

Генетика, 2023, T. 59, № 2, стр. 209-216
Характеристика нового интронного варианта в гене F5, ассоциированного с аберрантным сплайсингом и тяжелой формой наследственного дефицита фактора V
О. С. Пшеничникова 1, *, Е. В. Яковлева 1, Н. И. Зозуля 1, Ю. М. Познякова 1, Е. Ю. Демидова 1, В. Л. Сурин 1
1 Национальный медицинский исследовательский центр гематологии
125167 Москва, Россия
* E-mail: pshenichnikovaolesya@gmail.com
Поступила в редакцию 04.04.2022
После доработки 24.06.2022
Принята к публикации 07.07.2022
- EDN: KYDKCD
- DOI: 10.31857/S0016675823020108
Аннотация
Наследственный дефицит фактора свертывания крови V (FV) – это редкое аутосомное наследственное заболевание, которое вызывается дефектами в гене F5 и проявляется кровотечениями разной степени тяжести. Цель данного исследования – молекулярная и функциональная характеристика нового варианта гена F5, приводящего к аберрантному сплайсингу и предельному снижению активности белка, выявленного у пациента с тяжелой формой дефицита FV. Были проведены мутационный скрининг гена F5 и функциональное исследование для пробанда (FV:C 0.4%), с детства страдающего желудочно-кишечными и посттравматическими кровотечениями, гематомами, экхимозами и дискомфортом в голеностопных суставах. Секвенирование выявило наличие нового гомозиготного варианта гена F5 NC_000001.10:169519985G>C (или NM_000130.5:c.1297 –8C>G). По результатам биоинформатического анализа данный вариант должен приводить к потере акцепторного сайта на границе интрона 8 и экзона 9. Однако анализ мРНК FV показал, что помимо этого он активирует аберрантный сайт сплайсинга на дистанции в семь нуклеотидов от сайта дикого типа и приводит к образованию аномального транскрипта, в котором остается семь нуклеотидов от интрона 8 и формируется преждевременный стоп-кодон. Нормальный транскрипт у исследуемого пациента не выявлялся. Результаты данного исследования свидетельствуют в пользу того, что изменения не только в канонических динуклеотидах сайтов сплайсинга могут в значительной мере нарушать процессинг пре-мРНК.
Фактор свертывания V (FV, проакцелерин или лабильный фактор) представляет собой гликопротеин размером в 330 кДа, который в активированном состоянии (FVa) выступает в качестве кофактора активированного FX (FXa) в протромбиназном комплексе, ускоряя превращение протромбина в тромбин [1–4]. Он также проявляет антикоагулянтные свойства, участвуя в инактивации FVIII, опосредованной активированным протеином C [2].
Наследственный дефицит FV (OMIM #227400) – это аутосомное рецессивное заболевание крови, частота встречаемости которого в общей популяции составляет 1 : 1 000 000. Он развивается в результате нарушений в гене F5, кодирующем фактор свертывания крови V, и проявляется в виде кровотечений разной степени тяжести (от носовых кровотечений, гематом, легкой синячковости, меноррагий и т.д. при легкой форме до серьезных желудочно-кишечных кровотечений и кровоизлияний в центральную нервную систему при тяжелой форме) [1, 2, 4–6].
Ген F5 занимает участок длиной в 74.68 тпн на хромосоме 1q24.2 (GRCh38/hg38) и состоит из 25 экзонов. На данный момент описано 165 различных патогенных вариантов F5 (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php, данные на март 2022 г.), 140 из которых ассоциированы с дефицитом FV. Из них 17 (12%) вариантов относятся к нарушениям сайта сплайсинга. Восемь вариантов, нарушающих сплайсинг, расположены вне консервативных динуклеотидов (±1–2) донорных и акцепторных сайтов сплайсинга, но не для всех из них была изучена функциональность нарушений [7–14].
В данном исследовании мы даем молекулярную и функциональную характеристику нового варианта гена F5, который приводит к аберрантному сплайсингу и практически полной потере активности белка FV.
МАТЕРИАЛЫ И МЕТОДЫ
В качестве биологического материала использовали цельную венозную кровь, взятую на 6%-ной ЭДТА от пациента с дефицитом FV, его матери и донора без дефицита FV (здоровый контроль) после получения информированного согласия. ДНК выделяли из ядерных клеток периферической крови после селективного лизиса эритроцитов 0.8%-ным раствором хлорида аммония по стандартной методике, включающей обработку додецилсульфатом натрия (0.5%) и протеиназой К (200 мкг/мл) с последующей фенол-хлороформной депротеинизацией и переосаждением этанолом.
Все 25 экзонов, экзон-интронные сочленения и промотор (около 600 пн) гена F5 амплифицировали в системе PCR Master Mix (ThermoScientific, США) с праймерами, опубликованными ранее [15] или разработанными нами в ходе данного исследования на основании нуклеотидной последовательности гена F5 GenBank Acc. No. NG_011806 (табл. 1), с 0.01–0.02 мкг геномной ДНК при следующих условиях: 94°C – 1 мин, 62°C – 1 мин, 72°C – 3 мин, 30 циклов. Самый длинный экзон 13 амплифицировали с применением методики long-distance PCR (LD-PCR) в системе Promega GoTaq®Long PCR Master Mix (Promega Corporation, США). Визуализацию продуктов ПЦР проводили с помощью регистрации в УФ-свете после разделения электрофорезом в 6%-ном полиакриламидном геле (ПААГ) и прокрашивания бромистым этидием. Все праймеры, использованные в работе, синтезировали в ЗАО “Синтол” (Москва, Россия).
Таблица 1.
Праймеры, использованные для мутационного скрининга гена F5
| Целевой фрагмент | Праймер | Последовательность (5'–3') | Размер фрагмента, пн | Источник |
|---|---|---|---|---|
| Промотор | HCVP-F | CTATGCTGCAGCTTAGCTGG | 597 | [15] |
| HCVP-R | GCTGCAATGAGCTCTAGAGG | |||
| Экзон 1 | F5-1D | GACGCTGCCACCCACAGCCT | 368 | Наш дизайн |
| F5-1R | GCCATGACATTGCAAAGGGA | |||
| Экзон 2 | F5-2D | CTGGTATTCTACTAGAAGTCTGA | 269 | Наш дизайн |
| F5-2R | CATGTGAATGCCAAATTACCCA | |||
| Экзон 3 | F5-3D | CTTGCTTAGCAACTCTGCCT | 307 | Наш дизайн |
| F5-3R | CCAGTTGCAAGAGATTTCCCT | |||
| Экзон 4 | HCFV-4F | ACTGCCCACATGTCTTGATGG | 311 | [15] |
| HCFV-4R | TGACAGAACTCCTGACCATTCC | |||
| Экзон 5 | HCFV-5F | CTGCAGTGCTACTGAAAACATG | 306 | [15] |
| HCFV-5R | TCCTTCTTGATAGGGAGTTGC | |||
| Экзон 6 | HCFV-6F | GCCTAATCCTTTAGCAATCCCTG | 547 | [15] |
| HCFV-6R | CATTGAGAAGCAAGACTGTCAGG | |||
| Экзон 7 | HCFV-7F | GAGTTATTTCATTGTCTTTCTGTCC | 241 | [15] |
| HCFV-7R | GTCTTGAACCTTTGCCCAG | |||
| Экзон 8 | HCFV-8F | GCAGAATGTTTAAGCACAAGG | 306 | [15] |
| HCFV-8R | CTATGTAATTTCTCCCATGATTCTG | |||
| Экзон 9 | HCFV-9F | GATGACTTCAAAGACAGTGTCC | 550 | [15] |
| HCFV-9R | GGATTCAGTAGAAGTGAAAGATTC | |||
| Экзон 10 | HCFV-10F | ATGACAAGTTAATGGGTGCAGC | 541 | [15] |
| HCFV-10R | CTTGAAGGAAATGCCCCATTA† | |||
| Экзон 11 | HCFV-11F | TGGTCTATGCGTCTGTTCTTGTAC | 250 | [15] |
| HCFV-11R | CAACCACAGGAATGAAAAACTG | |||
| Экзон 12 | HCFV-12F | CATAGACTTGGAATTTTAACAG | 286 | [15] |
| HCFV-12R | CAAGCTTCCTCTGTGAGTGTC | |||
| Экзон 13 | F5-13D | ATGTCTTTTCCCAGACTTCCA | 2961 | Наш дизайн |
| F5-13R | TGCCCACAATTATCCCCCTGA | |||
| F5-13d1S* | CCCCACTGAGACACCTCATT | |||
| F5-13r1S* | CGTATCCCTGTGACATCTGGCTGTA | |||
| F5-13d2S* | CAGCCCCCAGAATGCCTCAC | |||
| F5-13d3S* | CAGACAGTGCCCCCAGAGGAACA | |||
| F5-13d4S* | GTCAGACAAACCTTTCTCCAGCCCT | |||
| F5-13d5S* | GTCCCTTTCCCCAGACCTCAGCCA | |||
| Экзон 14 | HCFV-14F | CTGACCTCATGGCACTTATACC | 615 | [15] |
| HCFV-14R | CCGAAGATCTTAGCAGTGCTC | |||
| Экзон 15 | HCFV-15F2 | GGCCATATCTCACAGGATGG | 600 | [15] |
| HCFV-15R | GTCATCTGAAGAGCTGCATGG | |||
| Экзон 16 | HCFV-16F | AGTGCATGGTAAGCACTTGG | 761 | [15] |
| HCFV-16R2 | ACCTGCCAGATTACATCAGC | |||
| Экзон 17 | HCFV-17F2 | CCTTTCCATGGCTAGGTAGG | 356 | [15] |
| HCFV-17R | TCTTAGCAGGGACCTCTTCC | |||
| Экзон 18 | HCFV-18F | GAAAGCCTCTTGTGAAGCAGG | 388 | [15] |
| HCFV-18R2 | TTCAATGCAATCAGACCATGG | |||
| Экзон 19 | HCFV-19F | ATTGAGTCAGAAACATAATCCC | 222 | [15] |
| HCFV-19R | GCATGCTGCACAACTGTAGG | |||
| Экзон 20 | HCFV-20F | AAGGATCTGGTTTTCCACTGG | 366 | [15] |
| HCFV-20R2 | ACCTCAGAGGGTTGATTTTAAGG | |||
| Экзон 21 | HCFV-21F | GCAGTGTGTGACTTGTTGAC | 241 | [15] |
| HCFV-21R | AGATTCAGATAGAAATATGCACAC | |||
| Экзон 22 | HCFV-22F | TCTTCCTGGAACTGGAATTATCC | 360 | [15] |
| HCFV-22R | TCTTGATTCTTTGAGTGGCAGTG | |||
| Экзон 23 | HCFV-23F2 | TGAGAACAGTATTTGGCACTTGG | 398 | [15] |
| HCFV-23R | CCAGATCCTCCATGTTTGTGG | |||
| Экзон 24 | HCFV-24F | AAGCAAAGGTTTTAACATCTTCC | 258 | [15] |
| HCFV-24R | TCTTTGCCCAGATGCCAC | |||
| Экзон 25 | HCFV-25F | CAGTCATACAGCTAATACAGACG | 630 | [15] |
| HCFV-25R | GGTCTTAAAGAGTCTCTTCCAGG |
ПЦР-фрагменты для секвенирования очищали на колонках Wizard (Promega Corporation). Секвенирование по Сэнгеру проводили в ЦКП “Геном” ИМБ РАН с помощью набора реактивов BigDye Terminator v3.1 (Applied Biosystems, США) и с последующим анализом продуктов реакции на автоматическом секвенаторе ABI PRISM 3100Avant (Applied Biosystems).
Названия выявленным вариантам присваивали согласно рекомендациям HGVS (Human Genome Variation Society, http://varnomen.hgvs.org/), используя в качестве референса последовательность мРНК NM_000130.5.
Для получения кДНК F5 использовали в качестве матрицы образцы тотальной РНК, выделенной из ядерных клеток пациента и здорового донора с использованием TRIzol® Reagent (Invitrogen, Нидерланды). Реакцию обратной транскрипции проводили с применением M-MLV Reverse Transcriptase, RNasin® Ribonuclease Inhibitor (Promega Corporation) и праймера F5RT 5'-GGTTTCCCCTGGTTGAACTGCT-3'. Полученный фрагмент кДНК F5, покрывающий часть экзона 8 и полностью экзон 9, амплифицировали при стандартных условиях в системе PCR Master Mix (Thermo Fisher Scientific, США) с использованием праймеров F5D 5'-CACAGTACGAAGATGAGTCCTT-3' и F5R 5'-GGTGTTGTTCCTGCCTGAGGTGA-3'. Секвенирование по Сэнгеру выполняли так же, как было описано выше. Для проведения количественной оценки нормального и мутантного транскрипта выполняли фрагментный анализ того же фрагмента кДНК, амплифицированного с тем же обратным праймером и с прямым праймером (F5DFAM), меченным флуоресцентным красителем (FAM). Длины амплифицированных фрагментов определяли на автоматическом генетическом анализаторе Нанофор 5 (ЗАО “Синтол”, Москва, Россия) со стандартом длин Liz 500 (Applied Biosystems) в программе GeneMapper v4.0 (Applied Biosystems).
Для определения потенциального влияния выявленного варианта на сайт сплайсинга проводили in silico анализ с помощью четырех предсказательных программ: Human Splicing Finder (HSF) [16], NetGene2 [17], Splice Site Prediction by Neural Network (SSPNN) [18] и SpliceAI [19].
РЕЗУЛЬТАТЫ
История болезни
Пробанд – мужчина, 34 года, страдающий желудочно-кишечными и посттравматическими кровотечениями, гематомами, экхимозами и дискомфортом в голеностопных суставах с детства. Был госпитализирован в ФГБУ “НМИЦ гематологии” Минздрава РФ в 2012 г. со спонтанной напряженной гематомой левого предплечья и с коагулопатией неясного генеза. При обследовании в коагулограмме было выявлено: АЧТВ 71 с, фибриноген 3.0 г/л, протромбин по Квику 48%, FV:C 5%, FII:C 98%, FVII:C 90%; FVIII:C 84%, FIX:C 78%, FX:C 96%, FXI:C 79.9%, vWF:C 130%, FXII:C 101%, XIIа-зависимый фибринолиз 6 мин, агрегация тромбоцитов с ристомицином 85%, агрегация тромбоцитов с коллагеном 73%, агрегация тромбоцитов с адреналином 62%, агрегация тромбоцитов с АДФ 69%. Никто из родственников проблем с кровотечениями не имел. С 1992 г. пациенту по требованию проводили трансфузии свежезамороженной плазмы, криопреципитата и эритроцитной массы. Дальнейшее обследование выявило значительный дефицит FV (FV:C в плазме составляла 0.4%) при отсутствии ингибитора к FV. Учитывая анамнестические, клинические и лабораторные данные, у больного был диагностирован наследственный дефицит FV. Мать пробанда симптомов не имела, активность FV в плазме была в пределах нормы (78%), клинические и лабораторные данные отца пробанда, к сожалению, были недоступны.
Мутационный анализ
Мутационный анализ с применением секвенирования по Сэнгеру фрагментов, охватывающих все 25 экзонов, экзон-интронные сочленения и около 600 пн 5'-промоторной области гена F5, позволил выявить три известных нейтральных SNP (single nucleotide polymorphism) в кодирующих областях: rs1800594 (c.3804 T>C, p.Ser1268Ser) CC; rs549332286 (c.4056 C>T, p.Leu1352Leu) TT; rs6030 (c.5290 A>G, p.Met1764Val) GG, а также множество интронных полиморфизмов по всей протяженности гена, все в гомозиготном состоянии. Кроме того, был обнаружен ранее не описанный гомозиготный вариант c.1297 –8C>G. В международных базах данных (GnomAD, http://gnomad.broadinstitute.org/; HGMD, http://www.hgmd.cf.ac.uk/ac/all.php; ClinVar NCBI, https://www.ncbi.nlm.nih.gov/clinvar; dbSNP, https://www.ncbi.nlm.nih.gov/snp/) он отсутствовал. У матери пробанда данный вариант (c.1297 –8C>G) был найден в гетерозиготном состоянии.
In silico анализ варианта c.1297 –8C>G
Так как вариант c.1297 –8C>G приходился не на консервативные динуклеотиды сайта сплайсинга (±1–2), его влияние на правильное созревание пре-мРНК F5 оставалось под вопросом. В связи с этим мы провели in silico анализ с помощью предсказательных программ. Все четыре использованные программы предсказали его потенциальное влияние на акцепторный сайт на стыке интрона 8 и экзона 9. По оценкам SpliceAI данный вариант с вероятностью 0.84 приводит к потере акцепторного сайта в пределах 50 нуклеотидов от места замены. NetGene2 и SSPNN выявили значительное уменьшение вероятности формирования акцепторного сайта при замене c.1297 –8C>G по сравнению с диким типом (с 0.17 до 0.07 и с 0.94 до 0.74 соответственно). HSF зафиксировал изменение в акцепторном сайте сплайсинга дикого типа, с большой вероятностью влияющее на сплайсинг (изменение значения между диким и мутантным типом составило –97.69%), а также активацию криптического донорного сайта в положении GRCh38:1:169550742-169550750 (изменение значения между диким и мутантным типом составило 56.68%).
Анализ мРНК F5
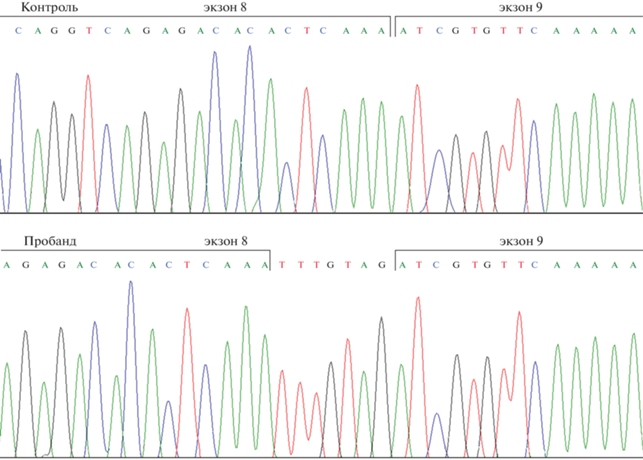
Для того чтобы оценить реальный эффект варианта c.1297 –8C>G на сплайсинг пре-мРНК F5, мы амплифицировали и секвенировали по Сэнгеру фрагмент кДНК, покрывающий целиком экзон 9 и часть экзона 8. По результатам секвенирования было показано, что рассматриваемый вариант приводит к возникновению и активации аберрантного сайта сплайсинга, смещенного на семь нуклеотидов в 5'-направлении относительно нормального (рис. 1). Активация этого сайта приводит к формированию более короткого белка FV за счет включения в зрелую мРНК семи концевых нуклеотидов интрона 8, приводящего к сдвигу рамки считывания и возникновению преждевременного стоп-сигнала через 13 кодонов от конца экзона 8. Следов нормальной мРНК у пробанда обнаружено не было.
Рис. 1.
Полученные с секвенатора электрофореграммы фрагмента кДНК F5 здорового донора и пробанда, демонстрирующие влияние варианта c.1297 –8C>G на сплайсинг.
Для количественной оценки соотношения мутантной и нормальной мРНК F5 у пробанда был проведен фрагментный анализ, также показавший полное отсутствие нормального транскрипта (рис. 2).
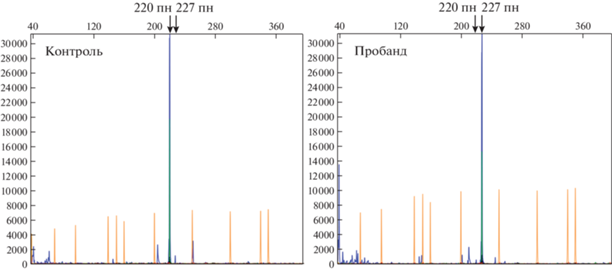
ОБСУЖДЕНИЕ
Данная работа посвящена молекулярно-генетическому исследованию, проведенному для российского пациента с тяжелой формой дефицита FV (уровень активности FV 0.4%). По результатам секвенирования промоторной области, экзонов и экзон-интронных сочленений гена F5 пробанд был гомозиготен практически по всем известным полиморфным позициям. Единственный выявленный потенциально патогенный вариант – c.1297 –8C>G, отсутствующий в базах данных, также находился в гомозиготном состоянии. Согласно рекомендациям ACMG (the American College of Medical Genetics and Genomics) [20] вариант c.1297 –8C>G относится к вероятно патогенным (критерии PM2, PS3, PP3). С помощью четырех программ был выполнен in silico анализ возможного влияния найденного варианта на сплайсинг. Все они указывали на нарушение нормального акцепторного сайта сплайсинга, но ни одна из них не выявила формирования нового аберрантного сайта. Согласно предсказаниям программ исследуемый вариант должен приводить к потере экзона 9. Однако результаты анализа мРНК свидетельствуют об активации возникающего вследствие однонуклеотидной замены c.1297 –8C>G аберрантного сайта сплайсинга, приводящего к включению в нормальный транскрипт F5 семи концевых нуклеотидов интрона 8, сдвигу рамки считывания и формированию преждевременного стоп-кодона. В результате в белке FV остаются только домен А1 и небольшая часть домена А2. В то же время мРНК, содержащие аномальные терминирующие кодоны, могут подвергаться нонсенс-опосредованному распаду (NMD, nonsense-mediated decay) [21], поэтому не исключено, что в нашем случае не синтезируется даже такой укороченный белок. У большинства пациентов с дефицитом FV, несущих гомозиготные варианты, нарушающие сплайсинг, болезнь сопровождается тяжелыми кровотечениями [9, 13, 14, 22, 23], которые были характерны и для исследованного в настоящей работе пациента. Гомозиготность по всем полиморфным позициям может указывать на делецию второй копии гена F5 или на близкородственный брак родителей пациента, хотя последнее пробанд отрицает. Биоматериал от отца пациента и какие-либо медицинские сведения о нем, к сожалению, были для нас недоступны.
Таким образом, результаты настоящего исследования подтверждают, что замены не только в канонических динуклеотидах сайтов сплайсинга могут в значительной степени нарушать процессинг пре-мРНК. Алгоритмы программ, предсказывающих эффект нуклеотидных замен на сплайсинг, иногда дают не полностью корректные заключения, что подчеркивает важность проведения анализа кДНК для правильной оценки патогенности варианта.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Mann K.G., Kalafatis M. Factor V: A combination of Dr Jekyll and Mr Hyde // Blood. 2003. V. 101. P. 20–30. https://doi.org/10.1182/blood-2002-01-0290
Duga S., Asselta R., Tenchini M.L. Coagulation factor V // Int. J. Biochem. Cell Biol. 2004. V. 36. P. 1393–1399. https://doi.org/10.1016/j.biocel.2003.08.002
Paraboschi E.M., Menegatti M., Peyvandi F. et al. Understanding the impact of aberrant splicing in coagulation factor V deficiency // Int. J. Mol. Sci. 2019. V. 20. № 4. P. 910. https://doi.org/10.3390/ijms20040910
Paraboschi E.M., Menegatti M., Rimoldi V. et al. Profiling the mutational landscape of coagulation factor V deficiency // Haematologica. 2020. V. 105. № 4. P. e180–e185. https://doi.org/10.3324/haematol.2019.232587
Tabibian S., Shiravand Y., Shams M. et al. A comprehensive overview of coagulation factor V and congenital factor V deficiency // Semin. Thromb. Hemost. 2019. V. 45. № 5. P. 523–543. https://doi.org/10.1055/s-0039-1687906
Asselta R., Tenchini M.L., Duga S. Inherited defects of coagulation factor V: The hemorrhagic side // J. Thromb. Haemost. 2006. V. 4. № 1. P. 26–34. https://doi.org/10.1111/j.1538-7836.2005.01590.x
Peyvandi F., Duga S., Akhavan S., Mannucci P.M. Rare coagulation deficiencies // Haemophilia. 2002. V. 8. № 3. P. 308–321. https://doi.org/10.1046/j.1365-2516.2002.00633.x
Nuzzo F., Bulato C., Nielsen B.I. et al. Characterization of an apparently synonymous F5 mutation causing aberrant splicing and factor V deficiency // Haemophilia. 2015. V. 21. № 2. P. 241–248. https://doi.org/10.1111/hae.12554
Dall’Osso C., Guella I., Duga S. et al. Molecular characterization of three novel splicing mutations causing factor V deficiency and analysis of the F5 gene splicing pattern // Haematologica. 2008. V. 93. № 10. P. 1505–1513. https://doi.org/10.3324/haematol.12934
Castoldi E., Duckers C., Radu C. et al. Homozygous F5 deep-intronic splicing mutation resulting in severe factor V deficiency and undetectable thrombin generation in platelet-rich plasma // J. Thromb. Haemost. 2011. V. 9. № 5. P. 959–968. https://doi.org/10.1111/j.1538-7836.2011.04237.x
Guella I., Paraboschi E.M., van Schalkwyk W.A. et al. Identification of the first Alu-mediated large deletion involving the F5 gene in a compound heterozygous patient with severe factor V deficiency // Thromb. Haemost. 2011. V. 106. № 2. P. 296–303. https://doi.org/10.1160/TH11-03-0149
Kuang S.Q., Hasham S., Phillips M.D. et al. Characterization of a novel autosomal dominant bleeding disorder in a large kindred from east Texas // Blood. 2001. V. 97. № 6. P. 1549–1554. https://doi.org/10.1182/blood.v97.6.1549
Lunghi B., Pinotti M., Maestri I. et al. Evaluation of factor V mRNA to define the residual factor V expression levels in severe factor V deficiency // Haematologica. 2008. V. 93. № 3. P. 477–478. https://doi.org/10.3324/haematol.11952
Asselta R., Montefusco M.C., Duga S. et al. Severe factor V deficiency: Exon skipping in the factor V gene causing a partial deletion of the C1 domain // J. Thromb. Haemost. 2003. V. 1. № 6. P. 1237–1244. https://doi.org/10.1046/j.1538-7836.2003.00160.x
van Wijk R., Nieuwenhuis K., van den Berg M. et al. Five novel mutations in the gene for human blood coagulation factor V associated with type I factor V deficiency // Blood. 2001. V. 98. № 2. P. 358–367. https://doi.org/10.1182/blood.v98.2.358
Desmet F.O., Hamroun D., Lalande M. et al. Human Splicing Finder: An online bioinformatics tool to predict splicing signals // Nucl. Acids Res. 2009. V. 37. P. e67. https://doi.org/10.1093/nar/gkp215
Hebsgaard S.M., Korning P.G., Tolstrup N. et al. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information // Nucl. Acids Res. 1996. V. 24. P. 3439–3452. https://doi.org/10.1093/nar/24.17.3439
Reese M.G., Eeckman F.H., Kulp D., Haussler D. Improved splice site detection in Genie // J. Comput. Biol. 1997. V. 4. P. 311–323. https://doi.org/10.1089/cmb.1997.4.311
Jaganathan K., Kyriazopoulou Panagiotopoulou S., McRae J.F. et al. Predicting splicing from primary sequence with deep learning // Cell. 2019. V. 176. № 3. P. 535–548. e24. https://doi.org/10.1016/j.cell.2018.12.015
Richards S., Aziz N., Bale S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology // Genet. Med. 2015. V. 17. № 5. P. 405–424. https://doi.org/10.1038/gim.2015.30
Hug N., Longman D., Cáceres J.F. Mechanism and regulation of the nonsense-mediated decay pathway // Nucl. Acids Res. 2016. V. 44. № 4. P. 1483–1495. https://doi.org/10.1093/nar/gkw010
Schrijver I., Koerper M.A., Jones C.D., Zehnder J.L. Homozygous factor V splice site mutation associated with severe factor V deficiency // Blood. 2002. V. 99. P. 3063–3065. https://doi.org/10.1182/blood.v99.8.3063
Fu Q.H., Zhou R.F., Liu L.G. et al. Identification of three F5 gene mutations associated with inherited coagulation factor V deficiency in two Chinese pedigrees // Haemophilia. 2004. V. 10. P. 264–270. https://doi.org/10.1111/j.1365-2516.2004.00896.x
Дополнительные материалы отсутствуют.