

Генетика, 2023, T. 59, № 4, стр. 371-380
Литературный обзор NBAS-ассоциированных патологий. Все ли найдены?
Л. Р. Жожиков 1, *, Ф. Ф. Васильев 1, Н. Р. Максимова 1
1 Северо-Восточный федеральный университет им. М.К. Аммосова, Медицинский институт
677013 Якутск, Россия
* E-mail: leonid.zhozhikov@gmail.com
Поступила в редакцию 03.06.2022
После доработки 31.08.2022
Принята к публикации 02.09.2022
- EDN: AYGTUB
- DOI: 10.31857/S0016675823040124
Аннотация
Мутации в гене NBAS вызывают две группы заболеваний – SOPH-синдром и синдром детской печеночной недостаточности 2-го типа (ILFS2, RALF), отличающиеся клиническими проявлениями, течением и прогнозами. Проведен литературный обзор клинико-генетических аспектов данных патологий и молекулярной биологии белка NBAS, а конкретно ретроградного мембранного трафика и нонсенс-опосредованного распада РНК.
SOPH-синдром (SOPH, Short stature, Optic atrophy, Pelger-Huet anomaly of leucocytes – OMIM #614800) является моногенным аутосомно-рецессивным наследственным заболеванием, характеризующимся триадой признаков: постнатальной гипоплазией, атрофией зрительных нервов и пельгеровской аномалией лейкоцитов [1]. SOPH-синдром, впервые описанный в 2010 г., считался распространенным только в якутской популяции, тем не менее каждый год по литературным данным обнаруживаются новые случаи SOPH-подобных состояний по всему миру. Частота гетерозиготного носительства причинной мутации составила 1300 на 100 000 здоровых якутов, при этом в Республиканском генетическом регистре наследственной и врожденной патологии Якутии насчитывается более 84 обнаруженных случаев SOPH по Республике Саха (Якутия), что составляет частоту чуть более 18 пациентов на 100 000 якутской популяции [2].
Детская печеночная недостаточность 2-го типа (ILFS2, Infantile Liver Failure Syndrome type 2 – OMIM #616483) – аутосомно-рецессивное заболевание, характеризующееся повторяющимися эпизодами острой печеночной недостаточности во время лихорадки, описанное впервые в 2015 г. Данный синдром встречается повсеместно и манифестирует в младенческом, раннем детском возрасте и считается одной из основных причин детской смертности неизвестной этиологии [3, 4]. Особенностью является полное выздоровление между эпизодами печеночной недостаточности, триггером которой чаще всего выступает вирусная инфекция, вызывающая лихорадку.
Причинами обеих патологий являются мутации в гене NBAS. Белок NBAS участвует в ретроградном транспорте везикул и в нонсенс-опосредованном распаде РНК (NMD, nonsense mediated RNA decay) [5, 6]. Стоит отметить, что хоть SOPH и ILFS2 описаны при мутациях в одном гене фенотипы их сильно различаются. Например, при SOPH-синдроме не наблюдается печеночной недостаточности, а при ILFS2 не наблюдается триады SOPH. Эти данные наталкивают на вопросы относительно функции белка и механизмов его реализации, а также о патофизиологии данных заболеваний. В настоящей работе освещена современная молекулярная биология белка и описаны фенотипы двух синдромов, ассоциированных с мутациями в гене NBAS.
МОЛЕКУЛЯРНАЯ БИОЛОГИЯ NBAS
Ген NBAS (neuroblastoma amplified sequence), также известный как NAG (neuroblastoma amplified gene), был впервые обнаружен в конце прошлого века в клеточных линиях нейробластом как коамплифицирующийся ген вместе с MYCN, он считался предиктором течения нейробластомы [7]. Амплификация фрагментов ДНК при онкологических заболеваниях считается неблагоприятным признаком течения, и обнаруженный новый ген во фрагментарном бульоне опухолевого ДНК помог пролить свет на разнообразие клинических исходов нейробластом. Несмотря на то что коамплификация гена NBAS + MYCN была связана с более благоприятным течением заболевания у пациентов, функция гена оставалась неизвестной и никаких однозначных выводов тогда сделано не было [8–10].
Позже Т. Aoki с соавт. [5] на клеточных линиях HeLa и 273T было установлено, что белок NBAS является частью комплекса синтаксина 18 (STX18), участвующего в ретроградном транспорте везикул с аппарата Гольджи (аГ) до эндоплазматического ретикулума (ЭР). Также D. Longman с соавт. на примере эмбрионального развития рыбок D. rerio было предложено и впоследствии доказано на клеточных линиях HeLa участие NBAS в нонсенс-опосредованном распаде РНК (NMD) [11]. Этим же коллективом авторов впоследствии была опубликована модель механизма работы и взаимодействия NBAS с другими участниками NMD [12].
Свойства и структура NBAS
Основными доменами белка можно считать два: SEC39 и β-пропеллер. Регион с β-пропеллером перекрывается тремя схожими доменами: бета-цепью хинопротеинаминодегидрогеназы, повторами WD40/YVTN и одноименным доменом N-конца Neuroblastoma-amplified sequence. По данным исследований на археях β-пропеллер защищает клетку от воздействий экстремальных факторов, а повторы WD40 являются скаффолдом для белок-белковых взаимодействий различных белковых комплексов [13]. Считается, что домен SEC39 ассоциирован с мембранным транспортом везикул от аппарата Гольджи к ЭР (рис. 1).
Рис. 1.
Ген NBAS и белок с основными доменами с указанием миссенс-мутации SOPH (с.5741G>A, p.R1914H). NBAS расположен на коротком плече второй хромосомы, его длина 563278 пар нуклеотидов (пн), включает в себя 52 экзона. Каноничный транскрипт состоит из 7.3 тпн, из них 7.1 тпн являются кодирующими. Основными доменами считаются два: SEC39 и β-propeller. Регионы взаимодействия белка NBAS с основными партнерами указаны зеленым и красным цветом для ZW10, RINT1 и для USE1 соответственно. Трехмерная структура белка предсказана AlphaFold.
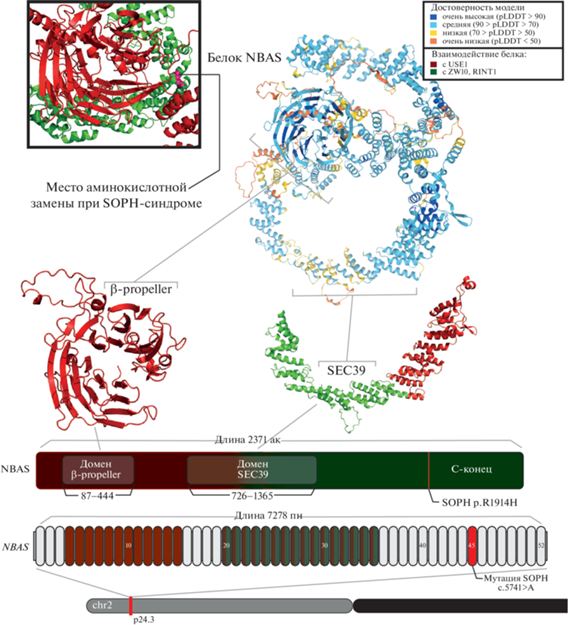
Эксперименты, проведенные с урезанными белками NBAS, показали отсутствие связи с белком-партнером по мембранному трафику р31 при укорочении с С-конца, а при укорочении с N-конца не наблюдалось взаимодействия с ZW10 и RINT-1 [5]. Белок NBAS не обладает тканевой специфичностью и обнаруживается во всех типах клеток. Тем не менее в исследованиях на эмбрионах мышей был обнаружен особый паттерн экспрессии NBAS, характеризующийся диффузной экспрессией на конечностях на начальных стадиях развития (11-й день) и затем постепенным смещением экспрессии больше к эктодерме и перихондриальной мезодерме (на 13-й день). Аналогичный паттерн наблюдался при окрашивании срезов всего тела: на ганглиях дорсальных корешков и в нервной трубке, а также в коронарном отделе головного мозга (на 13-й день). Также специфичное окрашивание было обнаружено на хондроцитах тел позвонков. Принимая это во внимание, можно сказать, что белок NBAS востребован на начальных этапах развития эмбрионов млекопитающих. Тем не менее эти данные, а именно участие NBAS в раннем физиологическом развитии не объясняет некоторые клинические наблюдения, связанные с нередкой поздней манифестацией ILFS2 (примерно в 6.5 лет), предполагая, что NBAS имеет постоянную роль в жизнедеятельности клетки и не ограничивается физиологическим развитием [14].
Роль NBAS в ретроградном транспорте
Мембранный трафик хорошо изучен у таких организмов как дрожжи и бактерии [15]. Вкратце органеллы, участвующие в секреторном и эндокринном путях клетки, коммуницируют между собой при помощи транзитных везикул (внутри которых, например, незрелые белки), отпочковывающихся от донорной органеллы и транспортирующихся к акцепторной. При отпочковании везикула покрывается специальным покрытием – белками-коатомерами COPI или COPII (Coat Protein), которое она теряет при слиянии с акцепторной органеллой. Сцепка везикул с целевой органеллой осуществляется семейством малых гуанозинтрифосфатгидролаз Rab (Rab-ГТФаз) и связывающими факторами, после чего следует слияние мембран везикулы и органеллы при помощи комплекса SNARE (Soluble NSF Attachment Receptor), локализующегося на мембране акцепторной органеллы. Связывающие факторы, которые регулируют не только сцепку, но и процесс слияния везикулы, можно поделить на две группы: группа белков, содержащих вытянутые суперспирали, и мультисубъединичные комплексы (МТС, Multisubunit Tethering Complex). Таким образом коммуницируют такие органеллы как аппарат Гольджи (аГ) и эндоплазматический ретикулум (ЭР): везикулы покрытые COPII отпочковываются от ЭР и транспортируются в аГ, где затем сливаются с его мембраной, – антероградный транспорт, а от аГ обратно в ЭР возвращаются везикулы покрытые COPI – ретроградный транспорт. Сцепка и слияние COPI с ЭР у дрожжей регулируется комплексом Dsl1. Этот комплекс, включающий Dsl1, Tip20 и Sec39/Dsl3, входит в подсемейство комплексов CATCHR (Complexes Associated with Tethering Containing Helical Rods), являющихся МТС. Механизм действия комплекса заключается в том, что Dsl1 как “лассо” связывается с COPI, в то время как Tip20 и Dsl3 держатся за комплекс SNARE, а конкретно за его компоненты Sec20 и Use1 соответственно. Комплекс SNARE далее, являясь предполагаемым донором механохимической энергии, завершает процесс слияния везикулы с органеллой [16].
Ортологом у млекопитающих комплекса Dsl1 является комплекс NRZ: NBAS – Sec39/Dsl3, RINT1 – Tip20 и ZW10, являющийся ортологом Dsl1 соответственно. Как и комплекс Dsl1, NRZ взаимодействует со SNARE: RINT1 с BNIP1(Sec20) и NBAS c p31(Use1), тем самым выполняя аналогичные функции сцепки и слияния везикулы с ЭР. Хотя белок NBAS является достаточно консервативным среди эукариотов, тем не менее размер белка очень сильно варьирует: от 82 кДа у дрожжей до 270 кДа у человека. Это связано с расширением N-конца – добавлением домена с β-пропеллером, и С-конца с пока что неясной функцией (кроме взаимодействия с белками-партнерами). Расширение гена может свидетельствовать о других функциях, которые выполняет данный ген помимо ретроградного транспорта везикул, например об участии в нонсенс-опосредованном распаде РНК.
Есть несколько работ, исследующих нарушения в мембранном трафике, вызванные мутациями в NBAS [14, 17, 18]. Функциональными исследованиями на клетках HEK293T методом иммунофлуоресценции было показано изменение распределения белка NBAS с усилением свечения патогенного варианта, с мутацией в области домена SEC39, на ЭР в отличие от дикого типа, в то время как в аппарате Гольджи не было замечено видимых изменений между диким типом и мутантом. Однако аналогичное исследование, проведенное на клетках остеосаркомы Saos-2, выявило обратную картину: мутантный вариант теперь показывал низкое свечение у аппарата Гольджи в отличие от дикого типа, а изменений свечения между диким типом и мутантом в области ЭР не наблюдалось. Существующие данные могут свидетельствовать лишь о возможном нарушении ретроградного мембранного трафика при наличии патогенных вариантов в области домена SEC39 белка NBAS.
Исследования взаимодействия с белками-партнерами по ретроградному транспорту в клетках с мутантным вариантом NBAS в области С-конца также не выявили однозначных результатов. Тем не менее методом двойной иммунофлуоресценции на клетках HEK293T было обнаружено частичное снижение связывания белка NBAS, имеющего пропуск 48-го экзона, с ЭР в отличие от дикого типа [14].
С. Staufner с соавт. было проведено функциональное исследование фибробластов 14 пациентов с молекулярно-генетически подтвержденным ILFS2 [18]. Хотя и мутации находились в различных регионах гена и уже сами фибробласты имели сниженный уровень NBAS по сравнению с контролем, было обнаружено кратное уменьшение уровня белка NBAS при смене температурного режима с 37 до 40°С у мутантных фибробластов, в то время как на контрольных образцах повышение температуры не повлияло на уровень белка NBAS. Более того, изменение температуры также повлияло и на уровень роста клеток, снизив его у фибробластов пациентов. Далее методом двойной иммунофлуоресценции после пермеабилизации было обнаружено значительное снижение свечения антител к ERGIC-53 и GPP130 у фибробластов пациентов по сравнению с контролем, данные мембранные белки локализуются на промежуточных компартментах между ЭР-аГ и аГ-эндосомой соответственно. Аналогичное исследование с KDEL-R и GM130 также выявило схожий эффект, но менее выраженный. Данные белки также являются маркерами мембранного трафика.
Тем не менее остается неясным как меняется мембранный трафик при миссенс-мутации, вызывающей SOPH-синдром. Так как данная мутация не затрагивает домен SEC39, связанный с мембранным трафиком, можно спекулятивно ожидать, что возможно мембранный трафик не будет нарушен, но возможна и ситуация, когда при наличии работающего трафика у клетки при мутации SOPH будет снижена толерантность к внешним экстремальным факторам, например повышению температуры, которое особенно у детей с еще неразвитым иммунитетом часто развивается и в повседневном нормальном развитии. Повышение температуры, как было показано в исследованиях, уже являлось триггером острой печеночной недостаточности с нередким летальным исходом, возможно при SOPH-синдроме имеет место накопительный эффект от периодического повышения температуры. Учитывая относительно мягкие клинические проявления SOPH в отличие от ILFS2, исследования различий белковых взаимодействий NBAS при SOPH в этих двух температурных состояниях помогут найти подходы к терапии NBAS-ассоциированных патологий.
Роль NBAS в нонсенс-опосредованном распаде РНК
Nonsense Mediated Decay – это механизм контроля качества и регуляции мРНК на уровне трансляции белка на рибосомах, предотвращающий накопление неправильно собранных белков в клетке. NMD влияет на 5–10% экспрессирующихся генов у различных эукариот [19]. В основном считается, что NMD находит и предотвращает от трансляции транскрипты с преждевременными кодонами терминации (PTC, Premature Termination Codon), возникающие из-за мутаций, ошибок процессинга мРНК, приводящих, например, к ошибкам сплайсинга. Цели NMD не ограничиваются контролем PTC, также ими являются комплексы сращивания экзонов (EJC, Exon Junction Complex), расположенные ниже нетранслируемой области (табл. 1) Стоит отметить, что цели для деградации системой NMD принадлежат к эволюционно консервативным путям. Более того, были найдены вирусные транскрипты, являющиеся целями для распада, что наталкивает на более широкую зону ответственности данной системы, либо на ее низкую специфичность [19]. Составные части NMD – группа белков UPF (Up-Frameshift), их еще называют “коровыми” факторами NMD и белками, стимулирующими UPF к процессам распада транскриптов (SMG1, SMG6 и др.).
Таблица 1.
Механизмы активации и функции NMD (адаптировано со статьи [41])
| Роль NMD | Архитектура транскрипта | Механизм NMD |
|---|---|---|
| Контроль качества | Ошибка сплайсинга | Стимулированный комплексом сращивания экзонов (EJC) |
| PTC во внутреннем экзоне | ||
| PTC во внешнем экзоне | Стимулированный длиной нетранслируемой области (3' UTR) | |
| Нарушенный поли-А конец | ||
| Регуляция | Альтернативный экзон | Стимулированный комплексом сращивания экзонов (EJC) |
| Короткие открытые рамки считывания | ||
| Удлиненный 3' конец нетранслируемой области | Стимулированный длиной нетранслируемой области (3' UTR) |
Пример механизма работы выглядит так: UPF1 и SMG1 вместе с другими факторами NMD образуют комплекс SURF (SMG-1–Upf1–eRF1–eRF3), распознающий PTC, и этот комплекс начинает свой дозор вдоль мРНК. При обнаружении PTC комплекс SURF c помощью дополнительных белков UPF2 и UPF3B связывается с EJC, находящимся ниже PTC, и вместе они образуют комплекс, индуцирующий распад РНК (DECID, decay-inducing complex), который инициирует фосфорилирование хеликазы UPF1 с помощью PI3K-подобной киназы (Phosphatidylinositol 3-Kinase-like kinases) SMG1 (Suppressor of Morphogenesis in Genitalia 1), после чего происходит рекрутирование распад-ассоциированных белков и начинается сам процесс. Белки UPF могут действовать вместе как в данном примере, но также показывают одиночную активность. Существует также и альтернативный способ активации NMD без контакта с EJC, который нацелен на транскрипты с удлиненным 3' UTR.
D. Longman с соавт. был обнаружен локальный путь ER-NMD, звеном которого является белок NBAS, модулирующий трансляцию белков клеточного стресса на ЭР [12]. Последовательный нокдаун генов мембранного трафика и NMD не выявил взаимосвязи этих двух процессов, подтверждая двоякую и при этом независимую роль NBAS, принимающего участие в обоих процессах. РНК-профилирование генов, связанных с нокдауном NBAS и UPF1, выявило кратные изменения экспрессии генов, связанных с клеточным стрессом ЭР, что может свидетельствовать о том, что при нарушении работы NBAS нарушается толерантность клетки к стрессовым факторам. Локализация трансляции регулируемых NBAS и UPF1 транскриптов именно в районе ЭР, а не в цитоплазме, была подтверждена методом флуоресцентной гибридизации in situ (FISH). Прямая связь NBAS–UPF1 с ЭР, а именно с его белком SEC61, формирующим канал транслокона, была показана методом PLA (proximity ligation assay). Результаты данных исследований могут являться подтверждением того, что NBAS находится в непосредственной физической близости к процессу трансляции, являющемуся активирующим фактором NMD.
Для того чтобы узнать на каком этапе происходит участие NBAS в процессе NMD, были использованы два мутантных варианта UPF1: в гипофосфорилированном состоянии, имитирующем состояния UPF1 в комплексе SURF, и гиперфосфорилированном состоянии для имитации состояния в комплексе DECID. Более предпочтительной связью для NBAS оказался комплекс SURF, образующийся на начальных этапах NMD, что может также свидетельствовать о том, что большинство регуляторных шагов процесса распада РНК проходит на начальных этапах. Интересно, что NBAS показывает временное взаимодействие и с SMG5, SMG6, SMG7, участвующими на более поздних этапах NMD.
Результаты данных исследований подтверждают непосредственное участие NBAS в процессах нонсенс-опосредованного распада РНК. Сложно сказать, где кончается функция контроля качества и начинается функция регуляции транскриптов [20]. Неясно и то, как система NMD “видит” свои цели для деградации, тем не менее считается, что белки-участники NMD могут распознавать конформацию рибонуклеопротеинов (mRNP, Messenger Ribonucleoprotein), несущих нарушенные мРНК. Как известно, мРНК после синтеза не перемещается до рибосомы в чистом виде, а нуждается в несущих белках-переносчиках, которыми выступают mRNP. Данный комплекс представляет из себя гранулы самособирающихся белков, содержащих РНК и РНК-связывающие белки. Одной из основных функций данного комплекса является пространственно-временная организация мРНК для дальнейшей трансляции [21]. При возникновении нарушений в структуре мРНК предполагается, что меняется и архитектура крепления мРНК к mRNP, что впоследствии распознается системой NMD.
При отсутствии или снижении функции NMD происходит накопление неправильно собранных белков в клетке, ассоциированных, например, с такими заболеваниями как болезнь Альцгеймера, Паркинсона и др. Тем не менее нет научных исследований того, как мутации, например при SOPH-синдроме, могут нарушить процесс NMD. Учитывая фундаментальную роль данного клеточного механизма, можно спекулятивно сказать, что патогенные варианты в NBAS не только сами могут накапливаться в клетке, нарушая ее работу, но и нарушенный NBAS сам не может выполнять свою роль в NMD по контролю качества транскриптов, что приводит к еще большему ущербу клетки. Все же даже если это так, то остается не совсем ясным почему одни мутации вызывают при малейшем клеточном стрессе летальный исход, а другие развиваются только с возрастом и, по-видимому, не имеют смертельных рисков.
КЛИНИКА
Пациенты с мутацией в NBAS имеют большое фенотипическое разнообразие признаков, характеризующееся мультисистемным поражением органов и тканей. Ввиду относительной редкости данных наследственных патологий в мире в основном данные о пациентах из литературы можно получить от клинических случаев, которые не могут предоставить исчерпывающий систематический анализ спектра признаков при патогенных вариантах NBAS.
Тем не менее международный многоцентровой обзор имеющихся на тот момент научных данных о клинике мутаций NBAS 110 пациентов, проведенный С. Staufner с соавт., выявил генотип-фенотипическую зависимость локализации миссенс-мутаций или делеций от фенотипических проявлений [22]. Авторы выделили три генотип-фенотипические группы в зависимости от локализации мутации: группа с патогенными вариантами в регионе β-пропеллера (1), в регионе SEC39 (2) и на С-конце (3). Группа с мутациями на С-конце включает SOPH-подобные состояния, группа SEC39 включает пациентов с ILFS2. Стоит отметить, что регион с β-пропеллером характеризуется перекрытием фенотипических признаков SOPH и ILFS2 (табл. 2). Данные исследования могут быть использованы для поиска подходов к терапии подобных состояний, так как зная регион мутации можно спрогнозировать предрасположенность к развитию острой печеночной недостаточности и начать своевременную поддерживающую терапию у данных пациентов [4, 23].
Таблица 2.
Клинические проявления трех генотип-фенотипических групп (1, 2, 3). Перечислены отдельные фенотипические признаки, характерные для одних и отсутствующие для других состояний, ассоциированных с мутациями в гене NBAS (адаптировано с работы [22])
| Фенотипический признак | β-пропеллер (1) | SEC39 (2) | С-конец (3) |
|---|---|---|---|
| Эпизод острой печеночной недостаточности | Очень часто | Очень часто | Очень редко |
| Постоянно повышенные трансаминазы | Очень редко | Очень редко | Часто |
| Низкий рост | Часто | Редко | Очень часто |
| Остеопороз | Часто | Не обнаружено | Очень часто |
| Задержка закрытия родничка | Часто | Очень редко | Очень часто |
| Дисплазия тел позвонков, аномалии позвонка | Часто | Не обнаружено | Часто |
| Атрофия зрительного нерва | Очень редко | Не обнаружено | Очень часто |
| Задержка моторного развития | Часто | Очень редко | Очень часто |
| Нарушения интеллекта | Не обнаружено | Очень редко | Редко |
| Сенильная кожа | Редко | Очень редко | Часто |
| Низкий уровень IgG | Редко | Очень редко | У всех обнаруженных случаев |
| Низкий уровень NK-клеток | Часто | Часто | Очень часто |
| Пельгеровская аномалия лейкоцитов | Часто | Очень редко | Очень часто |
| Гипотония мышц | Редко | Очень редко | Очень часто |
| Скелетно-мышечная атрофия | Часто | Не обнаружено | Часто |
SOPH-синдром
В данный синдром изначально были включены пациенты с постнатальной гипоплазией, низким пропорциональным ростом, лицевыми дизморфиями с частичной атрофией зрительного нерва и пельгеровской аномалией лейкоцитов в общем анализе крови. При этом у данной группы были исключены эндокринологические патологии и отсутствовали интеллектуальные нарушения.
Дети с SOPH-синдромом рождаются в срок и с нормальными показателями роста и веса. Первые жалобы появляются после трехлетнего возраста на отставания в росте, частые простудные заболевания и снижение зрения. Фенотипические особенности характеризуются дряблой и старческой кожей, легким экзофтальмом, тонкими губами и пропорциональным туловищем при низком росте [1].
С появлением все более новых данных о пациентах с SOPH-подобными состояниями клинический спектр SOPH только расширяется, но основные фенотипические признаки, определенные в названии данного синдрома, как правило присутствуют [24, 25].
ILFS2
Синдром детской печеночной недостаточности 2-го типа является жизнеугрожающей патологией. По данным исследований в 50% всех случаев этиология острой печеночной недостаточности у детей остается неизвестной. Мутации в NBAS были ассоциированы с эпизодами RALF (Recurrent Acute Liver Failure). Прогноз для детей до года с RALF неблагоприятен. Зачастую единственный вариант лечения – трансплантация печени.
Клиника характеризуется частыми приступами печеночной недостаточности, тем не менее с возрастом частота эпизодов RALF спадает. Пациенты доставляются в клинику в тяжелом состоянии, характеризующемся рвотой и нарастающей вялостью. На лабораторных анализах повышены АЛТ и АСТ, обнаруживается тяжелая коагулопатия с печеночной желтухой. Интересно, что ЩФ и ГГТП как правило в пределах нормы [18]. Тем не менее если пациент выживает после приступа, то симптомы проходят через дни или недели с полным восстановлением лабораторных показателей. Как правило первые эпизоды ILFS2 самые тяжелые.
Дополнительно к печеночному фенотипу также могут присутствовать кардиомиопатия, неврологические симптомы, а также синдромы поражения ЖКТ вместе с иммунологическими нарушениями.
Другие состояния, ассоциированные с NBAS
Обнаружены также несколько компаунд-мутаций, связанных с несовершенным остеогенезом и первичным иммунодефицитом у детей [26–31]. Помимо данных патологий мутации в гене также обнаруживались при таких состояниях как сахарный диабет 2-го типа, эпилепсия, синдром прерывания ножки гипофиза, атопический дерматит, акрофронтофационазальный дизостоз 1-го типа и др. [14, 32–39]. Список новых состояний, при которых обнаруживаются новые варианты NBAS, пополняется с каждым годом благодаря распространению в клинической практике технологии массового параллельного секвенирования.
МОЛЕКУЛЯРНАЯ ДИАГНОСТИКА
На данный момент диагностика проводится методами ПЦР для скринингового поиска уже известных мутаций, в частности в диагностике SOPH. Секвенирование NGS используется при пограничных состояниях, когда поиск известных мутаций не дал положительных результатов и имеется перекрытие имеющихся симптомов с описанными ранее [17, 33, 40]. Функциональные исследования фибробластов также могут быть использованы для подтверждения дефицита белка NBAS, но по некоторым данным это может не обнаруживаться [24].
ЗАКЛЮЧЕНИЕ И ПЕРСПЕКТИВЫ
Белок NBAS играет двоякую роль в жизнедеятельности клетки, фенотипический спектр мутаций в гене NBAS также обладает широким полиморфизмом. Дальнейшие исследования, направленные на изучение патофизиологии каждого конкретного клинического синдрома, опираясь на имеющиеся фундаментальные знания о функции белка, помогут исследователям найти подходы к лечению данных и возможно на сегодняшний день еще неизвестных NBAS-ассоциированных состояний.
Коллектив авторов благодарит Анастасию Русанову за помощь и консультацию в графическом оформлении молекулярной структуры гена и белка NBAS.
Работа выполнена в рамках Государственного задания Министерства науки и высшего образования Российской Федерации на тему: “Геномика Арктики: эпидемиология, наследственность и патология” (номер проекта № FSRG-2020-0014).
Настоящая статья не содержит каких-либо исследований с использованием в качестве объекта животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Maksimova N., Hara K., Nikolaeva I. et al. Neuroblastoma amplified sequence gene is associated with a novel short stature syndrome characterised by optic nerve atrophy and Pelger-Huet anomaly // J. Med. Genet. 2010. V. 47. № 8. P. 538–548. https://doi.org/10.1136/jmg.2009.074815
Максимова Н.Р., Ноговицына А.Н., Куртанов Х.А., Алексеева Е.И. Популяционная частота и возраст мутации G5741 → A в гене NBAS, являющейся причиной SOPH-синдрома в республике Саха (Якутия) // Генетика. 2016. Т. 52. № 10. С. 1194–1201. https://doi.org/10.7868/S0016675816090101
Haack T.B., Staufner C., Köpke M.G. et al. Biallelic mutations in NBAS cause recurrent acute liver failure with onset in infancy // Am. J. Hum. Genet. 2015. V. 97. № 1. P. 163–169. https://doi.org/10.1016/j.ajhg.2015.05.009
Calvo P.L., Tandoi F., Haak T.B. et al. NBAS mutations cause acute liver failure: When acetaminophen is not a culprit // Ital. J. Pediatr. 2017. V. 43. № 1. P. 88. https://doi.org/10.1186/s13052-017-0406-4
Aoki T., Ichimura S., Itoh A. et al. Identification of the neuroblastoma-amplified gene product as a component of the syntaxin 18 complex implicated in golgi-to-endoplasmic reticulum retrograde transport // Mol. Biol. Cell. V. 2009. V. 20. № 11. P. 2639–2649. https://doi.org/10.1091/mbc.e08-11-1104
Longman D., Jackson-Jones K.A., Maslon M.M. et al. Identification of a localized nonsense-mediated decay pathway at the endoplasmic reticulum // Genes Dev. 2020. V. 34. № 15–16. P. 1075–1088. https://doi.org/10.1101/gad.338061.120
Scott D.K., Board J.R., Lu X. et al. The neuroblastoma amplified gene, NAG: Genomic structure and characterisation of the 7.3 kb transcript predominantly expressed in neuroblastoma // Gene. 2003. V. 307. P. 1–11. https://doi.org/10.1016/S0378-1119(03)00459-1
De Preter K., Speleman F., Combaret V. et al. Quantification of MYCN, DDX1, and NAG gene copy number in neuroblastoma using a real-time quantitative PCR assay // Mod. Pathol. 2002. V. 15. № 2. P. 159–166. https://doi.org/10.1038/modpathol.3880508
Kaneko S., Ohira M., Nakamura Y. et al. Relationship of DDX1 and NAG gene amplification/overexpression to the prognosis of patients with MYCN-amplified neuroblastoma // J. Cancer Res. Clin. Oncol. 2007. V. 133. № 3. P. 185–192. https://doi.org/10.1007/s00432-006-0156-y
Wimmer K., Zhu X.X., Lamb B.J. et al. Co-amplification of a novel gene, NAG, with the N-myc gene in neuroblastoma: 1 // Oncogene. Nature Publ. Group. 1999. V. 18. № 1. P. 233–238. https://doi.org/10.1038/sj.onc.1202287
Anastasaki C., Longman D., Capper A. et al. Dhx34 and Nbas function in the NMD pathway and are required for embryonic development in zebrafish // Nucl. Acids Res. 2011. V. 39. № 9. P. 3686–3694. https://doi.org/10.1093/nar/gkq1319
Longman D., Jackson-Jones K.A., Maslon M.M. et al. Identification of a localized nonsense-mediated decay pathway at the endoplasmic reticulum // Genes Dev. 2020. V. 34. № 15–16. P. 1075–1088. https://doi.org/10.1101/gad.338061.120
Jing H., Takagi J., Liu J. et al. Archaeal surface layer proteins contain β propeller, PKD, and β helix domains and are related to metazoan cell surface proteins // Structure. 2002. V. 10. № 10. P. 1453–1464. https://doi.org/10.1016/S0969-2126(02)00840-7
Palagano E., Zuccarini G., Prontera P. et al. Mutations in the neuroblastoma amplified sequence gene in a family affected by acrofrontofacionasal dysostosis type 1 // Bone. 2018. V. 114. P. 125–136. https://doi.org/10.1016/j.bone.2018.06.013
Tagaya M., Arasaki K., Inoue H. et al. Moonlighting functions of the NRZ (mammalian Dsl1) complex // Front. Cell Dev. Biol. 2014. V. 2. https://doi.org/10.3389/fcell.2014.00025
Travis S.M., DAmico K., Yu I.-M. et al. Structural basis for the binding of SNAREs to the multisubunit tethering complex Dsl1 // J. Biol. Chem. 2020. V. 295. № 30. P. 10125–10135. https://doi.org/10.1074/jbc.RA120.013654
Ritelli M., Palagano E., Cinquina V. et al. Genome-first approach for the characterization of a complex phenotype with combined NBAS and CUL4B deficiency // Bone. 2020. V. 140. P. 115571. https://doi.org/10.1016/j.bone.2020.115571
Staufner C., Haack T.B., Köpke M.G. et al. Recurrent acute liver failure due to NBAS deficiency: phenotypic spectrum, disease mechanisms, and therapeutic concepts // J. Inherit. Metab. Dis. 2016. V. 39. № 1. P. 3–16. https://doi.org/10.1007/s10545-015-9896-7
Kishor A., Fritz S.E., Hogg J.R. Nonsense-mediated mRNA decay: The challenge of telling right from wrong in a complex transcriptome // WIREs RNA. 2019. V. 10. № 6. https://doi.org/10.1002/wrna.1548
Isken O., Maquat L.E. The multiple lives of NMD factors: Balancing roles in gene and genome regulation // Nat. Rev. Genet. 2008. V. 9. № 9. P. 699–712. https://doi.org/10.1038/nrg2402
Buchan J.R. mRNP granules // RNA Biol. 2014. V. 11. № 8. P. 1019–1030. https://doi.org/10.4161/15476286.2014.972208
Staufner C., Peters B., Wagner M. et al. Defining clinical subgroups and genotype–phenotype correlations in NBAS-associated disease across 110 patients // Genet. Med. 2020. V. 22. № 3. P. 610–621. https://doi.org/10.1038/s41436-019-0698-4
Geem D., Jiang W., Rytting H.B. et al. Resolution of recurrent pediatric acute liver failure with liver transplantation in a patient with NBAS mutation // Pediatr. Transplant. 2021. V. 25. № 7. P. e14084. https://doi.org/10.1111/petr.14084
Fischer-Zirnsak B., Koenig R., Alisch F. et al. SOPH syndrome in three affected individuals showing similarities with progeroid cutis laxa conditions in early infancy // J. Hum. Genet. 2019. V. 64. № 7. P. 609–616. https://doi.org/10.1038/s10038-019-0602-8
Li X., Cheng Q., Li N. et al. SOPH syndrome with growth hormone deficiency, normal bone age, and novel compound heterozygous mutations in NBAS // Fetal Pediatr. Pathol. 2018. V. 37. № 6. P. 404–410. https://doi.org/10.1080/15513815.2018.1509406
Khoreva A., Pomerantseva E., Belova N. et al. Complex multisystem phenotype with immunodeficiency associated with NBAS mutations: reports of three patients and review of the literature // Front. Pediatr. 2020. V. 8. P. 577. https://doi.org/10.3389/fped.2020.00577
Lenz D., Pahl J., Hauck F. et al. NBAS variants are associated with quantitative and qualitative NK and B cell deficiency // J. Clin. Immunol. 2021. V. 41. № 8. P. 1781–1793. https://doi.org/10.1007/s10875-021-01110-7
Balasubramanian M., Hurst J., Brown S. et al. Compound heterozygous variants in NBAS as a cause of atypical osteogenesis imperfecta // Bone. 2017. V. 94. P. 65–74. https://doi.org/10.1016/j.bone.2016.10.023
Petukhova D.A., Gurinova E.E., Sukhomyasova A.L. et al. Identification of a novel compound heterozygous variant in NBAS causing bone fragility by the type of osteogenesis imperfecta // Bioinformatics Research and Applications / Eds Cai Z. et al. Cham: Springer Intern. Publ. 2020. V. 12304. P. 38–43. https://doi.org/10.1007/978-3-030-57821-3_4
Cotrina-Vinagre F.J., Rodríguez-García M.E., Martín-Hernández E. et al. Characterization of a complex phenotype (fever-dependent recurrent acute liver failure and osteogenesis imperfecta) due to NBAS and P4HB variants // Mol. Genet. Metab. 2021. V. 133. № 2. P. 201–210. https://doi.org/10.1016/j.ymgme.2021.02.007
Ricci S., Lodi L., Serranti D. et al. Immunological features of neuroblastoma amplified sequence deficiency: Report of the first case identified through newborn screening for primary immunodeficiency and review of the literature // Front. Immunol. 2019. V. 10. P. 1955. https://doi.org/10.3389/fimmu.2019.01955
Suzuki S., Kokumai T., Furuya A. et al. A 34-year-old Japanese patient exhibiting NBAS deficiency with a novel mutation and extended phenotypic variation // Eur. J. Med. Genet. 2020. V. 63. № 11. P. 104039. https://doi.org/10.1016/j.ejmg.2020.104039
Kim K.W., Myers R.A., Lee J.H. et al. Genome-wide association study of recalcitrant atopic dermatitis in Korean children // J. Allergy Clin. Immunol. 2015. V. 136. № 3. P. 678–684. https://doi.org/10.1016/j.jaci.2015.03.030
Zou J., Zhao Z., Zhang G. et al. MEFV, IRF8, ADA, PEPD, and NBAS gene variants and elevated serum cytokines in a patient with unilateral sporadic Meniere’s disease and vascular congestion over the endolymphatic sac // J. Otol. 2022. https://doi.org/10.1016/j.joto.2022.03.001
Mallakmir S., Nagral A., Bagde A. et al. Mutation in the neuroblastoma amplified sequence gene as a cause of recurrent acute liver failure, acute kidney injury, and status epilepticus // J. Clin. Exp. Hepatol. 2019. V. 9. № 6. P. 753–756. https://doi.org/10.1016/j.jceh.2019.03.008
Lipiński P., Greczan M., Piekutowska-Abramczuk D. et al. NBAS deficiency due to biallelic c.2809C > G variant presenting with recurrent acute liver failure with severe hyperammonemia, acquired microcephaly and progressive brain atrophy // Metab. Brain Dis. 2021. V. 36. № 7. P. 2169–2172. https://doi.org/10.1007/s11011-021-00827-z
Dayan R.R., Bignall O.N.R. II, Johnson S. et al. Neuroblastoma amplified sequence gene mutations inducing acute kidney and liver injury in an adolescent female // Case Rep. Nephrol. Dial. 2020. V. 10. № 3. P. 117–123. https://doi.org/10.1159/000508784
Costantini A., Valta H., Suomi A.-M. et al. Oligogenic inheritance of monoallelic TRIP11, FKBP10, NEK1, TBX5, and NBAS variants leading to a phenotype similar to odontochondrodysplasia // Front. Genet. 2021. V. 12. P. 680838. https://doi.org/10.3389/fgene.2021.680838
Brauner R., Bignon-Topalovic J., Bashamboo A. et al. Pituitary stalk interruption syndrome is characterized by genetic heterogeneity // PLoS One. 2020. V. 15. № 12. P. e0242358. https://doi.org/10.1371/journal.pone.0242358
Zhernakova D.V., Brukhin V., Malov S. et al. Genome-wide sequence analyses of ethnic populations across Russia // Genomics. 2020. V. 112. № 1. P. 442–458. https://doi.org/10.1016/j.ygeno.2019.03.007
Nasif S., Contu L., Mühlemann O. Beyond quality control: The role of nonsense-mediated mRNA decay (NMD) in regulating gene expression // Semin. Cell Dev. Biol. 2018. V. 75. P. 78–87. https://doi.org/10.1016/j.semcdb.2017.08.053
Дополнительные материалы отсутствуют.