

Генетика, 2022, T. 58, № 4, стр. 483-488
Клиническая и молекулярно-генетическая характеристика пациентов с глазокожным альбинизмом 1-го типа
С. А. Ионова 1, *, В. В. Кадышев 1, Н. В. Журкова 1, А. В. Марахонов 1, Р. А. Зинченко 1, 2
1 Медико-генетический научный центр им. академика Н.П. Бочкова
115522 Москва, Россия
2 Национальный научно-исследовательский институт общественного здоровья
им. Н.А. Семашко
105064 Москва, Россия
* E-mail: sofya.aydarovna.g@gmail.com
Поступила в редакцию 25.11.2021
После доработки 02.12.2021
Принята к публикации 06.12.2021
- EDN: BFODWQ
- DOI: 10.31857/S0016675822040051
Аннотация
Альбинизм – гетерогенная группа генетически обусловленных заболеваний, связанных с полным или частичным нарушением синтеза меланина. Выделяют различные формы наследственного альбинизма, включающие глазокожный альбинизм, глазной альбинизм и различные синдромальные формы. В работе проанализирована выборка пациентов (n = 90) с различными формами альбинизма на наличие патогенных вариантов нуклеотидной последовательности в гене TYR как наиболее распространенной причины глазокожного альбинизма 1-го типа. Данная клинико-генетическая форма выявлена у 32 пациентов. На основании полученных результатов изучены спектр и частота вариантов в гене TYR. Описаны клинические признаки, которые чаще всего встречаются среди пациентов, а также нетипичные для данного заболевания клинические особенности.
Изучение причин возникновения наследственных заболеваний и дальнейшее их описание – ведущее направление медицинской генетики. Несомненный интерес для врачей представляет альбинизм как гетерогенная группа генетически обусловленных заболеваний, связанных с полным или частичным нарушением синтеза меланина. Выделяют различные формы наследственного альбинизма, включающие глазокожный альбинизм, глазной альбинизм и различные синдромальные формы, которые представляют интерес для врачей разных специальностей. Клиническая картина и генетические причины альбинизма различаются и определяют поражение многих органов и систем, однако основным симптомом при всех формах заболевания являются гипопигментация кожи, волос и изменения со стороны зрительного аппарата. Наиболее часто встречающейся формой альбинизма является глазокожный альбинизм (ГКА) – аутосомно-рецессивное заболевание, поражающее в разной степени все пигментированные ткани организма. По литературным данным, распространенность ГКА составляет в среднем 1 на 17 000 человек (1 на 16 949 – по данным Orphanet) [1, www.orpha.net]. На сегодняшний день выделяют 12 клинико-генетических типов ГКА (фенотипическая серия OMIM PS203100 [http://omim.org]), которые ассоциированы с патогенными вариантами нуклеотидной последовательности восьми различных генов: TYR (OMIM #203100), OCA2 (OMIM #203200), TYRP1 (OMIM #203290), SLC45A2 (OMIM #606574), OCA5 (OMIM #615312), SLC24A5 (OMIM #113750), LRMDA (OMIM #615179), DCT (OMIM #619165). Наиболее распространенным типом ГКА в европейской популяции с частотой 1 на 40 000 (1/40 000) населения является первый тип (ГКА 1-го типа) [2]. Выделяют два подтипа – IA (OMIM #203100) и IB (OMIM #606952). Они связаны с полным или частичным отсутствием активности фермента тирозиназы соответственно.
Цель настоящего исследования – анализ частоты встречаемости патогенных вариантов нуклеотидной последовательности в гене TYR у пациентов с глазокожным альбинизмом 1-го типа.
От всех обследованных больных и их родителей (или законных представителей) получено информированное согласие на участие в исследовании. Исследование одобрено этическим комитетом ФГБНУ “МГНЦ”, протокол № 6/2 от 19.04.2021. Общая анализируемая выборка включает 90 пробандов из 90 неродственных семей с клиническим диагнозом ГКА.
Для молекулярно-генетического исследования у пациентов и их родителей собраны образцы периферической крови. Геномная ДНК выделена с использованием стандартных методик. Для выявления крупных вариаций числа копий локуса 11q14.3 использован метод мультиплексной лигазо-зависимой амплификации проб (MLPA).
Поиск вариантов нуклеотидной последовательности в гене TYR проведен с помощью прямого автоматического секвенированя по Сэнгеру кодирующих областей гена TYR и экзон-интронных соединений. Для амплификации экзонных и фланкирующих их интронных последовательностей в гене TYR подобраны пары праймеров. Последовательность праймеров:
1) (1F) TTAACTGGGTTTGCTTAGGTC, (1R) TATACCCTGCCTGAAGAAGTG; 2) (2F) CTCCTCAGGAGAAGTCTAAC, (2R) AACTCAGAAAATTCTGAATTC; 3) (3F) ACACACTGGGTATCCAGAATG, (3R) ACAATAGACTACCATAACTTCTTAGC; 4) (4F) TCAAGGCCTGAAAGAATAAACTA, (4R) GCCTATGTTAAGCAAAATGACC; 5) (5F) TGTCTACTCCAAAGGACTGT, (5R) ACTTAGCTGGATGTGTTATAGA.
Амплификация проводилась по стандартному протоколу. Секвенирование ДНК больных проводили в компании “Евроген” (Москва, Россия). Все варианты нуклеотидной последовательности гена TYR обозначены в соответствии с референсным транскрипционным вариантом NM_000372.4.
Метод MLPA для выявления крупных вариаций числа копий локуса гена TYR проведен с использованием набора SALSA MLPA P325-A2 (MRC-Holland, Нидерланды), в соответствии с рекомендациями производителя. Результаты MLPA проанализированы с помощью программы Coffalyser.Net (MRC-Holland). Границы делеций обозначены по сборке генома человека hg18.
За период 2020–2021 гг. ФГБНУ “МГНЦ” сформирована выборка из 90 человек. Всем пробандам проведен поиск вариантов нуклеотидной последовательности в гене TYR и вариаций числа копий локуса этого гена. У 32 пробандов выявлено по два патогенных генетических варианта и подтвержден диагноз ГКА 1 типа: 29 пробандов с ГКА 1A-типа, три пробанда с ГКА 1B-типа (табл. 1). В рассматриваемой выборке: восемь семейных, 22 изолированных случая и два пробанда, по которым нет информации (ребенок усыновлен). Соотношение по полу составило: 0.56 : 0.44 (17 мужчин, 15 женщин). Средний возраст пробандов в выборке – 4.5 года (от 9 мес. до 36 лет).
Таблица 1.
Обнаруженные генетические варианты в гене TYR у пробандов с ГКА 1 типа
| Пациент | Диагноз (тип ГКА) | Аллель 1 | Аллель 2 |
|---|---|---|---|
| 1 | 1A | c.140G>A,p.(Gly47Asp) | c.650G>A,p.(Arg217Gln) |
| 2 | 1A | c.140G>A,p.(Gly47Asp) | c.650G>A,p.(Arg217Gln) |
| 3 | 1A | c.650G>A,p.(Arg217Gln) | c.1037-3C>G,p.? |
| 4 | 1A | c.650G>A,p.(Arg217Gln) | c.650G>A,p.(Arg217Gln) |
| 5 | 1A | c.650G>A,p.(Arg217Gln) | c.1037-3C>G,p.? |
| 6 | 1A | c.650G>A,p.(Arg217Gln) | c.1204C>T, p.(Arg402*) |
| 7 | 1A | c.1037G>A,p.(Gly346Glu) | c.1037-7T>A,p.? |
| 8 | 1A | c.650G>A,p.(Arg217Gln) | c.650G>A,p.(Arg217Gln) |
| 9 | 1A | c.650G>A,p.(Arg217Gln) | c.650G>A,p.(Arg217Gln) |
| 10 | 1A | c.650G>A,p.(Arg217Gln) | c.650G>A,p.(Arg217Gln) |
| 11 | 1A | с.1026Т>А,(р.Asn342Lys) | с.1026Т>А,р.(Asn342Lys) |
| 12 | 1A | c.650G>A,p.(Arg217Gln) | c.1037-7T>A,p.? |
| 13 | 1B | c.1264C>T,p.(Arg422Trp) | chr11:g.(88563972_88601051)_(88601132_88657939)del |
| 14 | 1A | c.650G>A,p.(Arg217Gln) | c.1037-7T>A,p.? |
| 15 | 1A | c.650G>A,p.(Arg217Gln) | c.650G>A,p.(Arg217Gln) |
| 16 | 1A | с.1037G>A,p.(Gly346Glu) | с.1036+1G>A,p.? |
| 17 | 1A | c.1037G>A,p.(Gly346Glu) | c.766C>T,p.(His256Tyr) |
| 18 | 1A | c.1037-7T>A,p.? | c.1130T>G,p.(Val377Gly) |
| 19 | 1A | c.929dup,p.(Arg311Lysfs*7) | c.1193A>G,p.(Glu398Gly) |
| 20 | 1B | c.1064C>T,p.(Ala355Val) | c.[575C>A;1205G>A],p.[(Ser192Tyr;Arg402Gln)] |
| 21 | 1A | c.650G>A,p.(Arg217Gln) | c.650G>A,p.(Arg217Gln) |
| 22 | 1A | c.650G>A,p.(Arg217Gln) | c.650G>A,p.(Arg217Gln) |
| 23 | 1A | c.157G>T,p.(Gly53Cys) | c.1204C>T,p.(Arg402*) |
| 24 | 1A | chr11:g.(?_88550674)_(88550758_88563885)del | chr11:g.(?_88550674)_(88550758_88563885)del |
| 25 | 1B | c.1037-7T>A,p.? | c.1110G>A,p.(Met370Ile) |
| 26 | 1A | c.1037G>A,p.(Gly346Glu) | c.650G>A,p.(Arg217Gln) |
| 27 | 1A | c.1036+1G>A,p.? | chr11:g.(88563972_88601051)_(88601132_88657939)del |
| 28 | 1A | c.[575C>A;1205G>A], p.[(Ser192Tyr;Arg402Gln)] | c.1204C>T,p.(Arg402*) |
| 29 | 1A | c.650G>A,p.(Arg217Gln) | c.650G>A,p.(Arg217Gln) |
| 30 | 1A | c.1037-7T>A,p.? | c.605A>G,p.(His202Pro) |
| 31 | 1A | c.650G>A,p.(Arg217Gln) | c.[575C>A;1205G>A],p.[(Ser192Tyr;Arg402Gln)] |
| 32 | 1A | c.650G>A,p.(Arg217Gln) | c.1037G>A,p.(Gly346Glu) |
В результате молекулярно-генетического исследования определены спектр, частоты и типы генетических вариантов в гене TYR, ассоциированных с ГКА 1-го типа. Выявлено 17 вариантов нуклеотидной последовательности, из которых 14 известных и два незарегистрированных, а также один ранее описанный комплексный аллель c.[575C>A;1205G>A] (табл. 2). По литературным данным известно, что варианты c.575C>A и c.1205G>A, находясь в цис-положении, могут приводить к мягкому фенотипу альбинизма как в случае наличия патогенного варианта на другой хромосоме, так и в гомозиготном состоянии [3]. Кроме того, в гене TYR выявлено 11 миссенс-мутаций, один нонсенс-вариант (c.1204C>T), одна однонуклеотидная дупликация (c.929dup), три интронных варианта (с.1036+1G>A, c.1037-3C>G, c.1037-7T>A). Среди вариантов имеются те, которые встречаются в исследуемой выборке с наибольшей частотой в гене TYR. Частоты на всю рассматриваемую выборку 90 пациентов: c.650G>A – 0.144; c.1037-7T>A – 0.033; c.1037G>A – 0.028; c.1204C>T – 0.022. Если рассматривать вклад выявленных патогенных вариантов в ген TYR, частоты (на 32 пациента) составляют: c.650G>A – 0.406; c.1037-7T>A – 0.093; c.1037G>A – 0.078; c.1204C>T – 0.047.
Таблица 2.
Спектр и частота вариантов в гене TYR, ассоциированных с ГКА 1-го типа
| Геномная позиция (hg19) | Позиция по кодирующей части гена (NM_000372.4) | Позиция по белковой изоформе (NP_000363.1) | Число аллелей | Частота варианта в выборке (n = 32) (95%CI) | Новый/описанный (HGMDID) |
|---|---|---|---|---|---|
| chr11:88911771G>A | c.650G>A | p.(Arg217Gln) | 26 | 0.4063 (0.2851–0.5363) | CM930714 |
| chr11:88960984T>A | c.1037-7T>A | p.? | 6 | 0.0938 (0.0352–0.1930) | CS930868 |
| chr11:88960991G>A | c.1037G>A | p.(Gly346Glu) | 5 | 0.0781 (0.0259–0.1730) | CM942069 |
| chr11.[88911696C>A;89017961G>A] | c.[575C>A;1205G>A] | p.([Ser192Tyr;Arg402Gln]) | 3 | 0.0469 (0.0098–0.1309) | [3] |
| chr11:89017960C>T | c.1204C>T | p.(Arg402*) | 3 | 0.0469 (0.0098–0.1309) | CM941348 |
| chr11:88911261G>A | c.140G>A | p.(Gly47Asp) | 2 | 0.0313 (0.0038–0.1084) | CM930711 |
| chr11:88911887C>T | c.766C>T | p.(His256Tyr) | 2 | 0.0313 (0.0038–0.1084) | CM013054 |
| chr11:88924576T>G | с.1026Т>А | р.(Asn342Lys) | 2 | 0.0313 (0.0038–0.1084) | Новый |
| chr11:88924587G>A | с.1036+1G>A | p.? | 2 | 0.0313 (0.0038–0.1084) | CS1517800 |
| chr11:88960988C>G | c.1037-3C>G | p.? | 2 | 0.0313 (0.0038–0.1084) | CS172425 |
| chr11:88911278G>T | c.157G>T | p.(Gly53Cys) | 1 | 0.0156 (0.0004–0.0840) | CM1916714 |
| chr11:88924479dupC | c.929dup | p.(Arg311Lysfs*7) | 1 | 0.0156 (0.0004–0.0840) | Новый |
| chr11:88961018C>T | c.1064C>T | p.(Ala355Val) | 1 | 0.0156 (0.0004–0.0840) | CM135755 |
| chr11:88961064G>A | c.1110G>A | p.(Met370Ile) | 1 | 0.0156 (0.0004–0.0840) | CM115209 |
| chr11:88961084T>G | c.1130T>G | p.(Val377Gly) | 1 | 0.0156 (0.0004–0.0840) | CM115210 |
| chr11:89017949A>G | c.1193A>G | p.(Glu398Gly) | 1 | 0.0156 (0.0004–0.0840) | CM052937 |
| chr11:89018020C>T | c.1264C>T | p.(Arg422Trp) | 1 | 0.0156 (0.0004–0.0840) | CM033061 |
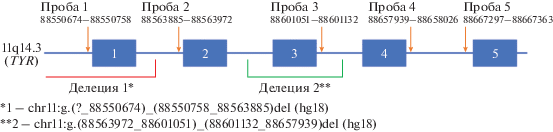
Также впервые были выявлены две крупные хромосомные делеции, захватывающие локус гена TYR: chr11:g.(?_88550674)_(88550758_88563885)del с минимальным размером 84 пары нуклеотидов (максимальный размер неизвестен), захватывающая промоторную область и один экзон, и chr11:g.(88563972_88601051)_(88601132_88657939)del размером от 81 до 93 967 пн, захватывающая 3-й экзон (рис. 1).
По всем больным, которые проанализированы, в исследуемой выборке собрана подробная информация о клинической картине (гипопигментация волос и кожи, глазные проявления). Абсолютно у всех больных (n = 32) выявлены гипопигментация кожи и волос и различные поражения глаз: гипопигментация радужки, сетчатки, снижение остроты зрения, косоглазие, нистагм, светобоязнь и аномалии рефракции (миопия, астигматизм). Помимо основных клинических признаков обнаружены нетипичные проявления, которые, однако, могут указывать как на вовлеченность в патологический процесс других систем и органов, так и на сочетанную патологию: признаки дисфункции срединных структур головного мозга, дисплазия соединительной ткани (n = 1), локальное расширение арахноидального пространства в левой лобной области головного мозга (n = 1), гиперактивность (n = 1), ишемическое поражение центральной нервной системы (n = 1), гипертрофия миндалин, атопический дерматит (n = 1), сколиоз грудного отдела позвоночника (n = 1), отставание в моторном развитии, пузырно-мочеточниковый рефлюкс 3–4-й степени, четыре острых пиелонефрита, лицевые дисморфии (n = 1), кровотечение после удаления зубов, единичные экхимозы (n = 1).
Молекулярный механизм патогенеза ГКА 1-го типа объясняется снижением или отсутствием фермента тирозиназы, которая кодируется геном TYR. Тирозиназа определяет несколько ключевых этапов в синтезе меланина, у больных при этом сохраняется нормальное количество меланоцитов в эпидермисе и волосяных фолликулах, но пигмент в них полностью или частично отсутствует [4]. Снижение содержания пигмента приводит к светлому цвету кожи, белым или светлым волосам, а также к специфическим глазным аномалиям по депигментации радужной оболочки и сетчатки (альбинотическое глазное дно). Офтальмологические изменения включают снижение остроты зрения, патологию зрительных нервов и нистагм [5]. По уровню активности тирозиназы различают несколько типов ГКА: тип 1A характеризуется полным отсутствием активности тирозиназы, тип 1B характеризуется сниженной активностью фермента. Клинически оба подтипа схожи, но подтип 1A считают более “тяжелым”. При ГКА типа 1B симптомы присутствуют с рождения, но могут улучшаться с возрастом [6]. В рассматриваемой нами выборке не удалось проанализировать данный факт в связи с малым количеством выявленных больных типа 1B.
В небольшой выборке пациентов (32 пробанда) при секвенировании кодирующих областей и экзон-интронных соединений гена TYR выявлено 17 вариантов, из них – два незарегистрированных раннее. На сегодняшний день описано значительное количество генетических вариантов в генах, ассоциированных с наследственными формами альбинизма, но наиболее часто патогенные варианты встречаются в гене TYR [6], что выявлено и в настоящем исследовании, проведенном на группе российских пациентов. Однако четыре частых для России варианта (c.650G>A, c.1037-7T>A, c.1037G>A, c.1204C>T) встречаются в странах Европы значительно реже (данные GnomAD). При составлении алгоритма диагностики глазокожного альбинизма необходимо принять во внимание несколько факторов: доступность анализа кодирующих областей гена TYR (пять экзонов) по сравнению с другими генами, ответственными за альбинизм, частоту встречаемости вариантов в гене TYR, а также основной молекулярный механизм развития.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Bashour M., Suh D.W. Albinism: Background, pathophysiology, epidemiology // Medscape Ophtalmology. 2020. Sep 30.
Grønskov K., Ek J., Brondum-Nielsen K. Oculocutaneous albinism // Orphanet J. Rare Dis. 2007. V. 2. 43. https://doi.org/10.1186/1750-1172-2-43
Jagirdar K., Smit D.J., Ainger S.A. et al. Molecular analysis of common polymorphisms within the human Tyrosinase locus and genetic association with pigmentation traits // Pigment Cell Melanoma Res. 2014. V. 27(4). P. 552–564. https://doi.org/10.1111/pcmr.12253
Marcon C.R., Maia M. Albinism: Epidemiology, genetics, cutaneous characterization, psychosocial factors // Bras. Dermatol. 2019. V. 94. P. 503–520. https://doi.org/10.1016/j.abd.2019.09.023
Brodsky M.C., Fray K.J. Positive angle kappa: A sign of albinism in patients with congenital nystagmus // Am. J. Ophthalmol. 2004. V. 137(4). P. 625–629. https://doi.org/10.1016/j.ajo.2003.11.066
Farney S.K., Dolinska M.B., Sergeev Y.V. Dynamic analysis of human tyrosinase intra-melanosomal domain and mutant variants to further understand oculocutaneous albinism type 1 // J. Anal. Pharm. Res. 2018. V. 7(6). P. 621–632. https://doi.org/10.15406/japlr.2018.07.00293
Дополнительные материалы отсутствуют.