитол (0–1.2 M), NaH2PO4-KOH буфер (5 мМ или 50 мМ, рН 7.0) и каталазу (80.0 мкг/мл) при 25°С. При проведении опытов в среде изменяли содержание только того вещества, влияние которого на показания электрода Кларка изучали. Содержание других компонентов среды при этом оставалось неизменным. Среда содержала 50 мМ NaH2PO4-KOH буфера при получении данных, представленных на рис. 1, и 5 мМ или 50 мМ NaH2PO4-KOH буфера при получении данных, представленных в табл. 1. Раствор H2O2 для каталазной реакции приготавливали из аптечного раствора H2O2 путем разведения последнего дистиллированной водой. Точное содержание H2O2 в добавляемом в O2-измерительную ячейку растворе H2O2 определяли фотометрически с использованием молярного коэффициента светопоглощения ɛ240 nm = = 43.6/(М см). Для проведения каталазной реакции в O2-измерительную ячейку вносили 3.79 мл реакционной среды, ячейку закрывали и ждали наступления устойчивости в показаниях электрода Кларка. Затем в закрытую ячейку добавляли микрошприцом в зависимости от опыта 10–20 мкл раствора H2O2 (74–295 мМ) и записывали обусловленное каталазной реакцией увеличение содержания O2 в ячейке (ΔO2). При вычислении окончательного ΔO2 к показанному прибором ΔO2 прибавляли количество O2, поглощенное электродом Кларка за время измерения ΔO2.
Таблица 1.
Влияние NaCl, сахарозы и фосфатного буфера на показания электрода Кларка
| № опыта | Содержание в среде изменяемых веществ* | Ответ электрода Кларка на нормированную добавку O2, %** |
|---|---|---|
| 1 | 5 мМ NaH2PO4-KOH 50 мМ NaH2PO4-KOH |
100.0 107.1 |
| 2 | 0 мМ сахарозы + 50 мМ NaCl 0 мМ сахарозы + 200 мМ NaCl 200 мМ сахарозы + 50 мМ NaCl |
100.0 104.2 108.7 |
Примечание. * Нормированное увеличение содержания O2 в средах производили посредством разложения H2O2 каталазой в опыте 1 и посредством реакции Хилла в изолированных хлоропластах в опыте 2, как описано в разделе “Материалы и методы”. В таблице указаны только те компоненты сред, содержание которых изменялось и, соответственно, которые изменяли показания электрода Кларка. ** Приведены данные двух разных единичных опытов в виде средних значений 4–7 измерений в опыте 1 и 6–8 измерений в опыте 2. При определении ответа электрода Кларка содержание O2 в среде в O2-измерительной ячейке увеличивали на одно и то же количество молекул O2: 200.0 и 64.8 нмоль/мл в опытах 1 и 2, соответственно.
Изменение содержания O2 в среде с помощью O2-насыщенной микродобавки воды. O2-насыщенную воду приготовляли в стеклянном сосуде объемом 14 мл и диаметром 20 мм. Для этого сосуд заполняли дистиллированной водой (~9 мл) и закупоривали резиновой пенициллиновой пробкой. Чтобы сосуд случайно не открылся, пробку прижимали к сосуду винтовым прижимным устройством. Через пробку вводили две одинаковые шприцевые иглы для продувки воды в сосуде O2-газом. Через одну иглу, конец которой опускали примерно на 2 см вглубь воды в сосуде, воду в сосуде для ее насыщения кислородом продували с умеренной (чтобы не вызвать повышения давления в сосуде) скоростью O2-газом из баллона со сжатым O2. Через вторую иглу, конец которой располагался в верхней газовой части сосуда, выходящий из воды газ уходил во внешнюю среду. Насыщение воды кислородом, как и собственно O2-измерения, производили при 25°С.
Для определения ответа электрода Кларка на добавку O2 в виде O2-насыщенной воды, в O2-измерительную ячейку вносили 3.7 мл насыщенных воздухом дистиллированной воды или водного раствора сахарозы, ячейку закрывали и ждали наступления устойчивости в показаниях электрода Кларка. После этого из непрерывно продуваемого чистым O2 сосудика через пробку в газовый микрошприц (“Hamilton Co”, Швейцария) размеренно набирали 100 мкл O2-насыщенной воды, шприц из сосуда вынимали и набранную шприцем воду добавляли в закрытую O2-измерительную ячейку для записи ответа электрода Кларка (ΔO2) на добавленный вместе с водой O2. При вычислении окончательного значения ΔO2 к показанному прибором ΔO2 прибавляли количество O2, поглощенное электродом Кларка за время измерения ΔO2.
В работе, наряду с веществами отечественного производства, использовали вещества иностранного производства: трис (биохимический буфер для систем с низким содержанием металлов, “Koch-Light Laboratories Ltd”, Англия), каталазу из бычьей печени (“Sigma Chemical Co”, США), сорбитол (“Panreac Química SLU”, Испания) и АДФ (“Reanal”, Венгрия).
Статистические сведения приведены в подписях к рисункам и таблицам.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
На рис. 1 представлены данные по влиянию растворенных в водных средах разного состава сахарозы и сорбитола на показания погруженного в эти среды электрода Кларка. При получении этих данных содержание O2 в использованных средах изменяли тремя способами. Первый способ (рис. 1а) состоял в нормированном повышении содержания O2 в среде посредством проведения реакции Хилла в изолированных хлоропластах с ФЦ, как акцептором электронов (см. “Материалы и методы”). Второй способ (рис. 1б) состоял в нормированном увеличении содержания O2 в среде посредством проведения каталазной реакции (см. “Материалы и методы”). Третий способ (рис. 1в) состоял в нормированном повышении содержания O2 в среде посредством добавки в нее микропорции воды, насыщенной чистым O2 (см. “Материалы и методы”). Можно видеть, что, несмотря на постоянство количества добавляемого O2 в среды с электродом Кларка, увеличение содержания сахарозы и сорбитола в этих средах с 0 до 1.2 М приводило к существенному линейно зависимому увеличению сигнала O2-датчика. Более того, несмотря на большую разнородность сред (суспензия хлоропластов (рис. 1а), среда для каталитической реакции (рис. 1б), просто водные растворы сахарозы (рис. 1в)), зависимости сигнала электрода Кларка от содержания сахарозы и сорбитола в средах для каждого из этих веществ в отдельности получились очень похожими. А именно, при увеличении содержания сахарозы и сорбитола в средах с 0 до 1.2 М сигнал электрода Кларка увеличивался на 52–56% и 32–34%, соответственно. Преобразование данных Robinson and Cooper [7] в графический вид показало, что сигнал электрода Кларка увеличивается линейным образом также и при увеличении содержания NaCl в водной среде снаружи O2-датчика с 0.5 до 5.43 М.
Хотя исследование влияния NaH2PO4-KOH буфера и NaCl на показания электрода Кларка первоначально не входило в задачу настоящей работы, такое исследование со вспомогательной целью в ходе работы все же было проведено. Результаты этого исследования, которые следует рассматривать как приблизительные с точки зрения статистических требований, приведены в табл. 1. Можно видеть, что при увеличении содержания NaH2PO4-KOH буфера в среде в O2-измерительной ячейке с 5 до 50 мМ (опыт №1) сигнал электрода Кларка увеличился на 7.1%, при увеличении содержания сахарозы с нуля до 200 мМ (опыт №2) сигнал увеличился на 8.7%, а при увеличении содержания NaCl с 50 до 200 мМ сигнал увеличился только на 4.2%. Что касается влияния NaCl на показания электрода Кларка, то его значение 4.2% в табл. 1 не очень существенно отличается от значения ~7%, полученного автором данной статьи для тех же концентраций NaCl при обработке данных Robinson and Cooper [7]. То же самое можно сказать о влиянии сахарозы. Данные в табл. 1 по влиянию сахарозы на сигнал электрода Кларка хорошо согласуются с данными, представленными на рис. 1. В обоих случаях сигнал электрода Кларка при 200 мМ сахарозы увеличился на ~9%.
Из данных, представленных на рис. 1 и в табл. 1, следует, что растворы, содержащие сахарозу, сорбитол, NaH2PO4-KOH буфер и NaCl в обычных для биологических исследований количествах, неравносильно, но при этом довольно существенно увеличивают сигнал электрода Кларка. О подобном действии реакционной среды для изолированных хлоропластов, а также растворов NaCl, моносахаридов, дисахаридов, длинноцепочечных спиртов, пирофосфатного буфера на показания электрода Кларка сообщалось ранее [2–4, 7, 10, 11]. Причина такого действия жидких биологических сред и простых химических растворов на сигнал электродов Кларка состоит в том, что данные O2‑измерительные устройства измеряют активность растворенных в жидких средах молекул O2, значение которой возрастает при увеличении содержания в этих средах других веществ. Если данное явление при проведении количественных O2-измерений не учитывать, то результаты таких измерений получатся тем более ошибочными, чем сильнее находящиеся в среде вещества изменяют активность растворенного O2. Не учитывать влияние исследуемых сред на показания электродов Кларка можно только в трех одновременно выполняющихся случаях. Первое, когда значения видимого и истинного содержания O2 в средах равны или почти равны друг другу, т.е. при проведении O2-измерений в средах, содержимое которых не влияет или слабо влияет на активность растворенного в этих средах O2. Второе, когда в средах не содержатся вещества, способные изменять проницаемость мембраны электрода Кларка для O2. Третье, когда в средах не содержатся вещества, способные наряду с O2 проникать внутрь электрода Кларка и взаимодействовать электрохимически или химически с анодом и/или катодом O2-датчика [6].
Поскольку растворимость, активность и диффузия O2 в жидких средах часто непредсказуемо зависят от природы (электролиты, не электролиты, биологический материал, не биологические нерастворимые частицы), концентрации и физико-химического состояния компонентов среды [3, 8, 15–17], вопрос влияния жидких сред на показания электродов Кларка тесно связан с вопросом калибровки этих O2-датчиков. Правильно выбранный способ калибровки используемого электрода Кларка является залогом правильности произведенных этим датчиком количественных измерений O2. Правильный подход к калибровке электродов Кларка при проведении биологических исследований важен не только потому, что из-за насыщенности биологических сред веществами и биологическими объектами значения активности и истинного содержания растворенного в них O2 заметно, а подчас сильно различаются. Он важен также потому, что точный состав биологических сред, а значит и растворимость в этих средах O2, часто не только неизвестны, но и изменяются прямо в ходе проведения O2-измерений (это может иметь место, например, при проведении измерений в природных условиях, в ферментируемых средах или в ходе культивирования растений, макро- и микроводорослей, грибов, бактерий). Важностью калибровки электродов Кларка для O2-измерений, трудностями, связанными с ее правильным проведением, и наличием препятствий для создания коммерческих стандартных калибровочных растворов O2 [18] объясняется непрекращающийся поиск новых и как можно более простых и пригодных для всех или, по крайней мере, для большого количества случаев O2-измерений способов калибровки данных O2-измерительных устройств.
Несмотря на многообразие предложенных за время существования метода способов калибровки электродов Кларка (в общей сложности не менее двадцати трех разных по трудоемкости, стоимости и условиям применения способов), ни один из них не обладает полным набором таких необходимых для калибровочного дела качеств, как простота, точность, дешевизна, быстрота и достаточная универсальность с точки зрения разнообразия случаев O2-измерений. Значительные ограничения имеются и у самой широко применяемой калибровки электродов Кларка газонасыщающим способом (калибровка, основанная на насыщении калибровочных сред газовыми смесями с разным содержанием O2) [3, 4, 8, 9, 19]. Чаще всего при калибровке электродов Кларка этим способом калибровочные среды насыщают разным количеством O2 путем их продувки купленными или приготовленными собственными силами газовыми смесями. При этом содержание O2 в насыщенных газовыми смесями средах рассчитывают с использованием соответствующих справочных данных. Однако, из-за того, что растворимость, активность и диффузия O2, как уже было отмечено выше, во многих средах неизвестны, но хорошо известны для чистой воды, калибровку электродов Кларка газонасыщающим способом часто делают не в той среде, в которой измеряют O2, а в воде, нарушая, таким образом, главное правило калибровки: калибровать электрод Кларка в той среде и в тех условиях, в которых производят измерение O2 [9, 19]. В итоге, в результаты количественных измерений O2 (например, измерения истинного содержания O2 или скоростей газообмена O2 в тех или иных средах) электродами Кларка вкрадывается ошибка, которая оказывается тем значительнее, чем значительнее активность O2 в использованных для O2-измерений средах и в воде отличаются друг от друга. О размерах такой ошибки можно судить по литературным данным, показывающим, что различие между растворимостью O2 в чистой воде и в суспензии изолированных митохондрий составляет 9–12%, а различия между растворимостью O2 в чистой воде и микробиологических средах или различия между растворимостью O2 в бродильных средах в начале и в конце процесса брожения составляют 5–41% [8, 20–24]. Размеры такой ошибки можно представить также исходя из литературных данных, показывающих, что ответ электрода Кларка на одно и то же количество добавленного O2 в 50 мМ водном растворе пирофосфата [11] и обычной среде для изолированных хлоропластов [2] на 10–11% больше по сравнению с ответом в чистой воде. Наконец, возможные размеры этой ошибки позволяют представить приведенные на рис. 1 и в табл. 1 данные, а также расчеты, сделанные при подготовке настоящей статьи с использованием данных Robinson and Cooper [7]. Из этих расчетов следует, что ответ электрода Кларка на одно и то же количество добавленного O2 становится примерно на 23% и 45% больше при увеличении концентрации NaCl в растворе на 0.5 и 1.0 М, соответственно. При большем увеличении содержания (1.0–6.0 М) растворенных веществ (KCl, NaCl, MgCl2, и Na2SO4) ответ электрода Кларка на добавку O2 начинает различаться с ответом в воде или разбавленном растворе еще больше [4, 7]. К примеру, сигнал электрода Кларка на одну и ту же добавку O2 в 5.43 М растворе NaCl становится на 228% больше, чем в 0.5 М растворе NaCl [7]. Данное обстоятельство дало основание Delieu and Walker высказать мнение, что из-за трудности учета влияния состава исследуемых сред на показания электродов Кларка калибровку O2-датчиков обычно производят в чистой воде независимо от состава исследуемых сред, из-за чего в усредненном целом измерения электродами Кларка получаются с ошибкой ~10% [25]. Подобного мнения придерживаются и другие исследователи [23, 26].
Чтобы рассмотренные выше особенности метода не были причиной неверного определения истинного содержания O2 в биологических средах, следует отказаться от калибровки O2-датчика в воде и независимо от выбранного способа калибровки калибровать его в используемых для O2-измерений средах. В этом случае, если выбор делается в пользу газонасыщающего способа калибровки, то прежде, чем его применить, придется удостовериться, что в справочниках или текущей литературе имеются проверенные данные по растворимости O2 в той среде или тех средах, в которой/которых производятся O2-измерения и калибровка O2-датчика. Если таковых там нет, то растворимость O2 в некоторых средах можно узнать путем вычислений, используя уравнения и модели, разработанные для расчетов растворимости O2 в этих средах. При этом надо иметь в виду, что, во-первых, и тот и другой подходы к определению растворимости O2 при калибровке электродов Кларка газонасыщающим способом ограничены средами определенного состава. Во-вторых, эти подходы далеко не всегда могут дать ту точность калибровки, которую ожидают от проводимых O2-измерений. В-третьих, при использовании этих подходов к определению растворимости O2 следует учитывать, что биологический материал и некоторые используемые в биологических исследованиях вещества (например, гемоглобин- и миоглобин-содержащие структуры, белковые молекулы, клетки живых существ, короткоцепочечные спирты, перфторуглероды, LiF, LiNO3, NiNO3, Na2B4O7, HNO3, NaBO2, NH4OH, Н3РО4) могут оказывать необычное и даже обратное влияние, по сравнению с обычным влиянием, на растворимость и активность O2 в жидкой среде [6, 15, 16].
В связи с тем, что, как было отмечено выше, у калибровки электродов Кларка газонасыщающим способом имеются значительные ограничения в применении, особое значение приобретают способы, позволяющие, в отличие от газонасыщающего способа, калибровать электроды Кларка в единицах истинного содержания O2 независимо от состава калибровочной среды. Существенно, что все три использованные в настоящей работе способа изменения содержания O2 в жидкой среде (рис. 1) могут быть применены как раз для такого рода калибровки. Возможность использования двух из этих трех способов, “хлоропластного” и “каталазного” (рис. 1а, 1б), в таком качестве уже была высказана и опробована ранее [2, 11, 27, 28]. Оценивая степень применимости всех трех способов изменения содержания O2 в жидкой среде, как калибровочных способов, приходится признать, что два последних способа, “хлоропластный” и “каталазный”, не могут иметь широкого применения. Это связано с тем, что и тот и другой способ работоспособны только в тех средах и условиях, в которых хлоропласты и каталаза сохраняют свою живучесть. При этом применимость первого способа гораздо меньше по сравнению со вторым способом по причине того, что изолированные хлоропласты гораздо менее доступны для использования и более требовательны к условиям измерений, чем каталаза. Слабым местом “хлоропластного” способа калибровки является также то, что изолированные хлоропласты способны не только выделять, но и поглощать O2. Причем, как показывают данные, представленные на рис. 2 и в табл. 2, хлоропласты поглощают O2 как в темноте, так в отсутствии ФЦ и на свету. Если вдруг используемые хлоропласты поглощают O2 на свету не только в отсутствии, но и в присутствии ФЦ, пусть даже с небольшой скоростью, то это плохо скажется на точности производимой с их помощью калибровки электродов Кларка. В связи с этим, поскольку O2-поглощающая способность у разных препаратов хлоропластов может быть в зависимости от метода их получения разной, использовать имеющиеся хлоропласты для точной калибровки электродов Кларка желательно лишь после проверки их на пригодность для такого точного применения. В добавление к отмеченной выше особенности, еще одной неудобной особенностью калибровки электродов Кларка “хлоропластным” способом является то, что она требует предпринимать меры (см. раздел “Материалы и методы”), предотвращающие взаимодействие ФЦ в суспензии хлоропластов с другими, чем вода, донорами электронов. Что касается калибровки электродов Кларка каталазным способом, то и у этой калибровки есть свои освещенные в литературе “подводные камни” и тонкости применения [27–30].
Таблица 2.
Поглощение O2 изолированными хлоропластами гороха в отсутствие феррицианида
| Содержание сахарозы/сорбитола в суспензии хлоропластов |
Поглощение O2 в темноте*, мкмоль/(мг Хл ч) |
Поглощение O2 на свету**, мкмоль/(мг Хл ч) |
|---|---|---|
| 0.0 М сахарозы | 0.4 ± 0.1 | 7.1 ± 0.2 |
| 0.8 М сахарозы | 0.4 ± 0.2 | 7.8 ± 0.5 |
| 1.2 М сахарозы | 0.5 ± 0.2 | 6.1 ± 0.8 |
| 0.6 М сорбитола | 0.7 ± 0.3 | 7.6 ± 0.5 |
| 1.2 М сорбитола | 0.4 ± 0.2 | 8.3 ± 0.3 |
Примечание. В таблице приведены средние значения результатов трех опытов и их стандартная ошибка. В каждом опыте измерения сделаны в 3–5 кратной повторности. Плотность хлоропластов в опытах составляла 32–38 мкг Хл/мл. * Измерения сделаны сразу после добавления хлоропластов в O2-измерительную ячейку. ** Измерения сделаны на красном свету интенсивностью 45 Вт/м2 после остановки реакции Хилла из-за исчерпания феррицианида. Представлены начальные, т.е. наибольшие (как показывает кривая 1 на рис. 2, поглощение O2 хлоропластами после исчерпания феррицианида уменьшалось во времени) скорости поглощения O2.
Что касается третьего способа изменения содержания O2 в жидкой среде, состоящего из добавления в среду нужного количества O2 в виде микропорции O2-насыщенной воды (рис. 1в) или, в случае необходимости, микропорции другой O2-насыщенной жидкости, то он, как калибровочный способ, очень выгодно отличается не только от двух других примененных в настоящей работе способов, но, по-видимому, и от всех вообще описанных к настоящему времени в литературе способов калибровки. Это связано с тем, что, во-первых, калибровка таким способом сравнима по простоте, доступности и точности с упомянутой выше широко применяемой калибровкой газонасыщающим способом. Во-вторых, у этого способа калибровки почти безграничные возможности применения. В-третьих, ошибка O2-измерений, связанная с изменением состава и свойств калибровочных сред, при таком способе калибровки электродов Кларка незначительна, поскольку само изменение этих сред под действием микродобавлений незначительно. Так, например, в случае, представленном на рис. 1в, объем добавляемой O2-насыщенной воды составлял всего 2.5% от общего объема среды в O2-измерительной ячейке. Но даже это незначительное количество добавляемой O2-насыщенной воды могло быть, учитывая возможности усиления сигнала использованной в настоящей работе O2-измерительной установкой, многократно уменьшено (по крайней мере, в 30 раз) без значительного ухудшения надежности измерений электродом Кларка. Кроме того, значительно увеличить сигнал и, следовательно, значительно уменьшить количество добавляемой O2-насыщенной воды в опытах, представленных на рис. 1в, можно было также за счет замены у электрода Кларка использованной мембраны на более O2-проницаемую мембрану, т.е. за счет увеличения чувствительности самого O2-измерительного устройства.
Таким образом, завершая рассмотрение вопроса об особенностях применения электродов Кларка в биологических исследованиях, представляется полезным кратко повторить ключевое положение изложенного материала. Оно состоит в том, что, поскольку электроды Кларка измеряют активность, а не истинное содержание растворенного O2, показания этих измерительных устройств, как и активность O2, зависят не только от содержания O2 в исследуемых средах, но также от качественного и количественного состава исследуемых сред в целом. Данное обстоятельство может вызвать затруднения в O2-измерениях либо сделать O2-измерения неправильными соответственно в тех случаях, когда соотношение между концентрацией и активностью O2 в исследуемых средах очень трудно или невозможно оценить, либо когда отмеченное выше свойство электродов Кларка ошибочно не учитывается. Тем не менее, как показывает, в том числе, и настоящее исследование, упомянутые затруднения и неопределенности при хорошем знании вопроса могут быть в подавляющем большинстве случаев успешно преодолены посредством правильного применения подходящего случаю способа калибровки используемого электрода Кларка.
Настоящая статья не содержит каких-либо исследований с участием людей и животных в качестве объектов исследований. Автор заявляет об отсутствии конфликта интересов.
Список литературы
Clark L.C.Jr. Monitor and control of blood and tissue oxygen tensions // Trans. – Am. Soc. Artif. Intern. Organs. 1956. V. 2. P. 41.
Delieu T., Walker D.A. An improved cathode for the measurement of photosynthetic oxygen evolution by isolated chloroplasts // New Phytol. 1972. V. 71. P. 201.
Fatt I. Polarographic Oxygen Sensor. Its Theory of Operation and Its Application in Biology, Medicine, and Technology. United States: CRC Press, Inc., 1976. 290 p.
Hitchman M.L. Measurement of Dissolved Oxygen. (Chemical analysis, V. 49). New York: John Wiley & Sons Inc., 1978. 255 p.
Severinghaus J.W., Astrup P.B. History of blood gas analysis. IV. Leland Clark’s oxygen electrode // Journal of Clinical Monitoring. 1986. V. 2. P. 125.
Бородин В.Б. Полярография. Применение электродов Кларка для O2-измерений // Ин-т фундаментальных пробл. биол. РАН, Пущино, Моск. обл. – Деп. в ВИНИТИ 18.11.2016, 2016. № 154-В2016. 56 с.
Robinson J. and Cooper J.M. Method of determining oxygen concentrations in biological media, suitable for calibration of the oxygen electrode // Anal. Biochem. 1970. V. 33. P. 390.
Beechey R.B., Ribbons D.W. Oxygen electrode measurements // Methods in Microbiology / Eds. Norris J.R., Ribbons D.W. New York: Acad. Press, 1972. V. 6B. P. 25.
Lee Y.H. and Tsao G.T. Dissolved oxygen electrodes // Advances in Biochemical Engineering (Book Series: Advances in Biochemical Engineering/Biotechnology). V. 13. Berlin/Heidelberg: Springer. 1979. P. 35.
Hale J.M. Factors influencing the stability of polarographic oxygen sensors // Polarographic Oxygen Sensors: Aquatic and Physiological Applications / Eds. Gnaiger E., Forstner H. New York: Springer-Verlag, 1983. P. 3.
Dixon M. and Kleppe K. D-amino acid oxidase. I. Dissociation and recombination of the holoenzyme // Biochim. Biophys. Acta. 1965. V. 96. P. 357.
Ramamoorthy R., Dutta P.K., Akbar S.A. Oxygen sensors: materials, methods, designs and applications // J. Mater. Sci. 2003. V. 38. P. 4271.
Jones S.T., Heindel T.J. A review of dissolved oxygen concentration measurement methods for biological fermentations // Mechanical Engineering Conference Presentations, Papers, and Proceedings (2007 ASAE Annual Meeting). Paper number: 077117. Minneapolis, 2007. 15 p. https://lib.dr.iastate.edu/me_conf/200
Borodin V.B. The Kok effect in pea. Comparative study of electron transport in washed and unwashed chloroplasts // Soviet Plant Physiology. 1992. V. 39. P. 217.
Baburin L.A., Shvinka J.E., Viesturs U.E. Equilibrium oxygen concentration in fermentation fluids // Eur. J. Appl. Microbiol. Biotechnol. 1981. V. 13. P. 15.
Ho C.S., Ju L.-K. Effects of microorganisms on effective oxygen diffusion coefficients and solubilities in fermentation media // Biotechnol. Bioeng. 1988. V. 32. P. 313.
Pourplanche C., Larreta-Garde V., and Thomas D. Comparison of polarographic and chemical measurements of oxygen uptake in complex media: the example of lipoxygenase reaction // Anal. Biochem. 1991. V. 198. P. 160.
Fisicaro P., Adriaens A., Ferrara E., Prenesti E. Assessment of the uncertainty budget for the amperometric measurement of dissolved oxygen // Anal. Chim. Acta. 2007. V. 597. P. 75.
Hitchman M.L. Calibration and accuracy of polarographic oxygen sesnsors // Polarographic Oxygen Sensors. Aquatic and Physiological Applications / Eds. Gnaiger E., Forstner H. New York: Springer-Verlag, 1983. P. 18.
Popović M., Niebelschütz H., Reuß M. Oxygen solubilities in fermentation fluids // Eur. J. Appl. Microbiol. Biotechnol. 1979. V. 8. P. 1.
Slininger P.J., Petroski R.J., Bothast R.J., Ladisch M.R. and Okos M.R. Measurement of oxygen solubility in fermentation media: a colorimetric method // Biotechnol. Bioeng. 1989. V. 33. P. 578.
Doran P.M. Bioprocess Engineering Principles. New York: Acad. Press, 1995. 439 p.
Gros J.B., Dussap C.G., and Catté M. Estimation of O2 and CO2 solubility in microbial culture media // Biotechnol. Prog. 1999. V. 15. P. 923.
Vendruscolo F., Rossi M.J., Schmidell W., Ninow J.L. Determination of oxygen solubility in liquid media // ISRN Chem. Eng. 2012. V. 2012. 5 p. https://doi.org/10.5402/2012/601458
Delieu T., Walker D.A. Polarographic measurement of photosynthetic oxygen evolution by leaf discs // New Phytol. 1981. V. 89. P. 165.
Rasmussen H.N., Rasmussen U.F. Oxygen solubilities of media used in electrochemical respiration measurements // Anal. Biochem. 2003. V. 319. P. 105.
Wingo W.J. and Emerson G.M. Calibration of oxygen polarographs by catalase-catalyzed decomposition of hydrogen peroxide // Anal. Chem. 1975. V. 47. P. 351.
Halbach S. Catalase activity measured with a micro oxygen electrode in a pressurized reaction vessel // Anal. Biochem. 1977. V. 80. P. 383.
Hickey C.W. Quantitative addition of dissolved oxygen to in situ benthic chamber systems by use of catalase and hydrogen peroxide // Appl. Environ. Microbiol. 1985. V. 49. P. 462.
Aebi H.E. Catalase in vitro // Methods Enzymol. 1984. V. 105. P. 121.
Дополнительные материалы отсутствуют.
Инструменты
Физиология растений