

Генетика, 2022, T. 58, № 10, стр. 1138-1154
Х-сцепленные CNV в патогенетике интеллектуальных расстройств
Е. Н. Толмачева 1, *, Е. А. Фонова 1, И. Н. Лебедев 1
1 Научно-исследовательский институт медицинской генетики, Томский национальный исследовательский медицинский центр Российской академии наук
634050 Томск, Россия
* E-mail: kate.tolmacheva@medgenetics.ru
Поступила в редакцию 04.04.2022
После доработки 25.05.2022
Принята к публикации 31.05.2022
- EDN: YMXSHE
- DOI: 10.31857/S0016675822100095
Аннотация
В обзоре рассматриваются моногенные и хромосомные мутации, ассоциированные с Х-сцепленной умственной отсталостью. Описаны особенности развития клинического фенотипа в случаях различных мутаций. Основное внимание уделяется Х-сцепленным CNV – микроделециям и микродупликациям. Представлены наиболее часто встречающиеся у пациентов с умственной отсталостью хромосомные микроперестройки. Обсуждается модифицирующее влияние инактивации Х-хромосомы на фенотип носительниц Х-сцепленных мутаций. Рассматриваются проблемы интерпретации клинической значимости Х-сцепленных CNV.
Сцепленная с Х-хромосомой умственная отсталость (УО) относится к группе наследственных заболеваний, характеризующихся различной степенью умственной отсталости, вызванной мутациями в различных генах, локализованных на Х-хромосоме. Диагноз УО устанавливается на основе трех следующих критериев: появление симптомов умственной отсталости до 18 лет; интеллектуальные функции значительно ниже среднего, с коэффициентом интеллекта (IQ) равным или ниже 70; плохие адаптивные навыки в следующих областях: общение, самообслуживание, социальные/межличностные контакты, самоуправление, успеваемость в школе, работа, отдых и здоровье. Познавательные способности обычно оцениваются с помощью определения IQ личности. УО может быть подразделена на четыре степени тяжести: легкая (уровень IQ от 50–55 до 70), умеренная (уровень IQ от 35–40 до 50–55), тяжелая (IQ уровень от 20–25 до 35–40) и глубокая (уровень IQ ниже 20–25).
Распространенность УО в западных странах оценивается примерно 2–3% [1]. В большинстве клинических случаев причины умственной отсталости остаются неизвестными, особенно в группе лиц с IQ более 50. В этой группе основную роль играет взаимодействие между генетическими факторами и факторами окружающей среды. В группе больных с “тяжелой” умственной отсталостью (IQ ниже 50) чаще присутствует один узнаваемый и идентифицируемый фактор. Выяснение причин умственной отсталости важно для прогноза, лечения и генетического консультирования. В 25–35% случаев умственная отсталость может быть следствием генетических причин. Следовательно, умственную отсталость следует рассматривать как симптом, а не как болезнь, глубинные причины которого крайне разнородны, и в большинстве случаев причина умственной отсталости неизвестна [1].
Еще в 1938 г. Пенроуз сообщил о наблюдении, что УО значительно чаще встречается у мужчин, чем у женщин [2]. Последующие исследования на многочисленных больших родословных подтвердили это наблюдение и поддержали вывод о том, что средняя частота интеллектуальной недостаточности у мужчин примерно на 30% выше, чем у женщин [3]. Эти наблюдения заложили обоснование концепции о том, что дефекты генов, локализованных на Х-хромосоме, играют важную роль в этиологии УО [3]. Клинические и генетические наблюдения показали, что Х-сцепленная умственная отсталость (X-linked intellectual disability, XLID) представляет собой очень разнородный набор состояний, ответственных за 10–15% всех наследственных случаев УО у мужчин, и встречается с частотой 1 на 600 индивидов мужского пола [4].
Полная последовательность Х-хромосомы была расшифрована в 2005 г., и оказалось, что необычно большое количество генов несут информацию о белках, важных для функционирования мозга [5]. Из 867 известных генов, кодирующих белок, которые локализованы на Х-хромосоме, продукты более 500 генов экспрессируются в мозге [6]. Считается, что большинство мутировавших генов, ответственных за развитие УО, влияют на развитие, миграцию клеток, формирование и поддержание нейронных сетей и межклеточной коммуникации в мозге. Гены, расположенные на Х-хромосоме, влияют не только на общий интеллект, но также имеют относительно специфический эффект на социальное познание и эмоциональную регуляцию [5].
Мутации, способствующие развитию XLID, могут быть генными, либо представляют собой микроструктурные хромосомные перестройки, такие как микроделеции и микродупликации [7]. В этом обзоре обсуждаются все виды мутаций на Х-хромосоме и факторы, влияющие на их манифестацию, но большее внимание уделяется хромосомным микроперестройкам, так как данные об их клинических эффектах остаются в недостаточной степени систематизированными.
XLID И МОНОГЕННЫЕ МУТАЦИИ
По данным Генетического Центра Гринвуда на декабрь 2021 г. идентифицировано 162 гена, связанных с Х-сцепленными формами интеллектуальной недостаточности [8]. Одним из первых описанных генов, вовлеченных в XLID, является FMR1 (Xq27.3, OMIM 309550), экспансия тринуклеотидного CAG-повтора в котором ответственна за синдром ломкой Х-хромосомы (OMIM #300624). Синдром ломкой Х-хромосомы составляет 1–2% всех случаев умственной отсталости в целом у мужчин, и эта мутация является наиболее частой причиной XLID [9, 10]. Помимо гена FMR1 среди пациентов с Х-сцепленной УО наиболее часто встречаются мутации в генах ARX (OMIM #300382) – 5–6% от всех случаев XLID, MECP2 (OMIM 300005), OPHN1 (OMIM 300127), PQBP1 (OMIM 300463), KDM5C (OMIM 314690) (от 1 до 4% для каждого от всех случаев XLID) [11].
XLID подразделяется на синдромальные и несиндромальные формы. Эта классификация была предложена впервые Kerr et al. (1991) [6]. Синдромальные XLID – это состояния, связанные с поражением других органов и наличием пороков развития или характерных клинических признаков. Некоторые синдромальные формы связаны с грубыми пороками развития головного мозга или нарушениями миграции нейронов.
У больных с синдромальной формой УО сопровождается изменениями роста, характерными черепно-лицевыми аномалиями, нервно-мышечными нарушениями, поведенческими аномалиями или метаболическими нарушениями. Эти сопутствующие аномалии позволяют разделить синдромальные XLID на четыре класса: синдромы пороков развития, нервно-мышечные расстройства, метаболические и доминантные состояния [12].
Синдромы пороков развития относительно редки и характеризуются УО и множественными врожденными аномалиями. Wilson et al. (1992) описали “новый” тип неспецифической Х-сцепленной умственной отсталости в трех поколениях семьи. Трое мужчин имели тяжелую форму УО (IQ 20–30), аутизм, задержку роста, частые инфекции, судороги и другие незначительные аномалии: брахицефалия, завиток волос на лобной части, квадратное лицо, большой рот, толстые губы и прогнатия [13]. Этот синдром был определен как Х-сцепленная умственная отсталость № 12 (OMIM 309545).
Самыми распространенными являются нервно-мышечные синдромы. У пациентов обычно присутствуют спастичность, атаксия, атетоз, тремор, гипотония или, напротив, гипертонус, ригидность и другие нервно-мышечные симптомы. Кроме того, часто встречаются дефицит специфических органов чувств, нарушение зрения или слуха. Одним из примеров является синдромальное нарушение умственного развития, сцепленное с Х-хромосомой, синдром Кабезаса (OMIM 300354), который помимо УО характеризуется низким ростом, гипогонадизмом и аномальной походкой, а также другими более вариабельными признаками, такими как задержка речи, выпуклая нижняя губа и тремор.
Метаболические синдромы возникают из-за дефектов в специфических биохимических путях. Одно из метаболических Х-сцепленных заболеваний – мукополисахаридоз типа II (MPS2), также известный как синдром Хантера (OMIM 309900), вызывается мутацией в гене, кодирующем идуронат-2-сульфатазу, локализованном в Xq28. MPS2 является мультисистемным заболеванием. Клинические проявления включают тяжелую обструкцию дыхательных путей, деформации скелета, кардиомиопатию и у большинства пациентов ухудшение неврологического статуса. Смерть обычно наступает на втором десятилетии жизни, хотя некоторые пациенты с менее тяжелым заболеванием доживают до пятого или шестого десятилетия [14, 15].
Доминантные синдромы имеют Х-сцепленное доминантное наследование с почти полным отсутствием пораженных мужчин из-за внутриутробной летальности, жизнеспособны только пораженные женщины [11]. Самым известным и частым доминантным синдромом является синдром Ретта (RTT, OMIM 312750) – тяжелое нарушение развития нервной системы, одна из наиболее частых причин умственной отсталости у женщин. Причиной синдрома являются мутации в гене MECP2. Хотя сначала считалось, что мутации MECP2, вызывающие RTT, были летальными для плодов мужского пола, в более поздних сообщениях было выявлено, что мальчики с RTT все-таки выживают, но характеризуются тяжелой неонатальной энцефалопатией. Затем несколько дополнительных сообщений подтвердили тяжелый фенотип у мужчин с RTT-ассоциированными мутациями гена MECP2 [10, 16, 17].
Несиндромальные формы XLID определяются как непрогрессирующие состояния, влияющие на когнитивную функцию при отсутствии других отличительных особенностей [18]. При несиндромальной УО часто встречается коморбидность с аутизмом, эпилепсией и нервно-мышечными нарушениями (например, атаксия, спастическая параплегия, сенсорная/моторная невропатия и мышечная дистрофия) [19]. Подсчитано, что две трети XLID являются несиндромальными [11]. Однако один и тот же ген или даже одна и та же мутация в гене может привести как к синдромальной, так и к несиндромальной форме заболевания [6]. Наличие “мягких” мутаций и/или неполная пенетрантность специфических клинических признаков у некоторых лиц, несущих мутации в генах, связанных с синдромальной умственной отсталостью, может стереть различие между синдромальной и несиндромальной умственной отсталостью [20]. Например, рекуррентная мутация, представляющая собой дупликацию вставки в 24 пн в экзоне 2 гена ATRX и приводящая к расширению полиаланинового тракта, связана и c синдромальными, и с несиндромальными формами, характеризующимися широким спектром фенотипических нарушений, начиная от лиссэнцефалии (LISX2; OMIM 300215) до синдрома Прауда (OMIM 300004), синдрома Партингтона (OMIM 309510) и несиндромальной формы (XLID29, OMIM 300419) умственной отсталости [21, 22]. Мужчины с мутациями ARX часто страдают более тяжело, женщины-носители мутаций также могут быть поражены [21, 22].
Продукты генов, ответственных за XLID, делятся на четыре общие группы по биологическим функциям, которые считаются критически важными для морфологии и целостности нейронов: везикулярный цикл в пресинаптическом нервном окончании; динамика цитоскелета; клеточная адгезия и транссинаптическая передача сигналов; регуляция трансляции [23]. Примерно 18% известных в настоящее время генов, ассоциированных с XLID, кодируют мембранные белки. Среди них классические CAMs (L1 Cell Adhesion Molecule – L1CAM; Neuroligin-3 – NL3; Neuroligin-4 – NL4; Protocadherin19 – PCDH19) [24], а также дополнительный белок рецептора интерлейкина-1 (ген IL1RAPL1), который опосредует синаптическую адгезию [25], и Tetraspanin-7 (TSPAN7), который взаимодействует с интегриновым классом CAMs [26]. Мутации в генах NL3 (OMIM 300366) и NL4 (OMIM 300427) были идентифицированы в двух родственных парах с тяжелой умственной отсталостью и аутизмом. Молекулы адгезии представляют собой интегральные мембранные белки, которые внеклеточно связывают белки клеточной поверхности или компоненты внеклеточного матрикса и обеспечивают мост через синаптическую щель. Помимо обеспечения структурной связи между пре- и пост-синапсом молекулы адгезии также функционально координируют две стороны синапса посредством двунаправленной передачи сигнала. Следовательно, роль молекул адгезии варьирует от синаптогенеза до созревания и пластичности синапсов [27]. Молекулы адгезии имеют общую структуру с внеклеточной частью, которая опосредует их адгезионные свойства, трансмембранным доменом и цитоплазматическим доменом, которые связывают цитоскелетные и сигнальные молекулы.
Х-СЦЕПЛЕННАЯ УМСТВЕННАЯ ОТСТАЛОСТЬ И CNV
Разработки в технологии геномных микрочипов произвели революцию в изучении изменчивости числа копий генов человека. Использование сравнительной геномной гибридизации в исследованиях XLID привело к идентификации новых генов XLID, изменений количества копий в известных генах XLID и описанию вариации числа копий (CNV) в генах, которые ранее не считались ответственными за развитие XLID. Было установлено, что субмикроскопические делеции и дупликации на Х-хромосоме могут объяснить около 5% идиопатических случаев XLID [28]. В результате многочисленных исследований был выявлен ряд особенностей, связанных с локализацией CNV на Х-хромосоме [28–30]:
1) на Х-хромосоме наблюдается больше дупликаций, чем делеций;
2) размер дупликаций на Х-хромосоме больше, чем размер делеций, из-за потенциальной нежизнеспособности имеющего большие делеции гемизиготного индивида;
3) большинство Х-сцепленных CNV (при различных патологиях от 30 до 80%) унаследованы от фенотипически здоровой матери.
Эти характеристики связаны с гемизиготным состоянием X-сцепленных генов у мужчин, что делает анализ CNV на X-хромосоме особенно значимым [28].
Субмикроскопические перестройки Х-хромосомы у мужчин можно разделить на внутригенные и те, которые затрагивают весь ген или несколько генов. Для делеций это, скорее всего, не имеет значения, поскольку делеции нескольких экзонов гена на Х-хромосоме у мужчин приведут к такому же отсутствию функционального продукта, как и потеря всего гена. Однако для дупликаций внутригенные перестройки, включая экзоны, могут привести к образованию неправильных транскриптов и удлиненных белков. Когда точка разрыва в дупликации находится внутри гена, окончательный результат зависит от того, является ли дупликация тандемной или инвертированной, либо дупликация представляет собой инсерцию в другое место в геноме. Далее будут обсуждены наиболее распространенные либо клинически значимые микроперестройки, связанные с XLID, а большее число Х-сцепленных CNV приведено в таблице (Приложение) и на рис. 1.
Рис. 1.
Локализация Х-сцепленных CNV на Х-хромосоме. Синий цвет – дупликации, красный цвет – делеции. Указана относительная протяженность хромосомных микроперестроек и входящие в их состав гены.
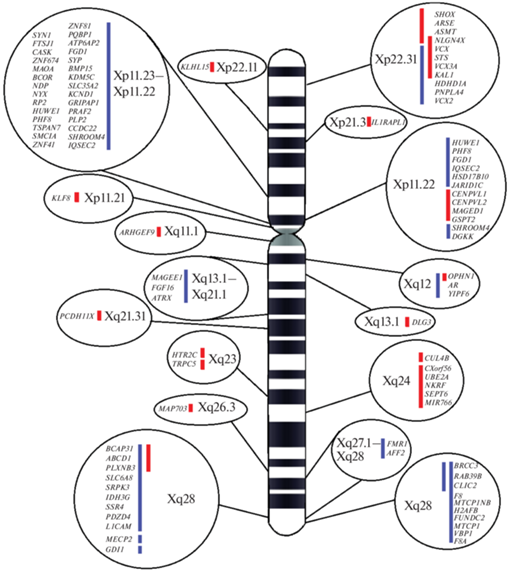
Делеция региона Xp21 с вовлечением гена IL1RAPL1
Миссенс-мутации и делеции в гене IL1RAPL1 (OMIM 300206) являются причиной формирования Х-сцепленной формы УО – XLID21 (OMIM 300143). Эта форма характеризуется спектром когнитивных неврологических нарушений, варьирующих от умеренных нарушений интеллектуального развития до симптомов растройства аутистического спектра (РАС). Мужчины обычно поражены тяжело, а у некоторых женщин-носительниц могут проявляться более легкие нарушения [31]. У мужчин с делецией размером 635 тпн, охватывающей экзоны 2–5 гена IL1RAPL1, наблюдались низкий IQ, отклонения в поведении, включая импульсивность, оппозиционное расстройство и гиперактивность. Кроме того, у пациентов были зафиксированы различные дисморфии: гипотония, воронкообразная деформация грудины, выдающаяся челюсть, синофриз и гиперрастяжение суставов. Ни у одного больного не было выявлено аутизма [32].
Делеция региона Xp11.4
У девочки с тяжелой формой УО, задержкой роста с микроцефалией, судорогами, прогрессирующим сколиозом, колобомой правой сетчатки и расщелиной нёба была обнаружена делеция de novo в регионе Xp11.4. Делеция размером 3.2 млн пн затрагивала восемь генов. В область делеции вошли три гена из списка генов, связанных с XLID: MAOA (OMIM 309850), NDP (OMIM 310600) и часть гена CASK (OMIM 300172). Гаплонедостаточность этих генов может объяснить тяжесть клинического фенотипа. У пациентки наблюдалась экстремальное смещение инактивации Х‑хромосомы (100/0) [33].
Делеция региона Xq23 с вовлечением гена CUL4B
Мутации гена CUL4B, расположенного в Xq23, вызывают синдром Кабезаса (OMIM 300354), который характеризуется УО, низким ростом и задержкой речи, наряду с другими более вариабельными признаками. Делеция в гене CUL4B у 10-летнего мальчика сопровождалась тяжелой формой УО и выраженной задержкой речи. У пациента присутствовали поведенческие проблемы, такие как тревожность, аутизм, а также агрессивный и склонный к самоповреждениям характер. У него был низкий рост, небольшая макроцефалия, низко посаженные уши, закругленный кончик носа, косоглазие, выступающая нижняя губа, скученность зубов, маленькие стопы и широкие пальцы ног. Он также страдал судорогами, легкой нейросенсорной тугоухостью, нарушением походки, задержкой мелкой моторики и истощением мышц голени [34].
Делеция в регионе Xq24
В 90% случаев всех крупных делеций этого региона задействованы пять генов: CXorf56, UBE2A, NKRF, SEPT6 и MIR766. Кандидатным геном для проявления клинических признаков заболевания, вероятнее всего, является ген UBE2A. Мутации в гене UBE2A и затрагивающие его делеции приводят к синдрому Насименто или дефицита UBE2A (OMIM 300860) [35]. Основные клинические симптомы данного синдрома: задержка психомоторного и речевого развития, гипотония, судороги, врожденный порок сердца (дефект межжелудочковой перегородки) и аномалии половых органов (крипторхизм, уменьшенный пенис) [36–38]. При анализе гено-фенотипических корреляций всех пациентов с синдромом дефицита UBE2A можно разделить на две основные группы: I группа из тех, у кого есть внутригенные мутации (либо точечные мутации, либо небольшие делеции), и II группа – имеющие большие делеции, включающие другие гены в дополнение к гену UBE2A. Пациенты с более крупными делециями, включающие ген UBE2A, имеют более высокую распространенность пороков белого вещества головного мозга и пороки развития мочеполовой системы, по сравнению с пациентами с внутригенными делециями и миссенс-мутациями. Более того, пороки сердца также чаще встречались у пациентов из второй группы по сравнению с пациентами первой [37]. Еще один ген, входящий в область делеции, – CXorf56 связан с задержкой интеллектуального развития (OMIM 301013). Ген экспрессируется в головном мозге. Инсерция в этом гене, приводящая к появлению преждевременного стоп-кодона и бессмысленной мРНК, приводит к развитию семейной формы УО [39].
Фенотипические последствия дупликации генов, связанных с Х-сцепленной умственной отсталостью, разнообразны. В первом случае дупликация может быть связана с фенотипом, идентичным фенотипу, связанному с мутацией потери функции или делецией гена. Так обстоит дело с дупликацией гена PLP1, которая приводит к синдрому Пелизеуса–Мерцбахера. Во втором случае дупликация гена XLID может привести к уникальному фенотипу. Дупликация MECP2, по-видимому, является наиболее распространенной дупликацией этого типа. Аналогичным образом проявляются дупликации генов STAG2, HUWE1 и OCRL [40]. Промежуточными между этими фенотипическими последствиями являются дупликации гена ATRX, которые связаны с вариабельными фенотипическими проявлениями синдрома умственной отсталости и альфа-талассемии (низкий рост, генитальные аномалии, умственная отсталость, гипотония), но лишены типичных черт лица, наблюдаемых при вариантах потери функций [40]. Кроме того, дупликации некоторых генов, связанных с XLID (IKBKG, ARX) и определенных областей Х-хромосомы (Xp21.33, Xq21.33), по-видимому, не связаны с аномалиями развития нервной системы, хотя они могут быть связаны с другими соматическими отклонениями [41–43].
Дупликация Xp11.23p11.22
Дупликация, затрагивающая область Xp11.23p11.22, очень редко встречается у представителей обоих полов. Известны наследуемые формы и формы de novo. Сообщалось о нерецидивных дупликациях размером 0.3–55 млн пн в дополнение к рецидивирующей форме размером около 4.5 млн пн, поэтому гены, участвующие в дупликациях у отдельных пациентов, могут быть совершенно разными. При этом у пациентов с разными вариантами этой дупликации, независимо от генного состава, проявляются весьма схожие симптомы [44]. В исследовании шести семей Froyen et al. (2008) [33] выделили минимальную перекрывающуюся область. Функциональный анализ вовлеченных генов выявил причинно-следственную связь между повышенной дозой генов и умственной отсталостью только в случае гена HUWE1 [45]. Мутации последовательности в HUWE1 были связаны с синдромом Юберга–Марсиди, синдромом Брукса (OMIM 309590), синдромом XLID-макроцефалии Тернера, а также были описаны в семье, в которой мужчины имели умеренную степень УО, но не имели лицевых дисморфий [33, 45, 46]. Дупликация HUWE1 связана с УО средней степени тяжести, ограниченной речью или дизартрией, лицевыми дисморфиями (гипертелоризм, косые глазные щели, синофриз, открытый рот) и, как правило, нормальными показателями пренатального и постнатального роста. В некоторых случаях пациенты не имели выраженных дисморфий и обладали нормальным ростом. Судороги выявлены у нескольких пациентов, у одного индивида была подслизистая расщелина нёба, а у двух мальчиков была гипоспадия первой степени [40]. Grems et al. (2015) [25] на основе их собственного анализа и ранее опубликованных случаев с дупликацией Xp11.2 подчеркнули важность двух субрегионов дупликации. Одна из них (область 1) содержит гены SHROOM4 и DGKK, другая (область 2) гены HUWE1, KDM5C и IQSEC2. Наиболее распространенными основными симптомами у пациентов с дупликацией Xp11.23p11.22 являются задержка развития, умственная отсталость различной степени тяжести, судороги и различные поведенческие аномалии.
Дупликация региона Xq22.2 c участием гена PLP1
Дефект гена PLP1 может вызывать широкий спектр клинических фенотипов, начиная от врожденной формы болезни Пелицеуса–Мерцбахера (PMD, OMIM 312080) и заканчивая формой Х-сцепленной спастической параплегии 2-го типа (SPG2, OMIM 312920). Симптомы варьируют от тяжелой картины, проявляющейся при рождении, со спастичностью, стридором и нистагмом, до относительно легкого парапареза без когнитивных нарушений. PMD/SPG2 наследуется как сцепленный с Х-хромосомой признак, ген PLP1 локализован в Xq22.2. Точечные мутации гена PLP1 ответственны за 20% случаев PMD/SPG2, вызывающих широкий спектр клинических фенотипов. Сообщалось, что редкие делеции целого гена PLP1 вызывают легкую форму PMD/SPG2 с умеренной спастической параплегией и легкой задержкой когнитивных функций [47]. Дупликация сегмента Х-хромосомы, содержащего ген PLP1, является наиболее частым генным дефектом, на который приходится 60–70% случаев PMD. У пациентов с дупликацией гена PLP1 обычно наблюдается классическая форма PMD с началом на первом году жизни, характеризующаяся нистагмом, УО и спастичностью. О наличии более двух копий гена PLP1 сообщалось у небольшого числа пациентов с тяжелой формой PMD [47]. Среди пациентов с классической формой заболевания и дупликацией гена PLP1 степень клинической тяжести может варьировать. Эта изменчивость не коррелирует с размером дупликации гена PLP1 [47].
Дупликация в регионе Xq25
Сообщалось о ряде дупликаций в Xq25, области обедненной генами, у мужчин и женщин. Ген STAG2, который кодирует субъединицу комплекса cohesin, в этих случаях полностью дуплицируется, а соседние гены (XIAP, THOC2, GRIA3, SH2D1A и TENM1) вариабельно дуплицируются полностью или частично [48]. В фенотипе преобладает УО различной степени тяжести. Другие общие черты включают нормальный рост и окружность головы, уплощенность скул, толстую красную кайму губ, прогнатию, лицевую гипотонию и поведенческие проблемы. Судороги и аутизм встречаются у трети или реже. Напротив, пациенты с делециями или вариантами точковых мутаций STAG2 имеют более серьезную задержку развития, нарушение роста, микроцефалию и пороки развития срединной линии, включая голопрозэнцефалию или другие аномалии ЦНС, расщелину лица и глазные колобомы.
Дупликация региона Xq26.1 (ген OCRL)
Синдром Лоу (OMIM 309000), вызванный мутациями последовательности или делециями гена OCRL, характеризуется ранним началом катаракты, снижением мышечного тонуса и рефлексов, аминоацидурией и УО. Дупликация OCRL была описана в трех семьях, у всех в сочетании с дупликациями соседних генов [49, 50]. В этих семьях у пораженных лиц наблюдались аномалии развития нервной системы, УО, аутизм и судороги, но среди этих мальчиков не наблюдалось фенотипа синдрома Лоу.
Синдром дупликации региона Xq28 (OMIM 300815)
На Х-хромосоме была обнаружена высокая частота патогенетически значимых микродупликаций, демонстрирующая, что повышенная экспрессия Х-сцепленных генов также может нарушать нормальное когнитивное развитие [51]. Рекуррентные перестройки обычно опосредованы неаллельной гомологичной рекомбинацией между фланкирующими сегментными дупликациями или низкокопийными повторами (LCR; геномные дупликации >1 тпн с более 90% идентичности), что приводит к делециям, дупликациям, амплификациям или инверсиям промежуточных геномных сегментов. Несколько повторяющихся аберраций из-за неаллельной гомологичной рекомбинации между двумя высокогомологичными повторяющимися единицами были описаны на Х-хромосоме, например в Xp22.3, которые приводили к Х-сцепленному ихтиозу (MIM 308100). Сегмент Xq28 представляет особый интерес, поскольку он содержит множество наборов LCR, расположенных в непосредственной близости друг от друга, что делает этот регион склонным к рекомбинации, которая может привести к заболеванию [51]. Дупликации Xq28 являются наиболее частыми хромосомными аберрациями, наблюдаемыми у пациентов с УО, особенно у мужчин. Эти дупликации происходят по вариабельным механизмам, включая интерстициальные дупликации, опосредованные сегментными дупликациями в этой области, и терминальные дупликации (функциональные дисомии), возникающие в результате транслокации с другими хромосомами. Наиболее часто дуплицированная область включает ген метил-CpG-связывающего белка 2 (MECP2) с минимальным размером дупликации 0.2 млн пн. У пациентов с дупликацией MECP2 наблюдаются тяжелая форма УО, некупируемые судороги и рецидивирующие инфекции. Дупликации в соседних теломерных областях, которые включают ген ингибитора диссоциации GDP 1 (GDI1) и ген ras-ассоциированного белка RAB39B (RAB39B), независимо связаны с УО, и многие сегментные дупликации, расположенные в этой области, могут опосредовать часто наблюдаемые интерстициальные дупликации.
Дупликации гена MECP2. Синдром Ретта, обусловленный делециями или мутациями последовательности в MECP2 и характеризующийся периодом нормального развития в младенчестве, сопровождаемым микроцефалией и эпизодическим, но неуклонным течением неврологического ухудшения, стереотипными движениями рук, судорогами и спастичностью, весьма существенно отличается от проявлений пациентов с дупликацией MECP2. Лица с дупликацией гена MECP2 имеют раннее начало гипотонии и прогрессирующую спастичность, поражающую нижние конечности. Кроме того, у 50% пораженных мужчин наблюдаются эпилептические припадки, и многие имеют предрасположенность к рецидивирующим инфекциям, в том числе и респираторным, которые часто требуют трахеостомии и искусственной вентиляции легких [52]. У них тяжелая форма УО, часто осложняющаяся судорогами, и большинство из них умирают в возрасте до 25 лет. Хотя первоначально дупликация описывалась как Х-сцепленный рецессивный синдром у мужчин, более поздние сообщения подтвердили возникновение ее у женщин, обычно выражающееся в инфантильной гипотонии, прогрессирующей до спастичности, тяжелой УО и проявлениях дефектов неврологического развития, включая расстройство аутистического спектра [40].
Дупликации с участием гена GDI1. В регионе Xq28 была идентифицирована дупликация размером 0.3 млн пн (chrX: 153.2–153.5 млн пн, NCBI36), содержащая не менее 11 генов и включающая кандидатный ген GDI1 (OMIM 300104). У мужчин, помимо УО, наблюдались аномалия Денди–Уокера с гипоплазией мозжечка и агенезией мозолистого тела, судороги, микроцефалия, брахицефалия, широкий лоб, гипотелоризм и обратный эпикантус. У пациентов из разных семей наблюдалось от двух до пяти копий гена GDI1, при этом число его копий строго коррелировало с тяжестью клинических проявлений [51].
Дупликации с участием гена RAB39B. Еще один вариант дупликации включает в себя дистальную область размером 0.5 млн пн, содержащую не менее восьми генов, в том числе RAB39B (OMIM 300774), мутации которого вызывают отдельную форму УО (XLID72, OMIM 300271). Мужчины с такой дупликацией имели когнитивные нарушения, агрессивное и/или гиперактивное поведение, рецидивирующие ушные инфекции или пневмонию и легкие дисморфичные черты лица, включая высокий лоб, нависающие верхние веки, широкую переносицу и толстую нижнюю губу [53].
МОДИФИЦИРУЮЩАЯ РОЛЬ ИНАКТИВАЦИИ Х-ХРОМОСОМЫ В ПРОЯВЛЕНИИ ФЕНОТИПИЧЕСКИХ ПРИЗНАКОВ X-СЦЕПЛЕННЫХ МУТАЦИЙ У ГЕТЕРОЗИГОТНЫХ НОСИТЕЛЬНИЦ
На манифестацию XLID у гетерозиготных носительниц мутаций в первую очередь влияет инактивация Х-хромосомы (X-chromosome inactivation, XCI) – эпигенетический процесс, позволяющий уровнять дозу Х-сцепленных генов. На очень раннем этапе развития эмбриона женского пола в каждой клетке происходит случайная инактивация либо отцовской, либо материнской Х-хромосом, и схема инактивации передается всем дочерним клеткам посредством митоза. Это приводит к мозаичной экспрессии генов, сцепленных с Х-хромосомой, у женщин, что может обеспечить защиту от болезней. Как правило, соотношение экспрессии материнских и отцовских аллелей у женщин составляет примерно 50 : 50; однако в некоторых случаях наблюдается отклонение, известное как асимметричная XCI (skewed XCI, sXCI). Обычно это соотношение составляет 80 : 20 или выше.
Женщины, как правило, менее восприимчивы к патогенным вариантам генов на активной Х‑хромосоме, поскольку этот вариант не экспрессируется во всех ее клетках (табл. 1). У большинства женщин Х-сцепленные заболевания не проявляются, потому что 1) женщины не являются гомозиготами по патогенному варианту и 2) их клетки, в которых экспрессируется мутантный аллель, получают от клеток, в которых транскрибируют нормальный аллель, достаточное количество генного продукта, необходимого для выполнения основной функции. Достаточное количество нормального белка обеспечивается одним из двух способов. Либо клетки передают его клеткам с дефицитом, либо, если этот перенос между клетками не происходит, недостаток функционального белка может привести к тому, что клетки с дефицитом будут делиться менее эффективно и поэтому в конечном итоге их число будет значительно меньше, чем клеток, вырабатывающих нормальный белок. Тем не менее в различных тканях клетки различаются по своей способности переносить генные продукты, поэтому в тканях организма могут быть различия в способности нормальных клеток делиться или аномальных клеток выживать. В большинстве случаев 50% активности более чем достаточно, а для многих генов достаточно и меньшего количества продукта. Это легко объясняет почему многие заболевания, сцепленные с Х-хромосомой, чаще всего не проявляются у женщин. Например, менее 5% фермента HPRT может изменить фенотип от тяжелой гиперурикемии, наблюдаемой у мужчин с синдромом Леша–Нихена (OMIM 300322), до подагры. У гетерозигот, несущих мутацию гена HPRT1, редко проявляются какие-либо признаки синдрома, включая подагру. В большинстве тканей этих женщин, кроме клеток крови, продукт реакции HPRT, инозиновая кислота, переносится из клеток, синтезирующих нормальный фермент, в клетки с дефицитом фермента благодаря щелевым контактам [54]. Недостаток инозиновой кислоты замедляет скорость деления клеток; нормальные клетки (поскольку вариант находится на их неактивной X) в конечном итоге заменяют дефектные [55]. Как следствие, гетерозиготные матери и сестры мужчин с синдромом Леша–Нихена имеют только нормальные клетки крови, а в других тканях мутантные клетки разделяют генные продукты, обеспечиваемые нормальными клетками. Совместное использование клетками белковых продуктов также происходит у женщин, гетерозиготных по вариантам, вызывающим Х-сцепленные лизосомные заболевания. Лизосомы содержат множество ферментов, расщепляющих белки и липиды. Варианты генов, кодирующих эти ферменты, вызывают заболевания из-за накопления нерасщепленного материала в лизосомах пораженных людей. Клетки, имеющие дефицит этих ферментов у гетерозигот, могут поглощать фермент, секретируемый нормальными клетками, посредством эндоцитоза. Следовательно, потенциальные проявления у носителей мутантных вариантов лизосомных ферментов, кодируемых Х-хромосомой, обычно блокируются за счет передачи этих ферментов от клеток, которые могут их производить, к тем, которые не могут [56].
Таблица 1.
Особенности проявления Х-сцепленных заболеваний у мужчин и женщин
| Ген | Х-сцепленное заболевание | Тип наследования | Проявление у мужчин | Проявление у женщин | sXCI | Ссылка |
|---|---|---|---|---|---|---|
| ABCD1 | Адренолейкодистрофия/ адреномиелоневропатия | Рецессивный | Смерть в первом десятилетии/ прогрессирующая скованность и слабость в ногах, развитие когнитивных и поведенческих расстройств, начиная со 2-го десятилетия | Здоровые носители, адреномиелоневропатия с поздним началом | Есть | [80] |
| AIFM1 | Спондилоэпиметафизарная дисплазия, Х-сцепленная, с гипомиелинизирующей лейкодистрофией | Рецессивный | Гипомиелинизирующая лейкодистрофия | Здоровые носители | Есть | [81] |
| ALG13 | Возрастная и эпилептическая энцефалопатия 36 | Эпилептическая энцефалопатия с ранним началом, тяжелая умственная отсталость | Развивающаяся и эпилептическая энцефалопатия-36; здоровые носители | Нет | [82] | |
| ATRX | Альфа-талассемия/УО | Доминантный | Тяжелая форма УО и дисморфические особенности | Мягкая форма УО | Нет | [83] |
| CASK | Умственная отсталость и микроцефалия с гипоплазией моста и мозжечка | Доминантный | УО, микроцефалия, мостик, гипоплазия мозжечка | Здоровые носители, РАС | Есть/ нет | [84] |
| CDKL5 | Ранняя детская эпилептическая энцефалопатия, ранняя смерть | Доминантный | Более мягкий фенотип, эпилепсия и глубокая идентификация | Тяжелая форма УО, эпилепсия с ранним началом, микроцефалия, менее тяжелая форма | Есть | [85] |
| DLG3 | Умственная отсталость, X-сцепленная 90 | Рецессивный | Средняя, серьезная степень УО | Пораженные/непораженные | Есть/нет | [86] |
| FMR1 | Синдром ломкой Х-хромосомы | Доминантный | УО | Слабая степень УО | Нет | [87] |
| IQSEC2 | Умственная отсталость, X-сцепленная 1/78 | Доминантный | Несиндромальная УО, эпилепсия | Некоторые трудности обучения, слабая форма УО с эпилепсией | Избегает XCI | [88] |
В общем можно идентифицировать четыре паттерна Х-инактивации у гетерозигот для Х‑сцепленных заболеваний: 1) случайная инактивация обычно связана с нормальным фенотипом; 2) случайная инактивация приводит к проявлениям заболевания, а нормальный фенотип проявляется только в случае смещения инактивации Х-хромосомы (sXCI) в пользу нормального аллеля в экспрессирующей ткани; 3) всегда sXCI, поскольку мутантные клетки гибнут или не развиваются, или мигрируют к месту назначения; 4) постепенное sXCI из-за пролиферативного преимущества клеток дикого типа (или мутантных) в экспрессирующей ткани (рис. 2).
Рис. 2.
Пути формирования различного характера инактивации Х-хромосомы и ее влияние на проявление клинического фенотипа гетерозиготных носительниц.
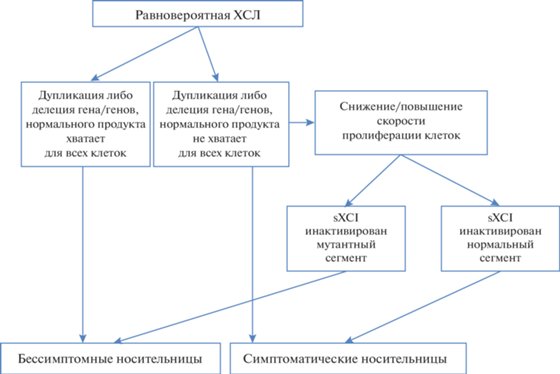
Смещение инактивации Х-хромосомы часто связано с патологическим фенотипом у человека и может возникать по различным причинам, но одной из наиболее вероятных являются летальные мутации или микроструктурные аберрации Х-хромосомы. Plenge et al. проанализировали характер инактивации при 20 различных формах XLID и обнаружили, что sXCI была характерна для ~50% семей [57]. Причем в случаях, когда sXCI составляет 80%, у женщины-носительницы не наблюдается даже минимальных клинических признаков заболевания. Авторы этого исследования сделали заключение, что мутации XLID представляют собой уникальный класс X-сцепленных мутаций, характеризующихся общим дефектом жизнеспособности или пролиферации клеток. Соответственно гены, связанные с развитием XLID как класс, могут влиять на жизнеспособность или пролиферацию клеток in vivo во многих типах тканей. В качестве примера можно привести описанный ранее ген ATRX, мутации которого ответственны за развитие нескольких форм XLID. Продукт этого гена участвует в ремоделировании хроматина, а значит и в регуляции транскрипции, пролиферации и развитии клеток [58]. В том случае, когда продукт Х-сцепленного мутантного гена не влияет на пролиферацию, либо жизнеспособность клеток, инактивация Х‑хромосомы у носительниц имеет стохастический характер и клинические признаки заболевания проявляются слабо [59].
Кроме того, на клинические проявления Х‑сцепленных заболеваний у женщин влияют еще несколько факторов, таких как избегание Х-инактивации и тип наследования мутантного варианта. Около 15% генов на Х-хромосоме избегают инактивации и экспрессируются как в активных, так и в неактивных хромосомах [OMIM. X-linked diseases. 2022. https://www.ncbi.nlm.nih.gov/omim/?term=X+ linked+diseases]. В табл. 1 показаны проявления клинического фенотипа некоторых Х-сцепленных заболеваний у мужчин и женщин, тип наследования мутантного аллеля и характер инактивации Х‑хромосомы при этих заболеваниях у гетерозигот. В случае доминантного наследования мутации вариант может оказаться настолько летальным, что большинство мужчин с тяжелым дефицитом продукта гена умирают внутриутробно. В этом случае выживают только женщины-носительницы мутации или мозаичные мужчины (синдром Клайнфельтера). Заболевания, сцепленные с Х-хромосомой, такие как пигментное недержание мочи или орофациальный дигитальный синдром типа 1, встречаются только у женщин или мозаичных мужчин [59]. Еще одним из классических примеров являются мутации гена MECP2. Известно, что этот ген преимущественно экспрессируется в головном мозге, где MeCP2 связывается с метилированной ДНК и действует как репрессор транскрипции [60]. MECP2 важен для пренатального нейрогенеза, постнатального развития синаптических связей и функций, синаптической пластичности и нервной функции взрослых [61]. Мутации или изменения копийности MECP2 могут приводить к синдрому дупликации MECP2, синдрому Ретта, тяжелой форме Х-сцепленной УО у мужчин или РАС. Хотя на тяжесть заболевания и изменчивость фенотипа влияют как локализация и тип мутации, так и генетический фон, показано, что пациенты с одним и тем же патогенным вариантом имеют клинические проявления, варьирующие от РАС, УО до синдрома Ретта, в связи с чем было предложено, что XCI является важным фактором для инициации заболевания и степени тяжести его течения. Zhang et al. описали китайскую семью с синдромом Ретта и тяжелой формой Х-сцепленной УО [62]. Они сообщили о восьми индивидах с вариантами MECP2 в шести семьях. Семья, состоящая из матери, дочери и сына, имела идентичный вариант MECP2 c.397C>T. У дочери был диагностирован сохраненный речевой вариант RTT, у сына – Х-сцепленная УО, а мать была здорова. Исследования XCI показали, что у матери было смещение инактивации Х в сторону нормального аллеля, а у дочери была случайная XCI. В другой семье у матери и дочери был обнаружен тот же вариант MECP2 c.397C>T. Однако, хотя у них обеих была случайная XCI, дочери был поставлен диагноз RTT, тогда как у матери были трудности с обучением в детстве и аутистическое поведение [62].
Одной из основных причин смещения инактивации Х-хромосомы является селекция клеток с одним из аллельных вариантов Х-сцепленного гена. Скорость потери дефицитных по продукту гена клеток может быть медленной или быстрой в зависимости от степени невыгодности варианта. Когда отбор интенсивен, т.е. когда он сильно неблагоприятен для клеток, экспрессирующих вариант, гетерозиготные организмы женского пола выигрывают, поскольку они быстро теряют все свои мутантные клетки. Иногда потеря вариантных клеток происходит так рано, что аномальные клетки никогда не могут проникнуть в ткань. В случаях иммунодефицитов, таких как синдром Вискотта–Олдрича (OMIM 301000), нарушение роста проявляется немедленно; мутантные предшественники В-клеток никогда не покидают костный мозг. К сожалению, иногда селективное преимущество имеет клетка с мутантным аллелем. По еще невыясненным причинам гетерозиготы с вариантом, вызывающим адренолейкодистрофию, медленно теряют свою популяцию клеток дикого типа [63]. Поэтому с возрастом у них обычно проявляются некоторые симптомы болезни.
Вероятно, CNV также могут быть одной из причин формирования смещения инактивации Х-хромосомы, особенно в тех случаях, когда в них задействованы гены, контролирующие процессы деления клетки и поддержания клеточного гомеостаза. Так, например, в случае делеции Xq24 с потерей генов SLC25A43, SLC25A5-AS1, SLC25A5, CXorf56, UBE2A, NKRF, SEPT6 и MIR766 у облигатных носительниц делеции наблюдалось экстремальное sXCI (от 98 до 100%) и инактивированной, вероятнее всего, была мутантная Х-хромосома [38].
Дупликация гена MECP2 приводит к фенотипу с приобретением функции, который наследуется по рецессивному типу, преимущественно поражает мужчин и характеризуется УО от тяжелой до глубокой степени и ограниченной или отсутствующей речью. Х-хромосома, несущая дупликацию, часто подавляется преимущественно у большинства бессимптомных носительниц, однако некоторые из них имеют мягкий фенотип, несмотря на инактивацию мутантной хромосомы. Симптоматические женщины со случайной XCI или смещением XCI с предпочтительной экспрессией дуплицированной хромосомы могут иметь различную степень тяжести и могут демонстрировать трудности в обучении в детстве, умственную отсталость, аутистические черты или психические симптомы [52].
В своем исследовании Di-Battista et al. выявили, что в случаях дупликации региона Xp11.23-p11.22, проявляющейся серьезными фенотипическими нарушениями, у больных гетерозигот наблюдается экстремально смещенная инактивация Х-хромосомы, причем активной остается именно мутантная Х-хромосома [64]. Авторы предположили, что увеличение дозы некоторых протоонкогенов (SSXP6, SSXP7, ERAS, OTUD5, WDR13, SUV39H1, PIM2, PRAF2, HDAC6 и WDR45), локализованных в регионе дупликации, дает селективное преимущество именно тому пулу клеток, который содержит активную Х-хромосому с дупликацией.
Среди заболеваний с Х-сцепленным наследованием и УО дупликация области Xp11.23p11.22 действительно является редким явлением, в литературе известно менее 90 случаев. Большинство из них были распознаны при рутинном применении методов aCGH, поскольку эти вариации числа копий (CNV) сильно различаются по размеру. Дупликации Xp11.22p11.23 выявлены у представителей обоих полов [44]. По мнению исследователей, при дупликации Xp11.22p11.23 причиной аномального фенотипа является функциональная дисомия генов, затронутых перестройкой [64]. Это кажется очевидным в случае пораженных мужчин; однако определение числа реально функционирующих копий у пациенток затруднено из-за Х-инактивации. Не во всех опубликованных отчетах приводятся данные о Х-инактивации, но примерно в половине исследованных случаев была обнаружена асимметричная Х-инактивация с преимущественной инактивацией нормальной Х-хромосомы в большинстве клеток. В случае дупликаций Xp11.23p11.22 все наоборот: у женщин с асимметричной Х-инактивацией Х-хромосома, несущая аллель дикого типа, замолкает, а аномальная активна в большинстве клеток. Следовательно, у женщин со случайной Х-инактивацией развивается более мягкий фенотип [44].
Определение характера смещения инактивации Х-хромосомы у гетерозиготных носительниц Х-сцепленных мутаций является важным диагностическим критерием, который необходимо использовать в генетическом консультировании семей, имеющих Х-сцепленные мутации.
ПРОБЛЕМЫ ДИАГНОСТИКИ И ИНТЕРПРЕТАЦИИ РЕЗУЛЬТАТОВ aCGH У ПАЦИЕНТОВ С Х-СЦЕПЛЕННОЙ УМСТВЕННОЙ ОТСТАЛОСТЬЮ
Использование сравнительной геномной гибридизации (CGH) резко изменило подход к идентификации генетических изменений, которые могут объяснить УО и/или врожденные аномалии. В первую очередь при анализе генного состава Х‑сцепленной CNV необходимо выявить кандидатные гены, ассоциированные с XLID. Для корректной интерпретации клинической значимости Х-сцепленных субмикроскопических перестроек следует обращать внимание на особенности их сегрегации по материнской линии. Кроме того, нельзя забывать о необходимости анализа статуса инактивации Х-хромосомы у гетерозиготных носительниц Х-сцепленной перестройки. Чаще всего у носительниц либо совсем не проявляются, либо проявляются минимальные фенотипические особенности, присущие конкретной хромосомной мутации из-за случайной или асимметричной XCI [28]. Но в некоторых случаях именно симптоматические носительницы имеют sXCI [59]. Поэтому анализ характера инактивации Х-хромосомы рекомендуется также проводить всем пациенткам с УО, имеющим Х-сцепленные CNV.
В отличие от делеций адекватная интерпретация клинической значимости дупликаций на Х‑хромосоме не всегда однозначна. Локализация в области дупликации генов, ассоциированных с XLID, не всегда означает, что дупликация является патогенетически значимой. В качестве примера можно рассмотреть несколько перестроек, интерпретация клинической значимости которых до настоящего времени остается проблематичной.
Субмикроскопическая дупликация региона Xp22.31 включает ген стероидсульфатазы (STS, OMIM 300747) (таблица в Приложении). Ген STS кодирует стероидную сульфатазу и дефицит транскрипта этого гена приводит к развитию Х-сцепленного ихтиоза. Делеция этого гена и соседних областей обнаруживается у 90% пораженных людей и происходит из-за рекомбинации между низкокопийными повторами (LCR), расположенными рядом с генами VCX, которые фланкируют STS [65]. Некоторые пациенты с делецией также имеют УО и, кроме того, показана связь между дефицитом транскрипции STS и предрасположенностью к синдрому дефицита внимания и гиперактивности, аутизму и дефициту социальной коммуникации. Патогенетическая роль дупликации этого региона до настоящего времени не подтверждена. Первоначальные исследования на больших группах индивидов выявили, что частота дупликации составляет 0.15% в здоровой контрольной популяции и примерно 0.41% в когорте людей с аномальными фенотипами, включающими УО [66, 67]. Размер дупликации Xp22.31 варьирует от 149 тпн до 1.74 млн пн. Соотношение мужчин и женщин, несущих эту перестройку, составляет 0.7. У всех пациентов наблюдалась задержка интеллектуального развития и трудности с обучением в детстве, у 4/9 наблюдалась судорожная активность, у 3/9 была косолапость, 2/9 испытывали трудности с кормлением, 2/9 имели в анамнезе РАС и у 2/9 наблюдалась гипотония [65]. Инактивация Х‑хромосомы у симптоматичных и бессимптомных носительниц дупликации была и равновероятной, и асимметричной, но у 80% бессимптомных гетерозигот наблюдалась асимметричная XCI с инактивацией мутантного аллеля [66]. Однако в недавнем исследовании [68], в котором оценивались множественные показатели физического и психического здоровья, когнитивных функций и нейроанатомии у мужчин (n = 414) и женщин (n = = 938) носителей 0.8–2.5 млн пн дупликаций, имеющих в своем составе ген STS, а также мужчин (n = 192 826) и женщин (n = 227 235) из Британского биобанка (UK Biobank), не подтвердили выводы более ранних исследований. Авторы этого исследования сделали выводы, что у мужчин-носителей дупликации наблюдается более высокая распространенность биполярного расстройства, снижение некоторых показателей, связанных с депрессией, и повышенное настроение. Когнитивные функции и успеваемость не различались между группами сравнения. Нейроанатомический анализ показал больший объем бокового желудочка и коры головного мозга у носителей дупликации [68]. Определение клинической значимости дупликации в регионе Xp22.31 является очень важным, так как эта дупликация часто встречается у пациентов с интеллектуальными расстройствами (1/470 мужчин и 1/240 женщин).
Еще одна дупликация, имеющая спорную патогенетическую значимость, – это внутригенная дупликация в гене TSPAN7 (OMIM 300096), локализованном в регионе Xq11.2. Ген TSPAN7 высоко экспрессируется в головном мозге, и некоторые генные мутации TSPAN7 связаны с формированием Х-сцепленной формы интеллектуальной недостаточности (XLID58; OMIM 300210). Связь моногенной микродупликации, затрагивающей ген TSPAN7, с патогенезом УО до сих пор остается неясной. Ранее Noor et al. показали, что внутригенная дупликация TSPAN7 у пациента с аутизмом не приводит к изменению последовательности кДНК и соответственно не может быть причиной этого заболевания [69]. Кроме того, микродупликации гена были выявлены у некоторых здоровых индивидов [70]. Вместе с тем накопился определенный массив данных в пользу того, что эта дупликация является патогенетически значимой. Были проанализированы клинические фенотипы 29 пациентов базы данных DECIPHER [https://www.deciphergenomics.org], двух пациентов из нашего исследования и двух братьев с такой же дупликацией на Х‑хромосоме, описанных в литературе ранее [71, 72]. Суммарные нарушения интеллектуальной сферы, включающие УО, задержку психического развития, задержку развития речи и расстройства аутистического спектра, наблюдались практически у 100% пациентов, мышечная гипотония в 15% случаев. Также были зарегистрированы поражения головного мозга (гетеротопия серого вещества, атрофия мозжечка, энцефалопатии), патология слуха, аномалии кожи, гипермобильность суставов, ожирение. Кроме того, у 20% гемизиготных носителей дупликации встречались такие дисморфические особенности как аномальные формы черепа (микроцефалия, брахицефалия, долихоцефалия), приросшая мочка уха, гипертелоризм, недоразвитые крылья носа, короткий сглаженный фильтр, высокое нёбо, тремы зубов.
Фенотип носителя субмикроскопической хромосомной перестройки может быть вариабельным из-за ряда генетических механизмов, таких как неполная пенетрантность, вариабельная экспрессивность и асимметричная Х-инактивация. Доброкачественный полиморфный вариант также может вести себя по-разному в разных популяциях или с разным геномным фоном и может вызывать патологический фенотип при различных условиях.
ЗАКЛЮЧЕНИЕ
Уникальность Х-хромосомы состоит в большом количестве локализованных на ней генов, связанных с развитием и функционированием мозга, особенностями передачи мутаций по материнской линии, гемизиготным состоянием мутаций у мужчин и модифицирующим влиянием Х-инактивации у гетерозигот. Все это делает Х-сцепленные CNV особенными. Было установлено, что субмикроскопические изменения числа копий ДНК на Х‑хромосоме могут объяснить около 5% идиопатических случаев XLID [28]. Высокая частота мутаций на Х-хромосоме в генах, ассоциированных с УО, подразумевает необходимость выявления максимального числа носителей мутаций в семьях для проведения инвазивной пренатальной диагностики и преимплантационного генетического тестирования. Однако для повышения эффективности молекулярно-генетической диагностики у пациентов с Х-сцепленными интеллектуальными расстройствами недостаточно использование только сравнительной матричной геномной гибридизации или массового параллельного секвенирования. Важно обращать внимание и на характер эпигенетической инактивации Х-хромосомы.
Исследование выполнено за счет гранта Российского научного фонда № 21-65-00017, https://rscf.ru/project/21-65-00017/.
Настоящая статья не содержит каких-либо исследований с использованием в качестве объекта животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Leonard H., Wen X. The epidemiology of mental retardation: Challenges and opportunities in the new millennium // Ment. Retard. Dev. Disabil. Res. Rev. 2002. V. 8. P. 117–134. https://doi.org/10.1002/mrdd.10031
Penrose L.S. A Clinical and Genetic Study of 1,280 Cases of Mental Defect. Issued by the Medical Research Council. London, 1938. 160 p.
Lehrke R.G. X-linked mental retardation and verbal disability // Birth Defects Orig. Artic. Ser. 1974. V. 10. P. 1–100.
Herbst D.S., Miller J.R. Nonspecific X-linked mental retardation II: The frequency in British Columbia // Am. J. Hum. Genet. 1980. V. 7. P. 461–469. https://doi.org/10.1002/ajmg.1320070407
Ross M.T., Grafham D.V., Coffey A.J. et al. The DNA sequence of the human X chromosome // Nature. 2005. V. 434. P. 325–337. https://doi.org/10.1038/nature03440
Gécz J., Shoubridge C., Corbett M. The genetic landscape of intellectual disability arising from chromosome X // Trends in Genetics. 2009. V. 25. № 7. P. 308–316. https://doi.org/10.1016/j.tig.2009.05.002
Stevenson R., Schwartz C., Rogers C. Atlas of X-Linked Intellectual Disability Syndromes. N.Y.: Oxford Press, 2012. 368 p.
XLID Genetic Research – Greenwood Genetic Center / SC. Greenwood Genetic Center. https://www.ggc.org/xlid-genetic-research. Publ. 2022. Accessed April 1, 2022.
Coffee B., Keith K., Albizua I. et al. Incidence of Fragile X syndrome by newborn screening for methylated FMR1 DNA // Am. J. Hum. Genet. 2009. V. 85. № 4. P. 503–514. https://doi.org/10.1016/j.ajhg.2009.09.007
Воинова В.Ю., Ворсанова С.Г., Юров Ю.Б., Юров И.Ю. Алгоритм диагностики Х-сцепленных форм умственной отсталости у детей // Рос. вестник перинатологии и педиатрии. 2016. Т. 61. С. 34–40. https://doi.org/10.21508/1027-4065-2016-61-5-34-41
Lisik M.Z. Health problems in females carriers of premutation in the FMR1 gene // Psychiatr. Pol. 2017. V. 51. № 5. P. 899–907. https://doi.org/10.12740/PP/65778
Chiurazzi P., Tabolacci E., Neri G. X-linked mental retardation (XLMR): From clinical conditions to cloned genes // Crit. Rev. Clin. Lab. Sci. 2004. V. 41. № 2. P. 117–158. https://doi.org/10.1080/10408360490443013
Wilson G., Richards C., Katz K., Brookshire G. Non-specific X linked mental retardation with aphasia exhibiting genetic linkage to chromosomal region Xp11 // J. Med. Genet. 1992. V. 29. № 9. P. 629–634. https://doi.org/10.1136/jmg.29.9.629
Wraith J., Scarpa M., Beck M. et al. Mucopolysaccharidosis type II (Hunter syndrome): A clinical review and recommendations for treatment in the era of enzyme replacement therapy // Eur. J. Pediatr. 2007. V. 167. № 3. P. 267–277. https://doi.org/10.1007/s00431-007-0635-4
Соловьёва Е.В., Минайчева Л.И., Склеймова М.М. и др. Преимплантационное генетическое тестирование синдрома Хантера: клинический случай // Мед. генетика. 2021. Т. 20. № 9(230). С. 34–44. https://doi.org/10.25557/2073-7998.2021.09.34-44
Moog U., Smeets E.E., van Roozendaal K.E.P. et al. Neurodevelopmental disorders in males related to the gene causing Rett syndrome in females (MECP2) // Eur. J. Paediatric Neurology. 2003. V. 7. № 1. P. 5–12. https://doi.org/10.1016/S1090-3798(02)00134-4
Villard L. MECP2 mutations in males // J. Med. Genet. 2007. V. 44. № 7. P. 417–423. https://doi.org/10.1136/jmg.2007.049452
Gécz J., Mulley J. Genes for cognitive function: developments on the X // Genome Res. 2000. V. 10. № 2. P. 157–163. https://doi.org/10.1101/gr.10.2.157
Chiurazzi P., Pirozzi F. Advances in understanding – genetic basis of intellectual disability // F1000Res. 2016. V. 5. P. 599. https://doi.org/10.12688/f1000research.7134.1
Piton A., Redin C., Mandel J. XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing // Am. J. Hum. Genet. 2013. V. 93. № 2. P. 368–383. https://doi.org/10.1016/j.ajhg.2013.06.013
Kato M., Saitoh S., Kamei A. et al. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome) // Am. J. Hum. Genet. 2007. V. 81. № 2. P. 361–366. https://doi.org/10.1086/518903
Wallerstein R., Sugalski R., Cohn L. et al. Expansion of the ARX spectrum // Clin. Neurol. Neurosurg. 2008. V. 110. № 6. P. 631–634. https://doi.org/10.1016/j.clineuro.2008.03.007
Schwartz C.E. X-linked intellectual disability genetics // eLS. Chichester: John Wiley & Sons, Ltd., 2015.
Bassani S., Zapata J., Gerosa L. et al. The neurobiology of X-linked intellectual disability // The Neuroscientist. 2013. V. 19. № 5. P. 541–552. https://doi.org/10.1177/1073858413493972
Pavlowsky A., Gianfelice A., Pallotto M. et al. A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation // Current Biology. 2010. V. 20. № 2. P. 103–115. https://doi.org/10.1016/j.cub.2009.12.030
Bassani S., Cingolani L.A., Valnegri P. et al. The X‑linked intellectual disability protein TSPAN7 regulates excitatory synapse development and AMPAR trafficking // Neuron. 2012. V. 73. № 6. P. 1143–1158. https://doi.org/10.1016/j.neuron.2012.01.021
Bukalo O., Dityatev A. Synaptic cell adhesion molecules // Adv. Exp. Med. Biol. 2012. V. 970. P. 97–128. https://doi.org/10.1007/978-3-7091-0932-8_5
Bauters M., Weuts A., Vandewalle J. et al. Detection and validation of copy number variation in X-linked mental retardation // Cytogenet. Genome Res. 2008. V. 123. № 1–4. P. 44–53. https://doi.org/10.1159/000184691
Froyen G., Van Esch H., Bauters M. et al. Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: Important role for increased gene dosage of XLMR genes // Hum. Mutat. 2007. V. 28. P. 1034–1042. https://doi.org/10.1002/humu.20564
Whibley A., Plagnol V., Tarpey P. et al. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability // Am. J. Hum. Genet. 2010. V. 87. № 2. P. 173–188. https://doi.org/10.1016/j.ajhg.2010.06.017
Piton A., Michaud J., Peng H. et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism // Hum. Mol. Genet. 2008. V. 17. № 24. P. 3965–3974. https://doi.org/10.1093/hmg/ddn300
Franek K.J., Butler J., Johnson J. et al. Deletion of the immunoglobulin domain of IL1RAPL1 results in nonsyndromic X-linked intellectual disability associated with behavioral problems and mild dysmorphism // Am. J. Med. Genet. Part A. 2011. V. 155. № 5. P. 1109–1114. https://doi.org/10.1002/ajmg.a.33833
Froyen G., Corbett M., Vandewalle J. et al. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation // Am. J. Hum. Genet. 2008. V. 82. № 2. P. 432–443. https://doi.org/10.1016/j.ajhg.2007.11.002
López M., Pérez-Grijalba V., García-Cobaleda I., Domínguez-Garrido E. A 22.5 kb deletion in CUL4B causing Cabezas syndrome identified using CNV approach from WES data // Clin. Case Rep. 2020. V. 8. № 12. P. 3183–3187. https://doi.org/10.1002/ccr3.3381
Nascimento R., Otto P., de Brouwer A., Vianna-Morgante A. UBE2A, which encodes a ubiquitin-conjugating enzyme, is mutated in a novel X-linked mental retardation syndrome // Am. J. Hum. Genet. 2006. V. 79. № 3. P. 549–555. https://doi.org/10.1086/507047
Honda S., Orii K., Kobayashi J. et al. Novel deletion at Xq24 including the UBE2A gene in a patient with X‑linked mental retardation // J. Hum. Genet. 2010. V. 55. № 4. P. 244–247. https://doi.org/10.1038/jhg.2010.14
Thunstrom S., Sodermark L., Ivarsson L. et al. UBE2A deficiency syndrome: A report of two unrelated cases with large Xq24 deletions encompassing UBE2A gene // Am. J. Med. Genet. Part A. 2014. V. 167. № 1. P. 204–210. https://doi.org/10.1002/ajmg.a.36800
Tolmacheva E., Kashevarova A., Nazarenko L. et al. Delineation of Clinical manifestations of the inherited Xq24 microdeletion segregating with sXCI in mothers: Two novel cases with distinct phenotypes ranging from UBE2A deficiency syndrome to recurrent pregnancy loss // Cytogenet. Genome Res. 2020. V. 160. № 5. P. 245–254. https://doi.org/10.1159/000508050
Verkerk A., Zeidler S., Breedveld G. et al. CXorf56, a dendritic neuronal protein, identified as a new candidate gene for X-linked intellectual disability // Eur. J. Hum. Genet. 2018. V. 26. № 4. P. 552–560. https://doi.org/10.1038/s41431-017-0051-9
Neri G., Schwartz C., Lubs H., Stevenson R. X-linked intellectual disability update 2017 // Am. J. Med. Genet. Part A. 2018. V. 176. № 6. P. 1375–1388. https://doi.org/10.1002/ajmg.a.38710
van Asbeck E., Ramalingam A., Dvorak C. et al. Duplication at Xq28 involving IKBKG is associated with progressive macrocephaly, recurrent infections, ectodermal dysplasia, benign tumors, and neuropathy // Clin. Dysmorphol. 2014. V. 23. № 3. P. 77–82. https://doi.org/10.1097/mcd.0000000000000038
Popovici C., Busa T., Boute O. et al. Whole ARX gene duplication is compatible with normal intellectual development // Am. J. Med. Genet. Part A. 2014. V. 164. № 9. P. 2324–2327. https://doi.org/10.1002/ajmg.a.36564
Maurin M., Arfeuille C., Sonigo P. et al. Large duplications can be benign copy number variants: A case of a 3.6-mb Xq21.33 duplication // Cytogenet. Genome Res. 2017. V. 151. № 3. P. 115–118. https://doi.org/10.1159/000460278
Czakó M., Till Á., Zima J. et al. Xp11.2 duplication in females: Unique features of a rare copy number variation // Front Genet. 2021. V. 12. https://doi.org/10.3389/fgene.2021.635458
Froyen G., Belet S., Martinez F. et al. Copy-number gains of HUWE1 due to replication- and recombination-based rearrangements // Am. J. Hum. Genet. 2012. V. 91. № 2. P. 252–264. https://doi.org/10.1016/j.ajhg.2012.06.010
Turner G., Gedeon A., Mulley J. X-linked mental retardation with heterozygous expression and macrocephaly: pericentromeric gene localization // Am. J. Med. Genet. 1994. V. 51. № 4. P. 575–580. https://doi.org/10.1002/ajmg.1320510456
Regis S., Grossi S., Corsolini F. et al. PLP1 gene duplication causes overexpression and alteration of the PLP/DM20 splicing balance in fibroblasts from Pelizaeus–Merzbacher disease patients // Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease. 2009. V. 1792. № 6. P. 548–554. https://doi.org/10.1016/j.bbadis.2009.04.002
Kumar R., Corbett M.A., Van Bon B.W. et al. Increased STAG2 dosage defines a novel cohesinopathy with intellectual disability and behavioral problems // Hum. Mol. Genet. 2015. V. 24. P. 7171–7181. https://doi.org/10.1093/hmg/ddv414
Møller R., Jensen L., Maas S. et al. X-linked congenital ptosis and associated intellectual disability, short stature, microcephaly, cleft palate, digital and genital abnormalities define novel Xq25q26 duplication syndrome // Hum. Genet. 2013. V. 133. № 5. P. 625–638. https://doi.org/10.1007/s00439-013-1403-3
Schroer R.J., Beaudet A.L., Shinawi M. et al. Duplication of OCRL and adjacent genes associated with autism but not Lowe syndrome // Am. J. Med. Genet. Part A. 2012. V. 158A. P. 2602–2605. https://doi.org/10.1002/ajmg.a.35566
Vandewalle J., Van Esch H., Govaerts K. et al. Dosage-dependent severity of the phenotype in patients with mental retardation due to a recurrent copy-number gain at Xq28 mediated by an unusual recombination // Am. J. Hum. Genet. 2009. V. 85. № 6. P. 809–822. https://doi.org/10.1016/j.ajhg.2009.10.019
Brand B., Blesson A., Smith-Hicks C. The impact of X‑chromosome inactivation on phenotypic expression of X-linked neurodevelopmental disorders // Brain Sci. 2021. V. 11. № 7. P. 904. https://doi.org/10.3390/brainsci11070904
El-Hattab A., Bournat J., Eng P. et al. Microduplication of Xp11.23p11.3 with effects on cognition, behavior, and craniofacial development // Clin. Genet. 2010. V. 79. № 6. P. 531–538. https://doi.org/10.1111/j.1399-0004.2010.01496.x
Cox R., Krauss M., Balis M., Dancis J. Evidence for transfer of enzyme product as the basis of metabolic cooperation between tissue culture fibroblasts of Lesch-Nyhan disease and normal cells // PNAS. 1970. V. 67. № 3. P. 1573–1579. https://doi.org/10.1073/pnas.67.3.1573
Migeon B.R. Studies of skin fibroblasts from 10 families with HGPRT deficiency, with reference in X-chromosomal inactivation // Am. J. Hum. Genet. 1971. V. 23. № 2. P. 199.
Migeon B.R., Sprenkle J.A., Liebaers I. et al. X-linked Hunter syndrome: The heterozygous phenotype in cell culture // Am. J. Hum. Genet. 1977. V. 29. № 5. P. 448.
Plenge R., Stevenson R., Lubs H. et al. Skewed X-chromosome inactivation is a common feature of X-linked mental retardation disorders // Am. J. Hum. Genet. 2002. V. 71. № 1. P. 168–173. https://doi.org/10.1086/341123
Gibbons R., McDowell T., Raman S. et al. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation // Nat. Genet. 2000. V. 24. № 4. P. 368–371. https://doi.org/10.1038/74191
Migeon B. X-linked diseases: susceptible females // Genetics in Medicine. 2020. V. 22. № 7. P. 1156–1174. https://doi.org/10.1038/s41436-020-0779-4
Mellén M., Ayata P., Dewell S. et al. MeCP2 binds to 5hmc enriched within active genes and accessible chromatin in the nervous system // Cell. 2012. V. 151. № 7. P. 1417–1430. https://doi.org/10.1016/j.cell.2012.11.022
Srivastava S., Sahin M., Prock L. Translational medicine strategies in drug development for neurodevelopmental disorders // Handbook Behavioral Neurosci. 2019. V. 29. P. 309–331. https://doi.org/10.1016/B978-0-12-803161-2.00022-9
Zhang Q., Zhao Y., Bao X. et al. Familial cases and male cases with MECP2 mutations // Am. J. Med. Genet. Part B: Neuropsychiatric Genetics. 2017. V. 174. № 4. P. 451–457. https://doi.org/10.1002/ajmg.b.32534
Migeon B.R., Moser H.W., Moser A.B. et al. Adrenoleukodystrophy: Evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells // PNAS. 1981. V. 78. № 8. P. 5066–5070.
Di-Battista A., Meloni V., da Silva M. et al. Unusual X‑chromosome inactivation pattern in patients with Xp11.23-p11.22 duplication: report and review // Am. J. Med. Genet. Part A. 2016. V. 170. № 12. P. 3271–3275. https://doi.org/10.1002/ajmg.a.37888
Esplin E., Li B., Slavotinek A. et al. Nine patients with Xp22.31 microduplication, cognitive deficits, seizures, and talipes anomalies // Am. J. Med. Genet. Part A. 2014. V. 164. № 8. P. 2097–2103. https://doi.org/10.1002/ajmg.a.36598
Li F., Shen Y., Köhler U. et al. Interstitial microduplication of Xp22.31: Causative of intellectual disability or benign copy number variant // Eur. J. Med. Genet. 2010. V. 53. № 2. P. 93–99. https://doi.org/10.1016/j.ejmg.2010.01.004
Liu P., Erez A., Sreenath Nagamani S. et al. Copy number gain at Xp22.31 includes complex duplication rearrangements and recurrent triplications // Hum. Mol. Genet. 2011. V. 20. № 10. P. 1975–1988. https://doi.org/10.1093/hmg/ddr078
Gubb S., Brcic L., Underwood J. et al. Medical and neurobehavioural phenotypes in male and female carriers of Xp22.31 duplications in the UK Biobank // Hum. Mol. Genet. 2020. V. 29 № 17. P. 2872–2881. https://doi.org/10.1093/hmg/ddaa174
Noor A., Gianakopoulos P.J., Fernandez B. et al. Copy number variation analysis and sequencing of the X‑linked mental retardation gene TSPAN7/TM4SF2 in patients with autism spectrum disorder // Psychiatric. Genet. 2009. V. 19. № 3. P. 154–155. https://doi.org/10.1097/YPG.0b013e32832a4fe5
Cai G., Edelmann L., Goldsmith J.E. et al. Multiplex ligation-dependent probe amplification for genetic screening in autism spectrum disorders: Efficient identification of known microduplications and identification of a novel microduplication in ASMT // BMC Med. Genomics. 2008. V. 1. № 1. P. 1–14. https://doi.org/10.1186/1755-8794-1-50
Utine G.E., Kiper P.Ö., Alanay Y. et al. Searching for copy number changes in nonsyndromic X-linked intellectual disability // Mol. Syndromology. 2011. V. 2. № 2. P. 64–71. https://doi.org/10.1159/000334289
Толмачева Е.Н., Кашеварова А.А., Беляева Е.О. и др. Клинические эффекты моногенной дупликации Xp11.4 с вовлечением гена TSPAN7 // Мед. генетика. 2021. Т. 20. № 9. С. 45–47. https://doi.org/10.25557/2073-7998.2021.09.45-47
Isrie M., Froyen G., Devriendt K. et al. Sporadic male patients with intellectual disability: Contribution of X‑chromosome copy number variants // Eur. J. Med. Genet. 2012. V. 55. № 11. P. 577–585. https://doi.org/10.1016/j.ejmg.2012.05.005
Diociaiuti A., Angioni A., Pisaneschi E. et al. X-linked ichthyosis: Clinical and molecular findings in 35 Italian patients // Exp. Dermatol. 2018. V. 28. № 10. P. 1156–1163. https://doi.org/10.1111/exd.13667
Pavone P., Corsello G., Marino S. et al. Microcephaly/trigonocephaly, intellectual disability, autism spectrum disorder, and atypical dysmorphic features in a boy with Xp22. 31 duplication // Mol. Syndromology. 2018. V. 9. P. 253–258. https://doi.org/10.1159/000493174
Mignon-Ravix C., Cacciagli P., Choucair N. et al. Intragenic rearrangements in X-linked intellectual deficiency: Results of a-CGH in a series of 54 patients and identification of TRPC5 and KLHL15 as potential XLID genes // Am. J. Med. Genet. Part A. 2014. V. 164. № 8. P. 1991–1997. https://doi.org/10.1002/ajmg.a.36602
Grau C., Starkovich M., Azamian M. et al. Xp11.22 deletions encompassing CENPVL1, CENPVL2, MAGED1 and GSPT2 as a cause of syndromic X-linked intellectual disability // PLoS One. 2017. V. 12. № 4. P. e0175962. https://doi.org/10.1371/journal.pone.0175962
Shimojima K., Sugawara M., Shichiji M. et al. Loss-of-function mutation of collybistin is responsible for X‑linked mental retardation associated with epilepsy // J. Hum. Genet. 2011. V. 56. № 8. P. 561–565. https://doi.org/10.1038/jhg.2011.58
Ramocki M.B., Tavyev Y.J., Peters S.U. The MECP2 duplication syndrome // Am. J. Med. Genet. Part A. 2010. V. 152A. № 5. P. 1079–1088. https://doi.org/10.1002/ajmg.a.33184
Wang Z., Yan A., Lin Y. et al. Familial Skewed X chromosome inactivation in adrenoleukodystrophy manifesting heterozygotes from a Chinese pedigree // PLoS One. 2013. V. 8. № 3. P. e57977. https://doi.org/10.1371/journal.pone.0057977
Pandolfo M., Rai M., Remiche G. et al. Cerebellar ataxia, neuropathy, hearing loss, and intellectual disability due to AIFM1 mutation // Neurology Genet. 2020. V. 6. № 3. P.e420. https://doi.org/10.1212/nxg.0000000000000420
Hamici S., Bastaki F., Khalifa M. Exome sequence identified a c.320A>G ALG13 variant in a female with infantile epileptic encephalopathy with normal glycosylation and random X inactivation: review of the literature // Eur. J. Med. Genet. 2017. V. 60. № 10. P. 541–547. https://doi.org/10.1016/j.ejmg.2017.07.014
Wada T., Sugie H., Fukushima Y., Saitoh S. Non-skewed X-inactivation may cause mental retardation in a female carrier of X-linked α-thalassemia/mental retardation syndrome (ATR-X): X-inactivation study of nine female carriers of ATR-X // Am. J. Med. Genet. Part A. 2005. V. 138A. № 1. P. 18–20. https://doi.org/10.1002/ajmg.a.30901
Seto T., Hamazaki T., Nishigaki S. et al. A novel CASK mutation identified in siblings exhibiting developmental disorders with/without microcephaly // Intractable & Rare Diseases Res. 2017. V. 6. № 3. P. 177–182. https://doi.org/10.5582/irdr.2017.01031
Zhao Y., Zhang X., Bao X. et al. Clinical features and gene mutational spectrum of CDKL5-related diseases in a cohort of Chinese patients // BMC Med. Genet. 2014. V. 15. № 1. https://doi.org/10.1186/1471-2350-15-24
Gieldon L., Mackenroth L., Betcheva-Krajcir E. et al. Skewed X-inactivation in a family with DLG3-associated X-linked intellectual disability // Am. J. Med. Genet. Part A. 2017. V. 173. № 9. P. 2545–2550. https://doi.org/10.1002/ajmg.a.38348
Kirchgessner C., Warren S., Willard H. X inactivation of the FMR1 fragile X mental retardation gene // J. Med. Genet. 1995. V. 32. № 12. P. 925–929. https://doi.org/10.1136/jmg.32.12.925
Wayhelova M., Ryzí M., Oppelt J. et al. Novel familial IQSEC2 pathogenic sequence variant associated with neurodevelopmental disorders and epilepsy // Neurogenetics. 2020. V. 21. № 4. P. 269–278. https://doi.org/10.1007/s10048-020-00616-3
Дополнительные материалы
- скачать ESM.docx
- Таблица. CNV, ассоциированные с Х-сцепленными формами умственной отсталости