

Генетика, 2021, T. 57, № 6, стр. 721-727
Важность накопления данных экзомного секвенирования российских больных при поиске причины наследственного заболевания в семье на примере аденоматозного полипоза
А. С. Цуканов 1, *, А. А. Баринов 1, В. П. Шубин 1, А. Н. Логинова 1, Т. А. Савельева 1, Д. Ю. Пикунов 1, А. М. Кузьминов 1, В. Н. Кашников 1, А. В. Поляков 2, Ю. А. Шелыгин 1
1 Национальный медицинский исследовательский центр колопроктологии им. А.Н. Рыжих
123423 Москва, Россия
2 Медико-генетический научный центр им. академика Н.П. Бочкова
115522 Москва, Россия
* E-mail: Tsukanov81@rambler.ru
Поступила в редакцию 11.08.2020
После доработки 14.09.2020
Принята к публикации 01.10.2020
Аннотация
Исследовали образцы ДНК шести членов одной семьи (три пациента с аденоматозным полипозом толстой кишки, двое здоровых и один человек с невыясненным статусом заболевания) методом полноэкзомного секвенирования. Предварительно у одной пациентки проводилась ДНК-диагностика генов APC/MutYH методом секвенирования по Сэнгеру, при которой герминальных мутаций найдено не было. В результате биоинформатического анализа данных у четырех кровных родственников (трое больных и один человек с неизвестным статусом болезни) была выявлена мутация в гене NSUN7, который ранее не описывался как ассоциированный с развитием аденоматозного полипоза толстой кишки. Однако в дальнейшем выяснилось, что популяционная частота этого варианта в данном гене именно в России существенно выше, чем встречаемость самого заболевания. При повторном биоинформатическом анализе только у трех больных выявлена ранее не описанная гетерозиготная делеция, включающая экзоны 2–16 гена APC, а также участки генов SRP19 и REEP5, которая впоследствии подтверждена методом мультиплексной проба-зависимой лигазной реакции с последующей амплификацией (MLPA, Multiplex Ligation-dependent Probe Amplification). Проведенное молекулярно-генетическое исследование продемонстрировало необходимость создания и постоянного пополнения отечественной базы данных вариантами, найденными при высокопроизводительном секвенировании. Также установлена целесообразность выполнения поиска больших делеций в гене APC у российских пациентов с аденоматозным полипозом толстой кишки.
Аденоматозные полипозные синдромы представляют собой целую группу наследственных заболеваний, проявляющихся развитием десятков, сотен, а иногда и тысяч полипов в толстой кишке пациента, вероятность злокачественной трансформации которых может достигать 100% в случае несвоевременного выполнения хирургического вмешательства. Наиболее частой причиной семейного аденоматоза толстой кишки являются гетерозиготные герминальные мутации в гене APC [1, 2]. Данный ген локализуется в регионе 5q22.2 и включает 16 экзонов, 15 из которых кодируют белок. Основную роль продукт гена APC выполняет в сигнальном WNT-пути [3]. Мутации в гене APC выявляются примерно у 70–80% пациентов, у которых к возрасту 40–45 лет в толстой кишке развивается более 100 аденоматозных полипов, что является классической формой семейного аденоматоза толстой кишки (FAP) [4, 5]. Как правило мутации у такого рода больных локализуются на участке 157–1595 кодонов (за исключением экзона 9) [6]. Если количество полипов у больного менее 100, то у пациента диагностируется ослабленная форма заболевания (AFAP), при которой мутации в гене APC целесообразно искать в области до 157-го кодона или после 1595-го кодона, а также в экзоне 9, однако вероятность их обнаружения составляет только 20% [7, 8].
В 2002 г. поиск других генов привел к открытию гена MutYH, который располагается в участке 1p34.1 и включает 16 кодирующих экзонов. Продукт этого гена принимает участие в эксцизионной репарации. К развитию полипоза приводят биаллельные мутации этого гена [9]. При этом клиническая картина MutYH-ассоциированного полипоза, как правило, сходна с ослабленной формой аденоматозного полипоза [10, 11]. Наиболее часто мутации у больных аденоматозным полипозом выявляются в генах APC и MutYH, однако описаны случаи обнаружения наследственных мутаций и в других генах.
В 2015 г. у семи родственников, пораженных аденоматозным полипозом, из трех различных семей были также выявлены биаллельные мутации в гене NTHL1, участвующем в эксцизионной репарации ДНК [12]. Данный ген локализован в регионе 16p13.3 и включает шесть кодирующих экзонов. В настоящий момент в мире описано около 20 семей с NTHL1-ассоциированным полипозом. Как правило, у пациентов с биаллельными мутациями в гене NTHL1 в толстой кишке выявляется до 100 аденоматозных полипов [13].
В 2013 г. C. Palles с соавт. описали наследственные гетерозиготные мутации в генах POLE и POLD1 у пациентов с полипами и случаями колоректального рака [14]. Гены POLE и POLD1 располагаются в участках 12q24.33 и 19q13.33 и включают 49 и 27 экзонов соответственно. Их продукты являются участниками системы репарации ошибочно спаренных нуклеотидов (mismatch repair, MMR). Однако в настоящее время накоплено недостаточно данных для того, чтобы можно было определенно говорить о преимущественном количестве полипов при PP-ассоциированном полипозе (Polymerase proofreading-associated polyposis) [15].
Необходимо отметить, что мутации в указанных генах не объясняют всех случаев аденоматозного полипоза у пациентов, поэтому нет сомнений, что с развитием и внедрением в клиническую практику технологии полноэкзомного (полногеномного) секвенирования в ближайшее время будут открыты и другие гены. В настоящей статье описывается поиск герминальных мутаций в экзонных областях генома человека.
МАТЕРИАЛЫ И МЕТОДЫ
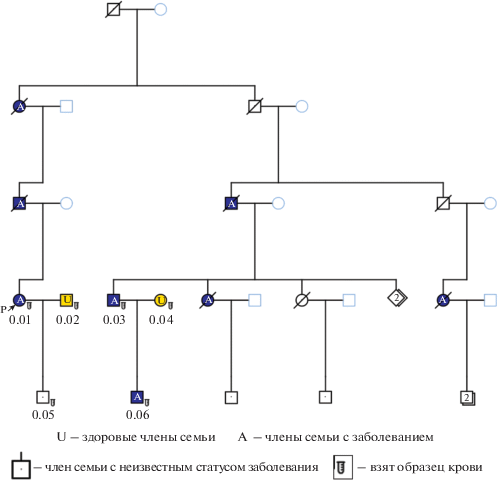
Члены семьи О.
В марте 2019 г. были собраны образцы крови 6 членов семьи O., в которой имелось большое количество родственников, пораженных семейным аденоматозом толстой кишки и колоректальным раком (рис. 1). У трех родственников диагностировано заболевание, у одного из которых нами ранее была проведена ДНК-диагностика генов APC и MutYH методом секвенирования по Сэнгеру (мутаций не выявлено) [5]; два члена семьи были здоровы; у одного родственника статус заболевания известен не был, поскольку он еще не выполнял эндоскопического обследования толстой кишки. От всех членов семьи, вовлеченных в данную работу, было получено информированное согласие.
Выделение ДНК
ДНК из лейкоцитов венозной крови выделяли с помощью набора QIAamp DNA Mini Kit (QIAGEN, Германия) из 200 мкл цельной крови согласно протоколу производителя, оценку количества ДНК проводили с помощью прибора DeNovix QFX (Denovix, США) и наборов для измерения ДНК Qubit dsDNA BR Assay Kit (ThermoFisher, США).
Массовое параллельное секвенирование
Для приготовления парно-концевых библиотек использовали 100 нг тотальной геномной ДНК. Проведены этапы пробоподготовки с первоначальной фрагментацией на приборе Covaris ME220 (Covaris, США) для получения целевого фрагмента 150 нктд с последующим обогащением экзомных регионов по комбинированному протоколу Illumina Truseq Exome совместно с зондами IDT xGen Exome v1 и секвенированием на платформе NextSeq550 с длиной прочтения 2*75 нктд (Illumina, США). Всего получено прочтений, прошедших фильтр (PF), в среднем 81 556 239 ± 5 780 081, среднее покрытие составило 97.4 ± 6.5, 50-кратное покрытие достигалось для 89.6% ± 3.7 нктд панели для исследуемых образцов.
Полученные прочтения были картированы на референсный геном GRCh37(hg19) с использованием BWA (v0.7.7) [16], SAMtools (v0.1.19) [17], варианты получены с помощью пакета программ GATK (v1.6) [18], аннотирование по базам данных (dpSNP v151, GnomAD v2.1, ExAC v0.3.1, 1000 Genomes Phase3 v5a, HGMD v2020.2), фильтрация и сегрегационный анализ проводили в облачном программном обеспечении BaseSpace Variant Interpreter (annotation engine v3.6.2.0). Анализ CNV-событий выполняли с помощью пакета СODEX [19] и визуализацией в IGV [20]. Полученные варианты оценивались по их встречаемости в популяции, влиянию на аминокислотную последовательность, нахождению в функциональном домене, оценке данных программ предсказателей патогенности (Sift, PolyPhen, DANN, MutationTaster, FAHMM-MKL) и других критериев согласно руководству по интерпретации данных последовательности ДНК человека, полученных методами массового параллельного секвенирования [21]. Оценку патогенности CNV проводили согласно опубликованным критериям “Technical standards for the interpretation and reporting of constitutional copy number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen)” [22].
Секвенирование по Сэнгеру
Найденную мутацию в гене NSUN7 подтверждали методом секвенирования по Сэнгеру. Подбор праймеров осуществляли с помощью программы PRIMER3. ПЦР проводили при следующих условиях: 95°C – 5 мин; 35 циклов (95°C – 10 с, 60°C – 10 с, 72°C – 10 с). Буфер для ПЦР: 2.5 мМ MgCl2 (ThermoFisher, США), 0.25 мМdNTP (ThermoFisher, США) (0.4 пМ F-праймер: TCAAGATTGTTCTGCCTCCA, 0.4 пM R-праймер: AGGGAGGAAGGTGTGTCATCT). Секвенирование выполняли с применением набора терминаторов BigDye 3.1 на генетическом анализаторе 3500 ABI PRISM (Applied Biosystems, США) согласно протоколу производителя. Полученные данные анализировали в программном обеспечении UGENE [23].
Метод мультиплексной проба-зависимой лигазной реакции с последующей амплификацией
Детекцию больших делеций в гене APC проводили методом мультиплексной проба-зависимой лигазной реакции с последующей амплификацией (MLPA) с использованием набора реактивов SALSA MLPA P043-APC, версия D1 (MRC-Holland, Нидерланды) по протоколу производителя. Фрагментный анализ осуществляли на генетическом анализаторе 3500 ABI PRISM (Applied Biosystems, США). При автоматической программной интерпретации результатов с помощью программы Coffalyser.Net (MRC-Holland, Нидерланды), данные картируются на референсный геном: hg18/mapview build 36.
Для преобразования координат генома и файлов аннотаций генома между сборками использовали ресурс открытого доступа: https://genome.ucsc.edu/cgi-bin/hgLiftOver.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Так как ранее у одной пациентки (O.01) с помощью секвенирования по методу Сэнгера в генах APC и MutYH наследственных мутаций выявлено не было, у нее и всех членов ее семьи провели полноэкзомное секвенирование.
В результате у всех трех больных (О.01, О.03, О.06), страдающих полипозом, а также у сына пациентки (О.04), чей статус заболевания не был известен, была найдена наследственная мутация c.2031_2032delinsAA (p.Cys677Ter) в гене NSUN7, которая была подтверждена с помощью секвенирования по методу Сэнгера (рис. 2). Ген NSUN7 (NOP2/Sun RNA methyltransferase family member 7; OMIM 617185) находится на хромосоме 4 и включает 12 экзонов, 11 из которых кодируют белок. В базе данных HGMD имеется описание только одной патогенной мутации в гене NSUN7. Мутация p.Thr141LeufsX17 (c.421delA) была обнаружена в 2015 г. иранскими исследователями N. Khosronezhad и соавт. среди пациентов с астеноспермией [24]. Однако исследование данного гена, проведенное H. Ren и соавт. на бóльшей выборке китайских пациентов, не выявило его связи с астеноспермией [25]. Стоит отметить, что астеноспермия является довольно частым заболеванием у мужчин, однако за последующие годы ни в одном исследовании у такого рода больных из других стран мутаций в гене NSUN7 найдено не было.
Рис. 2.
Сиквенс участка гена NSUN7: а – участок гена NSUN7 у здорового члена семьи O. (мутации нет), б – участок гена NSUN7 у больного члена семьи O. (мутация c.2031_2032delinsAA указана стрелками).
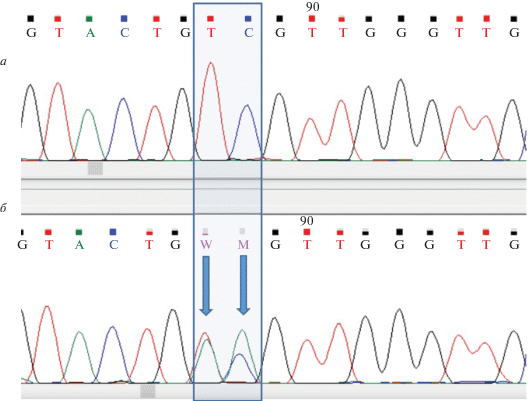
В найденном нами регионе гена NSUN7 ранее была выявлена мутация p.Cys677Ter (rs1338709562), которая в базе данных GnomAD описана с частотой 2 на 156 850 человек, при этом данных по ее клинической значимости не имеется. Также была обнаружена миссенс-мутация p.Arg678Ser (rs778600079) с частотами 3/21 796 и 6/125 568 человек в базах ExAC и TOPMED соответственно. Однако о ее клинической значимости также нет данных. Еще одной базой, в которой было решено проверить встречаемость данного наследственного варианта, прежде чем делать выводы об открытии нового гена, обусловливающего развитие полипоза, была база данных “NGS-DATA” ФГБНУ “МГНЦ”. Согласно данным этого, ресурса частота встречаемости варианта c.2031_2032delinsAA в гене NSUN7 в российской популяции составила 2/1223 человек, что существенно выше, чем частота самого заболевания (1/10 000 человек). Таким образом, стало очевидно, что мутация в гене NSUN7 не может являться причиной развития аденоматозного полипоза в данной семье.
В связи с этим поиск наследственной причины заболевания в данной семье продолжился, поскольку у нас имелись все генетические данные по каждому родственнику, был проведен повторный анализ результатов с помощью программы CODEX, позволяющей обнаруживать наличие больших вставок или делеций при проведении полноэкзомного секвенирования. В результате повторного биоинформатического анализа CNV у трех пораженных родственников была выявлена гетерозиготная делеция сегмента хромосомы 5 с захватом интервала 112 090 569–112 258 195 пн, включающего экзоны 2–16 гена APC, а также участки генов SRP19 и REEP5 (рис. 3,а). Из трех данных генов с развитием семейного аденоматоза толстой кишки ассоциирован только ген APC [1]. Наличие большой делеции в гене APC было подтверждено также с помощью метода MLPA (рис. 3,б).
Рис. 3.
Определение большой делеции в гене APC: а – схема делеции (указана красным цветом) участка 5 хромосомы, б – обнаружение делеции в гене APC методом MLPA.
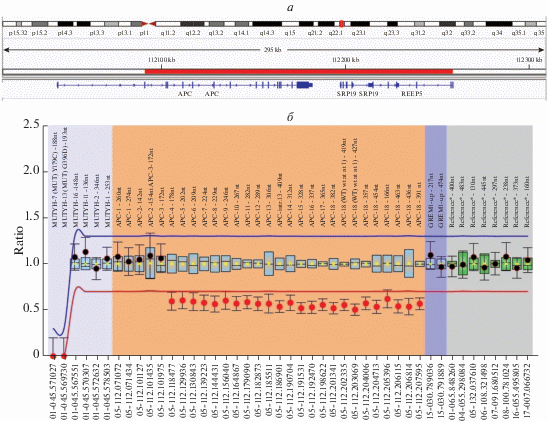
У сына пациентки, чей статус заболевания не был известен, мутации нет. Найденная у больных мутация ранее описана не была. Таким образом, в настоящей работе выявили, что частота герминальных мутаций в гене APC у российских больных аденоматозным полипозом выше, чем определенная нами ранее [5]. Это обусловлено тем, что на момент получения предыдущих результатов поиск больших делеций у пациентов не проводился. Необходимо уточнить, что встречаемость больших делеций в гене APC у пациентов из разных популяций варьирует от 0 в Словакии и Греции до 14% в Бельгии и Китае [26–29].
В заключение стоит отметить, что проведенный нами поиск молекулярно-генетической причины развития полипоза в обследованной семье, хотя и не позволил открыть новый ген, тем не менее привел к нескольким очень важным выводам. Прежде всего показана крайне высокая необходимость разработки и постоянного пополнения базы данных результатов высокопроизводительного секвенирования российских пациентов. Во-вторых, продемонстрирована возможность применения программ поиска больших вставок/делеций при анализе результатов полноэкзомного секвенирования. В-третьих, проведенное исследование позволило установить причину развития заболевания в конкретной семье и указало на более высокую частоту герминальных мутаций в гене APC у российских больных аденоматозным полипозом, чем предполагалось ранее. Наконец необходимо отметить, что в данной работе использование полноэкзомного секвенирования привело к обнаружению патогенной мутации в анализируемой семье; однако в тех случаях, когда наследственную причину с его помощью установить не удается, целесообразно применять метод полногеномного секвенирования.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Groden J., Thliveris A., Samowitze W. et al. Identification and characterization of the familial adenomatous polyposis coli gene // Cell. 1991. V. 66. № 3. P. 589–600. https://doi.org/10.1016/0092-8674(81)90021-0
Цуканов А.С., Шелыгин Ю.А., Фролов С.А., Кузьминов А.М. Семейный аденоматоз толстой кишки // Хирург. 2017. № 3. С. 14–24.
Näthke I.S. The adenomatous polyposis coli protein: The Achilles heel of the gut epithelium // Ann. Rev. Cell Dev. Biol. 2004. V. 20. № 1. P. 337–366. https://doi.org/10.1146/annurev.cellbio.20.012103.094541
Rivera B., González S., Sánchez-Tomé E. et al. Clinical and genetic characterization of classical forms of familial adenomatous polyposis: A Spanish population study // Ann. Oncol. 2011. V. 22. № 4. P. 903–909. https://doi.org/10.1093/annonc/mdq465
Цуканов А.С., Поспехова Н.И., Шубин В.П. и др. Мутации в гене APC у российских пациентов с классической формой семейного аденоматоза толстой кишки // Генетика. 2017. Т. 53 № 3. С. 356–363.
Nieuwenhuis M.H., Vasen H.F.A. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): A review of the literature // Crit. Rev. Oncol. Hematol. 2007. V. 61. № 2. P. 153–161. https://doi.org/10.1016/j.critrevonc.2006.07.004
Knudsen A.L., Bisgaard M.L., Bulow S. Attenuated familial adenomatous polyposis (AFAP): A review of the literature // Fam. Cancer 2003. № 2(1). P. 43–55. https://doi.org/10.1023/a:1023286520725
Kastrinos F., Syngal S. Inherited colorectal cancer syndromes // Cancer J. 2011. V. 17. № 6. P. 405–415. https://doi.org/10.1097/ppo.0b013e318237e408
Al-Tassan N., Chmiel N.H., Maynarde J. et al. Inherited variants of MYH associated with somatic G:C–T:A mutations in colorectal tumors // Nat. Genet. 2002. V. 30. № 2. P. 227–232. https://doi.org/10.1038/ng828
Цуканов А.С., Пикунов Д.Ю., Тобоева М.Х. и др. Трудности диагностики MUTYH-ассоциированного полипоза толстой кишки // Колопроктология. 2020. Т. 19. № 1 (71). С. 107–116. https://doi.org/10.33878/2073-7556-2020-19-1-107-116
Lubbe S.J., Di Bernardo M.C., Chandler I.P., Houlston R.S. Clinical implications of the colorectal cancer risk associated with MUTYH mutation // J. Clin. Oncol. 2009. V. 27. № 24. P. 3975–3980. https://doi.org/10.1200/JCO.2008.21.6853
Weren R.D.A., Ligtenberg M.J.L., Kets C.M. et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer // Nat. Genet. 2015. V. 47. № 6. P. 668–671. https://doi.org/10.1038/ng.3287
Altaraihi M., Gerdes A.-M., Wadt K. A new family with a homozygous nonsense variant in NTHL1 further delineated the clinical phenotype of NTHL1-associated polyposis // Hum. Genome Var. 2019. V. 6:46 (open access). https://doi.org/10.1038/s41439-019-0077-3
Palles C., Cazier J.-B., Howarth K.M. et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas // Nat. Genet. 2012. V. 45. № 2. P. 136–144. https://doi.org/10.1038/ng.2503
Talseth-Palmer B.A. The genetic basis of colonic adenomatous polyposis syndromes // Hered. Cancer Clin. Pract. 2017. V. 15. № 1. P. 5–12. https://doi.org/10.1186/s13053-017-0065-x
Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler Transform // Bioinformatics. 2009. V. 25. P. 1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li H., Handsaker B., Wysoker A. et al. The sequence alignment/map format and SAMtools // Bioinformatics. 2009. V. 25. № 16. P. 2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Van der Auwera G.A., Carneiro M.O., Hartl C. et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline // Curr. Protoc. Bioinformatics. 2013. V. 43. P. 1–33. https://doi.org/10.1002/0471250953.bi1110s43
Jiang Y., Oldridge D.A., Diskin S.J., Zhang N.R. CODEX: A normalization and copy number variation detection method for whole exome sequencing // Nucleic Ac. Res. 2015. V. 43. № 6. P. e39. https://doi.org/10.1093/nar/gku1363
Robinson J.T., Thorvaldsdóttir H., Winckler W. et al. Integrative Genome Viewer // Nat. Biotechnol. 2011. V. 29. № 1. P. 24–6. https://doi.org/10.1038/nbt.1754.Integrative
Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б. и др. Руководство по интерпретации данных последовательности ДНК человека, полученных методами массового параллельного секвенирования (Mps) (Редакция 2018, Версия 2) // Мед. генетика. 2019. Т. 18. № 2. С. 3–23. https://doi.org/10.25557/2073-7998.2019.02.3-23
Brandt T., Sack L., Arjona D. et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants // Genet. Med. 2020. V. 22. P. 336–344. https://doi.org/10.1038/s41436-019-0655-2
Okonechnikov K., Golosova O., Fursov M., Unipro U. GENE: A unified bioinformatics toolkit // Bioinformatics. 2012. V. 28. № 8. P. 1166–1167. https://doi.org/10.1093/bioinformatics/bts091
Khosronezhad N., Colagar A.H., Mortazavi S.M. The Nsun7 (A11337)-deletion mutation causes reduction of its protein rate and associated with sperm motility defect in infertile men // J. Assist. Reprod. Genet. 2015. V. 32. № 5. P. 807–815. https://doi.org/10.1007/s10815-015-0443-0
Ren H.Y., Zhong R., Ding X.P. et al. Investigation of polymorphisms in exon7 of the NSUN7 gene among Chinese Han men with asthenospermia // Genet. Mol. Res. 2015. V. 14. № 3. P. 9261–9268. https://doi.org/10.4238/2015.august.10.6
Sheng J.-Q., Wei-Jia Cui, Lei Fu et al. APC gene mutations in Chinese familial adenomatous polyposis patients // World J. Gastroenterol. 2010. V. 16. № 12. P. 1522–1526. https://doi.org/10.3748/wjg.v16.i12.1522
Stekrova J., Sulova M., Kebrdlova V. et al. Novel APC mutations in Czech and Slovak FAP families: Clinical and genetic aspects // BMC Med. Genet. 2007. V. 8. P. 16. https://doi.org/10.1186/1471-2350-8-16
Fostira F., Thodi G., Sandaltzopoulos R. et al. Mutational spectrum of APC and genotype-phenotype correlations in Greek FAP patients // BMC Cancer. 2010. V. 10. P. 389–398. https://doi.org/10.1186/1471-2407-10-389
Michils G., Tejpar S., Thoelen R. et al. Large deletions of the APC gene in 15% of mutation-negative patients with classical polyposis (FAP): A Belgian study // Hum. Mutat. 2005. V. 25. P. 125–134. https://doi.org/10.1002/humu.20122
Дополнительные материалы отсутствуют.