

Генетика, 2019, T. 55, № 12, стр. 1433-1441
Спектр мутаций в гене ATP7B у российских больных с болезнью Вильсона–Коновалова
Г. М. Баязутдинова 1, *, О. А. Щагина 1, А. С. Карунас 2, Н. В. Вялова 3, А. А. Соколов 4, А. В. Поляков 1
1 Медико-генетический научный центр
115522 Москва, Россия
2 Институт биохимии и генетики Уфимского федерального исследовательского центра
Российской академии наук
450054 Уфа, Россия
3 Дальневосточный государственный медицинский университет,
кафедра неврологии и нейрохирургии
680000 Хабаровск, Россия
4 Северо-Западный государственный медицинский университет им. И.И. Мечникова,
кафедра анестезиологии и реаниматологии
191015 Санкт-Петербург, Россия
* E-mail: bayazguln@yandex.ru
Поступила в редакцию 08.02.2019
После доработки 05.06.2019
Принята к публикации 24.06.2019
Аннотация
Болезнь Вильсона–Коновалова (БВК) – аутосомно-рецессивное заболевание, обусловленное избыточным накоплением меди в организме. Молекулярно-генетической причиной болезни являются повреждения в гене ATP7B, кодирующем медь-транспортирующую АТФазу. Определены спектр и частоты мутаций в гене ATP7B. Поиск мутаций в гене ATP7B проведен с использованием всего спектра современных методов: аллель-специфичного лигирования для поиска частой мутации, массового параллельного секвенирования для поиска редких изменений гена и количественного лигирования (MLPA) для анализа числа копий всех экзонов гена ATP7B. Исследование частот и спектра мутаций гена ATP7B проведено на выборке образцов ДНК 431 российского больного, направленных в лабораторию для диагностики болезни Вильсона–Коновалова. Для оценки популяционных частот использовались образцы ДНК, выделенные из крови 1000 необследованных жителей различных регионов РФ. В результате работы определен спектр мутаций гена ATP7B у больных, проживающих на территории РФ. Всего выявлено 66 различных вариантов нуклеотидной последовательности в гене АТР7В: 42 описанных ранее патогенных вариантов и 24 встретившихся впервые. На долю самого частого патогенного варианта в гене ATP7B – миссенс-замены c.3207C>A у российских больных БВК приходится 50% аллелей с мутацией. Также чаще других встречались патогенные варианты: c.2403insC, c.3204delC, c.3036insC, c.3190G>A. Установлено ожидаемое число больных болезнью Вильсона–Коновалова в исследуемой выборке, оно составило 208 человек (из 431). Установить причину болезни удалось у 78% реальных больных гепатолентикулярной дегенерацией. Определена частота носительства болезни Вильсона–Коновалова, которая составила 1 на 50 жителей РФ (доверительный интервал от 1 : 42 до 1 : 63). Расчетная частота заболевания составляет 1 на 10 000 россиян (доверительный интервал от 1 : 8333 до 1 : 12 500).
Болезнь Вильсона–Коновалова (гепатолентикулярная дегенерация, гепатоцеребральная дистрофия, OMIM 277900) – наследственная аутосомно-рецессивная болезнь. Заболевание характеризуется избыточным накоплением меди в организме, особенно в печени, мозге, роговице, почках, что приводит к нарушениям работы этих органов, неврологическим и психиатрическим расстройствам [1]. Частота болезни во всех популяциях мира составляет примерно 1 : 30 000–1 : 100 000, носительство болезни 1 : 90 [2].
Причиной болезни Вильсона–Коновалова (БВК) являются мутации в гене ATP7B, кодирующем медь-транспортирующую АТРфaзу P-типа [3]. По данным портала Human Gene Mutation Database, в этом гене на сегодняшний день описано более 900 патогенных вариантов (https://portal.biobase-international.com).
Спектр мутаций гена ATP7B различен у больных, принадлежащих к разным этническим группам. Для азиатов (Китай, Южная Корея, Япония) наиболее часта мутация p.R778L, ее доля составляет 14–49%, в зависимости от региона [4]. В Сардинии основной причиной БВК является делеция в промоторной области –441/–427del, которую содержали 92% хромосом в выборке пациентов с данным диагнозом [5]. В Бразилии и Венесуэле самой частой является мутация p.Ala1135Glnfs (c.3402delC), наряду с патогенным вариантом p.His1069Gln (с.3207С>А) [6]. Патогенный вариант p.His1069Gln – самая распространенная причина болезни Вильсона–Коновалова в Европе и Северной Америке, а также и в России [7–9].
Цель работы – изучение спектра мутаций гена ATP7B у российских больных гепатолентикулярной дегенерацией, определение частоты носительства болезни (носительства патогенных вариантов в гене ATP7B) у жителей Российской Федерации и расчет частоты данного заболевания на основе молекулярно-генетических данных.
МАТЕРИАЛЫ И МЕТОДЫ
Выделение ДНК из лейкоцитов периферической крови проводилось с помощью набора реактивов Wizard® Genomic DNA Purification Kit (Promega, США) по протоколу производителя. Выделение ДНК из сухих пятен крови на фильтровальной бумаге проводили при помощи набора реактивов DIAtom DNA Prep100 kit (IsogeneLab. Ltd., Россия) по протоколу производителя.
Поиск мутаций в гене ATP7B проведен на материале 431 неродственного пробанда. Все больные были направлены на исследование врачами-генетиками из разных регионов Российской Федерации (в исследование вошли пробанды, проживающие на территории Центральной России, а также в Приморском крае, Хабаровском крае, Кемеровской области, Томской области, Алтайском крае) с диагнозом “болезнь Вильсона–Коновалова” с марта 2006 по декабрь 2017 г. Информация о национальности, точном месте рождения больных отсутствовала.
Для определения популяционной частоты гетерозиготного носительства варианта c.3207C>A (p.His1069Gln) было проведено типирование 1000 неродственных необследованных жителей РФ, проживающих в различных регионах России. Об этнической принадлежности и о точном месте проживания людей доподлинно неизвестно.
Для детекции мутации с.3207С>А использовалась аллельспецифичная лигаза-зависимая реакция с последующей амплификацией (LPA) по методике, описанной в статье Баязутдиновой с соавт. [9].
Анализ ДНК методом массового параллельного секвенирования (MPS) проведен на секвенаторе нового поколения Ion S5. Для пробоподготовки использована технология ультрамультиплексной ПЦР, сопряженная с последующим секвенированием (AmpliSeq™). Анализ проведен с использованием кастомной панели “Обмен меди и железа”, включающей кодирующие последовательности генов: ATP7A, ATP7B, CP, TF, HEPC, PANK2, PLA2G6, ATP13A2, FTL, HFE, HJV, TFR2, SLC40A1, WDR45, C2ORF37. Расчетное покрытие гена ATP7B составило 99%.
Все выявленные варианты нуклеотидной последовательности подтверждены секвенированием по Сэнгеру. Для наименования выявленных вариантов использовалась номенклатура, представленная на сайте http://varnomen.hgvs.org/recommendations/DNA версия 2.15.11. Обработка данных секвенирования проведена с использованием стандартного автоматизированного алгоритма, предлагаемого TermoFisher Scientific (Torrent Suite™), а также программного обеспечения Gene-Talk [www.gene-talk.de]. Для оценки популяционных частот выявленных вариантов была применена база Genome Aggregation Database (gnomAD). Для оценки клинической значимости выявленных вариантов использованы база данных OMIM, база данных по патогенным вариантам HGMD® Professional версия 2018.4, специализированная база данных по болезни Вильсона–Коновалова http://www.wilsondisease.med.ualberta.ca, Руководство по интерпретации данных, полученных методами MPS, программы Polyphen-2, SIFT, Mutation Taster, UMD-Predictor, SpliceFinder, Netgene2 [10].
Для выявления экзонных делеций/дупликаций в гене АТР7В был использован набор фирмы MRC-Holland (P098-050R Wilson). Исследование проведено по протоколу производителя. Детекция проводилась с помощью фрагментного анализа на приборе Genetic Analyzer 3130xl фирмы Applied Biosystem.
Доверительный интервал рассчитывался с помощью программ STATISTICA 10.0 и Excel.
Расчет аллельной частоты мутации p.His1069Gln произведен по формуле на основании распределения Харди–Вайнберга p2 + 2pq + + q2 = 1, где 2pq – число гетерозигот в выборке, p – аллельная частота мутации p.His1069Gln.
Расчет гетерозиготного носительства мутаций в гене ATP7B проведен по формуле 2pq = m/N, где 2pq – доля гетерозигот, N – объем исследованной выборки, m – число выявленных носителей мутаций.
РЕЗУЛЬТАТЫ
На первом этапе работы образцы ДНК 431 пробанда с направляющим диагнозом “болезнь Вильсона–Коновалова” были исследованы на наличие самого частого патогенного варианта в гене ATP7B – p.His1069Gln (с.3207С>А). Мутация c.3207C>A в гомозиготном состоянии была обнаружена у 53 человек. Еще у 106 человек замена цитозина на аденин в положении 3207 была выявлена на одной из гомологичных хромосом. Зная это, возможно рассчитать долю частой мутации p.His1069Gln, основываясь на распределении Харди–Вайнберга (p2 + 2pq + q2 = 1). Если представить число гомозигот (n = 53) по частой мутации как Np2, где N – число реально больных БВК в исследуемой выборке, а p – доля частой мутации, то число гетерозиготных носителей частой мутации (n = 106) можно представить как 2Np(1 – p) (исходя из того, что q – доля мутаций в гене ATP7B, кроме p.His1069Gln, выражается как 1 – p). Отсюда доля мутации (p) p.His1069Gln равна 50%. Значит на долю всех остальных мутаций в гене ATP7B приходится также 50%. Зная это и исходя из того, что наблюдаемое соотношение генотипов должно соответствовать распределению Харди–Вайнберга, представляется возможным рассчитать реальное число больных БВК в исследуемой выборке (N = 53/p2), оно составило 212 человек. Так как ожидаемое количество больных БВК в выборке (n = 431) составило 212 человек, то 219 оставшихся пациентов в выборке не больны заболеванием, ассоциированным с геном ATP7B.
Была проведена оценка частоты популяционного носительства самого частого у европейцев патогенного варианта c.3207C>А гена ATP7B. Проведено типирование 1000 неродственных необследованных жителей России. Замена c.3207C>A (p.His1069Gln) была выявлена у 10 человек в гетерозиготном состоянии. Популяционная частота этой мутации составила 1 : 100 (доверительный интервал от 1 : 80 до 1 : 120). Данная величина является ключевой для последующего расчета частоты гепатолентикулярной дегенерации.
Учитывая популяционную частоту мутации c.3207C>A, получаем, что среди 219 человек выборки, причиной болезни у которых не являются мутации гена ATP7B, два человека – случайные популяционные носители данной замены. Таким образом, из 106 гетерозиготных носителей лишь 104 больны именно гепатолентикулярной дегенерацией. Используя соотношение Харди–Вайнберга и учитывая эту поправку, получаем расчетное ожидаемое число больных болезнью Вильсона–Коновалова в исследуемой выборке – 208 человек, а не 212, как было посчитано ранее. Дальнейший расчет долей обнаруженных вариантов нуклеотидной последовательности в гене ATP7B производился для этого ожидаемого числа больных. Из этих больных 25.5% являются гомозиготами по замене c.3207C>A (p.His1069Gln) и 50% гетерозиготами.
Зная долю частой мутации p.His1069Gln (0.5) и популяционную частоту этой мутации (1 : 100), мы определили частоту носительства гепатолентикулярной дегенерации (т.е. носительства дефектного гена ATP7B) в Российской Федерации, которая составила 1 : 50 (доверительный интервал от 1 : 42 до 1 : 63). Расчетная частота заболевания составляет 1 на 10 000 жителей РФ (доверительный интервал 1 : 8333 до 1 : 12 500).
На втором этапе образцы ДНК больных (кроме гомозигот по патогенному варианту с.3207С>А) были исследованы методом MPS с использованием панели генов обмена меди и железа, включающей в том числе ATP7B. Исследовались все экзоны и области экзон-интронных соединений этого гена. Из всех исследованных больных у 106 человек подтверждено наличие варианта с.3207С>А, из них у 92 гетерозигот по мутации с.3207С>А был выявлен второй патогенный вариант в гене ATP7B. Еще у 35 человек были зарегистрированы две другие мутации в гене ATP7B. Только один вариант в гене ATP7B был обнаружен у 19 человек, из них 14 – носители варианта с.3207С>А.
Далее у этих 19 пациентов с одной выявленной мутацией в гене АТР7В был проведен поиск крупных делеций/дупликаций методом количественной MLPA. Образцы ДНК пяти пациентов оказались непригодными для данного исследования. В образцах ДНК 14 пациентов с одним вариантом нуклеотидной последовательности в гене АТР7В крупных экзонных делеций/дупликаций не выявлено.
Спектр мутаций в гене АТР7В у российских больных представлен в табл. 1, а их схематичное расположение в гене – на рис. 1.
Таблица 1.
Варианты нуклеотидной последовательности в гене ATP7B, выявленные в данном исследовании
| Вариант (кДНК) | Эффект | Число хромосом | Доля мутации, % |
|---|---|---|---|
| c.3207C>A | p.His1069Glu | 212 | 50 |
| c.2304insC | p.Met769HisfsTer26 | 22 | 5.3 |
| c.3402delC | p.Ala1135GlnfsTer13 | 11 | 2.6 |
| c.3036insC* | p.Lys1013GlnfsTer13 | 11 | 2.6 |
| c.3190G>A | p. Glu1064Lys | 11 | 2.6 |
| c.[3942delCA;3947delG] | p.Lys1315_Arg1316del/insGlu | 8 | 1.9 |
| c.2998G>A | p.Glu1000R | 7 | 1.7 |
| c.3556+1G>T | Нарушение сплайсинга | 6 | 1.4 |
| c.4125-2A>G* | Нарушение сплайсинга | 5 | 1.2 |
| c.3809A>G | p.Asn1270Ser | 4 | 1 |
| c.1770insT* | p.Gly591TrpfsTer15 | 3 | <1 |
| c.3649del6 | p.Val1217_Leu1218del | 3 | <1 |
| c.2532delA | p.Val845SerfsTer28 | 3 | <1 |
| c.4022G>T | p.Gly1314Val | 3 | <1 |
| c.1847G>A | p.Arg616Gln | 3 | <1 |
| c.3659C>T | p.Thr1220Met | 3 | <1 |
| c.3121C>T | p.Arg1041Trp | 3 | <1 |
| c.2621C>T | p.Ala874Val | 3 | <1 |
| c.813C>A | p.Cys271Stop | 2 | <1 |
| c.2121+3A>G | Нарушение сплайсинга | 2 | <1 |
| c.2332C>G | p.Arg778Gly | 2 | <1 |
| c.2827G>A | p.Gly943Ser | 2 | <1 |
| c.2128G>A | p.Gly710Ser | 2 | <1 |
| c.3556G>A | p.Gly1186Ser | 1 | <1 |
| c.4103T>C | p.Leu1368Pro | 1 | <1 |
| c.3800A>T | p.Asp1267Val | 1 | <1 |
| c.2071G>A | p.Gly691Arg | 1 | <1 |
| c.3598C>T | p.Gln1200Ter | 1 | <1 |
| c.331C>T | p.Gln111Ter | 1 | <1 |
| c.2972C>T | p.Thr991Met | 1 | <1 |
| c.2230T>C | p.Ser744Pro | 1 | <1 |
| c.1630C>T | p.Gln544Ter | 1 | <1 |
| c.3646G>A | p.Val1216Met | 1 | <1 |
| c.3467G>A | p.Arg1156His | 1 | <1 |
| c.1877G>C | p.Gly626Ala | 1 | <1 |
| c.1883_1884delAT | p.His628ArgfsTer126 | 1 | <1 |
| c.3083_3085delAGAinsG | p.Lys1028SerfsTer40 | 1 | <1 |
| c.213_214delAT | p.Val73GlufsTer4 | 1 | <1 |
| c.4006delA | p.Ile1336TirfsTer55 | 1 | <1 |
| c.3412+1G>A | Нарушение сплайсинга | 1 | <1 |
| c.1745_1746delTA | p.Ile582ArgfsTer23 | 1 | <1 |
| c.1595A>G | p.Tyr532Cys | 1 | <1 |
| c.2510delG | p.Gln837GlufsTer34 | 1 | <1 |
| c.2078delC | p.Ser693SerfsTer2 | 1 | <1 |
| c.3888delC | p.Asp1296Glu | 1 | <1 |
| c.2107T>C | p.Cys703Arg | 1 | <1 |
| c.2998G>C | p.Gly1000Arg | 1 | <1 |
| с.3903+3G>T | Нарушение сплайсинга | 1 | <1 |
| c.2501T>A | p.Val834Asp | 1 | <1 |
| c.3459G>A | p.Trp1153Ter | 1 | <1 |
| c.2747T>C | p.Leu916Pro | 1 | <1 |
| c.1964T>G | p.Leu655Arg | 1 | <1 |
| c.2707G>T | p.Val903Leu | 1 | <1 |
| c.2164C>G | p.Leu722Val | 1 | <1 |
| c.2947T>A | p.Cys983Ser | 1 | <1 |
| c.3812A>C | p.Asp1271Ala | 1 | <1 |
| c.2558A>G | p.Asp853Gly | 1 | <1 |
| c.3892G>T | p.Val1298Phe | 1 | <1 |
| c.2383C>G | p.Leu795Val | 1 | <1 |
| c.1707+3del4 | Нарушение сплайсинга | 1 | <1 |
| c.3222_c.3243+21del43 | Эффект неизвестен | 1 | <1 |
| c.3663_3664delGG | p.Gly1221GlyfsTer35 | 1 | <1 |
| c.1897delC | p.Gln633ArgfsTer 13 | 1 | <1 |
| c.3973delC | p.Leu1325TrpfsTer3 | 1 | <1 |
| c.4246_4247insGG | p.Ala1386GlyfsTer6 | 1 | <1 |
| c.2122-3insC | Нарушение сплайсинга | 1 | <1 |
Рис. 1.
Варианты в гене АТР7В, вероятно имеющие отношение к диагнозу “гепатолентикулярная дегенерация”, обнаруженные у российских пациентов. Курсивом выделены неописанные ранее варианты; в процентах указаны суммарные доли вариантов в каждом экзоне гена АТР7В.
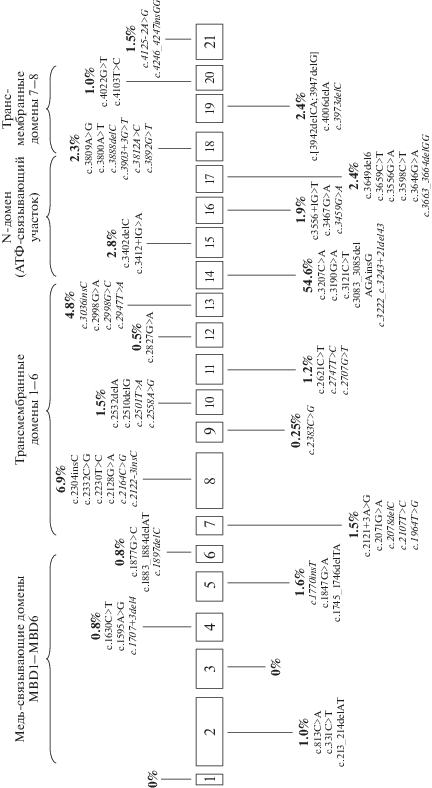
Как видно из табл. 1, всего у российских больных было выявлено 66 различных вариантов нуклеотидной последовательности в гене АТР7В. Из них 42 описанных в литературе и базах данных патогенных варианта и 24 новых варианта, ранее не описанных как причина болезни Вильсона–Коновалова.
Все впервые обнаруженные варианты в гене ATP7B оценены с помощью Руководства, данного в работе [10]. Результаты оценки представлены в табл. 2.
Таблица 2.
Анализ патогенности впервые выявленных и ранее не описанных как патогенные вариантов в гене АТР7В
| Выявленные варианты в гене АТР7В | Критерии патогенности | Результат | |
|---|---|---|---|
| кДНК | эффект | ||
| c.1707+3del4 | Нарушение сплайсинга | PM2, PP3 | Неопределенного значения |
| c.1770insT | p.Gly591TrpfsTer15 | PVS1, PM2 | Вероятно патогенный |
| c.1897delC | p.Gln633ArgfsTer 13 | PVS1, PM2 | » |
| c.1964T>G | p.Leu655Arg | PM2, PP3 | Неопределенного значения |
| c.2078delC | p.Ser693SerfsTer2 | PVS1, PM2 | Вероятно патогенный |
| c.2107T>C | p.Cys703Arg | PM2, PM5, PP3 | Неопределенного значения |
| c.2122-3insC | Нарушение сплайсинга | PM2, PP3 | » |
| c.2164C>G | p.Leu722Val | PM2, PM5, PP3 | » |
| c.2383C>G | p.Leu795Val | PM2, PM5, PP3 | » |
| c.2707G>T | p.Val903Leu | PM2, PP3 | » |
| c.2747T>C | p.Leu916Pro | PM1, PM2, PP3 | » |
| c.2947T>A | p.Cys983Ser | PM1, PM2, PP3 | » |
| c.2998G>C | p.Gly1000Arg | PS1, PM1, PM2, PP3 | Вероятно патогенный |
| c.3036insC | p.Lys1013GlnfsTer13 | PVS1, PM1, PM2 | » |
| c.3222_c.3243+21del43 | Эффект неизвестен | PM2 | » |
| c.3459G>A | p.Trp1153Ter | PVS1, PM1, PM2, PP3 | » |
| c.3663_3664delGG | p.Gly1221GlyfsTer35 | PVS1, PM1, PM2 | » |
| c.3812A>C | p.Asp1271Ala | PM1, PM2, PM5, PP3 | Неопределенного значения |
| c.3888delC | p.Asp1296GlufsTer32 | PVS1, PM1, PM2 | Вероятно патогенный |
| c.3892G>T | p.Val1298Phe | PM1, PM2, PM5, PP3 | Неопределенного значения |
| с.3903+3G>T | Нарушение сплайсинга | PM2, PP3 | » |
| c.3973delC | p.Leu1325TrpfsTer3 | PVS1, PM1, PM2 | Вероятно патогенный |
| c.4125-2A>G | Нарушение сплайсинга | PVS1, PM1, PM2 | » |
| c.4246_4247insGG | p.Ala1386GlyfsTer6 | PVS1, PM1, PM2 | » |
Как видно из табл. 2, среди выявленных ранее неописанных вариантов к вероятно патогенным отнесены 12 вариантов, к вариантам неопределенного значения – 12 вариантов. По мере накопления клинической информации о протекании болезни, появления информации о выявлении данных вариантов у неродственных больных, проведения функционального анализа эти варианты смогут быть переведены в разряд “патогенных” или “вероятно патогенных”, либо будет установлено, что они не имеют отношения к развитию болезни.
Суммируя вышесказанное, в исследованной выборке у 180 больных обнаружено по два варианта нуклеотидной последовательности в гене АТР7В. У 147 больных оба выявленных варианта являются патогенными, у 21 пробанда обнаружен патогенный вариант в сочетании с вероятно патогенным, у 10 человек патогенный вариант выявлен совместно с вариантом неопределенного значения, вероятно патогенный вариант в сочетании с вариантом неопределенного значения обнаружен у двух человек.
ОБСУЖДЕНИЕ
Таким образом, установить причину болезни и подтвердить клинический диагноз удалось у 78% российских больных гепатолентикулярной дегенерацией. У 19 пациентов выявлен лишь один патогенный вариант в гене АТР7В. Очевидно, что среди больных с одной выявленной мутацией находились и популяционные носители мутации в гене ATP7B, реальная причина заболевания которых носила не генетический характер, или больные, причиной заболевания которых являлись мутации в других генах. Так как ожидаемое число больных БВК в выборке составило 208 человек, то из 431 пациента 223 человека на самом деле не больны болезнью Вильсона–Коновалова, а имеют схожие с БВК клинические проявления. Из 223 человек, согласно рассчитанной частоте носительства болезни, четыре человека являются популяционными носителями патогенного варианта в гене АТР7В. У оставшихся носителей одного патогенного варианта в гене АТР7В (15 человек) второй вариант может локализоваться в некодирующих регуляторных участках гена АТР7В. Таким образом, доля выявленных патогенных аллелей в ожидаемой выборке больных БВК (n = 208) составила 90%, но 10% аллелей не удалось выявить, используя арсенал доступных нам методик.
Самым частым патогенным вариантом в гене АТР7В оказался вариант c.3207C>A, что соответствует результатам исследования европейских коллег [11]. Доля мутации c.3207C>A в Центральной, Восточной и Западной Европе варьирует в пределах 30–72%, а в России она составляет 50% [9].
Помимо мутации c.3207C>A в гене АТР7В у российских больных обнаружено большое разнообразие патогенных вариантов, многие из которых встретились лишь на одной хромосоме. Это свидетельствует о широкой генетической гетерогенности БВК у российских пациентов.
Самыми “горячими” экзонами гена ATP7B оказались экзоны 8 и 14 (рис. 1). В них расположены патогенные варианты, имеющие наиболее высокие доли. Мутации, расположенные в этих экзонах, затрагивают трансмембранные домены и N-домен (участок связывания молекулы АТФ), что ведет, соответственно, к нарушению переноса меди через канал и связывания АТФ со специфичным мотивом в АТФ-связывающем участке N-домена.
Анализ спектра мутаций в гене АТР7В у российских пациентов с БВК показал, что в целом причины БВК в России схожи с таковыми в большинстве европейских стран (Польша, Латвия, Литва, Болгария, Греция, Дания и др.), где самой частой причиной БВК являются мутации c.3207C>A, c.2304insC, c.3402delC [12]. Есть отличия от спектра мутаций в Северной Америке, где наряду с вариантом p.His1069Glu мутации p.Asn1270Ser и p.Gly1266Arg также часты. Сильно отличается от российского спектр мутаций в гене ATP7B в странах Азии (Китай, Корея, Япония, Индия), где самым частым является патогенный вариант p.R778L. Эта мутация в изучаемой выборке пациентов встретилась всего на двух хромосомах.
Несмотря на схожесть самых частых причин БВК в России и Европе, есть и существенные отличия. Варианты нуклеотидной последовательности c.3036insC, c.1770insT, c.4125-2A>G, встретившиеся неоднократно и впервые описанные как патогенные в лаборатории ДНК-диагностики МГНЦ ФГБНУ у пациентов с диагнозом “болезнь Вильсона–Коновалова”, вероятно, являются эндемичными для российских больных, так как ни разу не встречались в исследованиях других стран. Все три вышеописанных варианта нуклеотидной последовательности не встречаются в базе Genome Aggregation Database (gnomAD).
По последним данным разделяют два понятия: клиническая и генетическая распространенность болезни Вильсона–Коновалова. Под клинической частотой понимают число диагностированных случаев БВК, тогда как под генетической – расчетную частоту болезни, основанную на обширных данных полноэкзомного секвенирования. Так, в Европе клиническая распространенность БВК колеблется от 1.2 до 2 : 100 000, тогда как генетическая распространенность выше и составляет примерно 1 : 7000 [13]. По данным, приведенным в Федеральных клинических рекомендациях по диагностике и лечению болезни Вильсона–Коновалова [14], частота больных с клинически установленным диагнозом на территории Российской Федерации составляет в среднем 1 на 100 000 населения, в то время как полученная нами на основании молекулярно-генетических данных расчетная частота гепатолентикулярной дегенерации в России существенно выше и составляет 1 : 10 000. Разница клинической и генетической распространенности болезни Вильсона–Коновалова может быть следствием крайне широкой вариабельности клинических проявлений. Разнообразие не ограничивается лишь абдоминальной и церебральной формами болезни: каждая из этих форм делится еще на несколько клинических типов, в зависимости от преобладания той или иной клинической симптоматики. Для болезни Вильсона–Коновалова характерен крайне широкий возрастной диапазон манифестации: от периода новорожденности, когда дифференциальный диагноз приходится проводить с широким спектром других болезней накопления, до 60–70 лет жизни, когда неврологическая симптоматика, подобная БВК, может быть обусловлена различными нейродегенеративными болезнями как наследственной, так и ненаследственной природы [14]. Причиной данной вариабельности и неполной пенетрантности болезни может быть как различный патобиохимический эффект сочетания разных мутаций гена ATP7B в генотипе больного, так и существование генов-модификаторов [13].
Результатом определения спектра мутаций ATP7B у российских больных стало выявление 10 патогенных вариантов, встречающихся с частотой 1% и более. На основе полученных данных можно существенно оптимизировать молекулярно-генетическую диагностику, создав простые и недорогие мультиплексные системы. Повышение доступности молекулярно-генетической диагностики болезни Вильсона–Коновалова особенно актуально в связи с существованием эффективной и экономически доступной патогенетической терапии гепатолентикулярной дегенерации, способной снизить инвалидизацию и улучшить качество жизни больных [15].
Работа выполнена в рамках государственного задания ФАНО России.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Schilsky M.L. Wilson disease: genetic basis of copper toxicity and natural history // Semin. Liver Dis. 1996. № 16. P. 83–95. https://doi.org/10.1055/s-2007-1007221
Gollan J.L., Gollan T.J. Wilson disease in 1998: genetic, diagnostic and therapeutic aspects // J. Hepatol. 1998. Suppl. 1. P. 28–36.
Tanzi R.E., Petrukhin K., Chernov I. et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene // Nat. Genet. 1993. № 5. P. 344–350. https://doi.org/10.1038/ng1293-344
Ferenci P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing // Hum. Genet. 2006. № 120. P. 151–159. https://doi.org/10.1007/s00439-006-0202-5
Behari M., Pardasani V. Genetics of Wilsons disease // Parkinsonism Relat. Disord. 2010. № 16. P. 639–644. https://doi.org/10.1016/j.parkreldis.2010.07.007
Deguti M.M., Genschel J., Cancado E.L. et al. Wilson disease: novel mutation in the ATP7B gene and clinical correlation in Brazilian patients // Hum. Mutat. 2004. № 23(4). P. 398. https://doi.org/10.1002/humu.9227
Firneisz G., Lakatos P.L., Szalay F. et al. Common mutations of ATP7B in Wilson disease patientsfron Hungary // Am. J. Med. Genet. 2002. № 108(1). P. 23–28.
Figus A., Angius A., Loudianos G. et al. Molecular pathology and haplotype analysis of Wilson disease in Mediterranean population // Am. J. Hum. Genet. 1995. № 57. P. 1318–1324.
Баязутдинова Г.М., Щагина О.А., Поляков А.В. Мутация с.3207C>A гена ATP7B – наиболее частая причина гепатолентикулярной дегенерации в России: частота и причина распространения // Мед. генетика. 2018. № 4. С. 25–30.
Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б. и др. Руководство по интерпретации данных, полученных методами массового параллельного секвенирования // Мед. генетика. 2017. № 7. С. 4–17.
Gomes A., Dedoussis G.V. Geographic distribution of ATP7B mutations in Wilson disease // Ann. Hum. Biol. 2016. № 43(1). P. 1–8. https://doi.org/10.3109/03014460.2015.1051492
Chang I.J., Hahn S.H. The genetics of Wilson disease // Handb. Clin. Neurol. 2017. № 142. P. 19–34.
Poujois A., Woimant F. Wilson’s disease: A 2017 update // Clin. Res. Hepatol. Gastroenterol. 2018. № 42(6). P. 512–520. https://doi.org/10.1016/j.clinre.2018.03.007
Асанов А.Ю., Соколов А.А., Волгина С.Я. и др. Федеральные клинические рекомендации по диагностике и лечению болезни Вильсона–Коновалова. М., 2015.
Соколов А.А., Дембровский В.Н., Красильникова Е.Ю. Оказание медицинской помощи и лекарственное обеспечение пациентов, страдающих жизнеугрожающими и хроническими прогрессирующими редкими заболеваниями. Болезнь Вильсона (гепатолентикулярная дегенерация) // Проблемы стандартизации здравохранения. 2015. № 5–6. С. 30–36.
Дополнительные материалы отсутствуют.