

Геохимия, 2020, T. 65, № 5, стр. 452-463
Новая реакция при формировании состава нефтей: метилирование ароматических циклов
М. Б. Смирнов a, *, Р. С. Борисов a, Н. А. Ванюкова a
a Федеральное государственное бюджетное учреждение науки Ордена Трудового Красного Знамени
Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Ленинский просп., 29, Россия
* E-mail: m1952s@yandex.ru
Поступила в редакцию 05.02.2019
После доработки 14.05.2019
Принята к публикации 18.07.2019
Аннотация
Показано, что одним из процессов, формирующих состав ароматических углеводородов нефтей, в отдельных регионах является реакция электрофильного метилирования ароматических циклов. В нефтях до 40% от исходных соединений может оказаться преобразованными в продукты метилирования. Показано, что метилирование протекает, скорее всего, на стадии катагенеза, следующей после завершение генерации того флюида, из которого сформировалась изучаемая залежь нефти. Метилирование ароматических циклов является четким индикатором того, что нефтематеринскими породами являются либо глины, либо силициты с существенной долей глинистого материала. Установлено, что реакция метилирования не является общераспространенной даже в пределах одного бассейна. Следовательно, анализ на наличие продуктов электрофильного метилирования ароматических соединений может быть эффективно использован при геохимических реконструкциях. Отмечено, что следствием метилирования может стать изменение содержания изомеров ароматических соединений, используемых при расчетах коэффициентов зрелости, что приведет к некорректной интерпретации получаемых результатов.
ВВЕДЕНИЕ
В настоящее время утвердилось следующее представление о путях образования ароматических соединений нефтей. На первом этапе протекают реакции циклизации, элиминирования функциональных групп (в первую очередь – спиртовых) с образованием двойных С=С-связей и реакции переноса водорода (дегидрирования, диспропорционирования). Образование ароматических циклов может сопровождаться реакциями удаления метильных групп и их переноса в иные положения циклической системы. Далее имеют место реакции изомеризации, деструкции алкильных цепей и, в отдельных случаях, как, например, при трансформации исходных ароматических каротиноидов, специфические реакции элиминирования отдельных фрагментов молекул. На этой стадии могут протекать и реакции циклизации (Тиссо, Вельте, 1981; Петров, 1984; Peters et al., 2005; Остроухов и др., 1983; Остроухов, 2000; Koopmans et al., 1996; Kagi et al., 1990; Radke et al., 1984; Radke et al., 1986; Radke, 1988; Burnham et al., 1997; Burnham et al., 1998). Среди полиароматических углеводородов рассеянного органического вещества (РОВ) обнаружены соединения, которые могут являться продуктами радикального присоединения фенила к нафталину, фенантрену и антрацену (Rospondek et al., 2009). Кроме того, в качестве возможных рассматривали ряд реакций, например, алкилирование ароматических соединений альфа-олефинами или алифатическими радикалам, образующимися при крекинге алкильных цепей (Burnham et al., 1997; Burkle-Vitzthum et al., 2004; Domine et al., 2002; Гончаров и др., 2000), трансметилирование, при котором акцептором метильных групп являются фенантрены (Szczerba, Rospondek, 2010) или н-алкилтолуолы (Ellis et al., 1995), а донорами – полиметилзамещенные бензолы. Но однозначных свидетельств того, что именно эти процессы имеют место в природе, в литературе не найдено за исключением, быть может, единственной нефти (Гончаров и др., 2000). Эти представления лежат в основе всех проводимых на основе данных о составе ароматических соединений геохимических реконструкций, включая общеизвестные критерии зрелости органического вещества (Peters et al., 2005; Radke et al., 1984; Radke et al., 1986; Radke, 1988).
В настоящей работе представлены данные, свидетельствующие о том, что одним из процессов, формирующих состав ароматических углеводородов нефтей, в отдельных регионах является реакция электрофильного метилирования ароматических циклов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объектами изучения служили около 60 образцов битумоидов РОВ из отложений пашийского, семилукского, саргаевского горизонтов (верхний девон) и 40 нефтей (возраст коллекторов – от среднего девона до среднего карбона) Волго-Уральского нефтегазоносного бассейна России. Общая характеристика состава нефтей бассейна приведена в (Смирнов, Ванюкова, 2015); данные о составе биомаркеров нефтей и РОВ (в основном – для территории Татарстана) представлены в (Арефьев и др., 1994; Каюкова и др., 2006; Гордадзе, Тихомиров, 2007; Гордадзе, Тихомиров, 2005; Каткова и др., 2004; Aizenshtat et al., 1998; Смирнов, Полудеткина, 2018; Смирнов и др., 2018а). Почти все данные касаются состава насыщенных углеводородов.
После стандартного осаждения асфальтенов гексаном полученные из нефтей и битумоидов мальтены разделяли на силикагеле (Merck), импрегнированном AgNO3. При выделении фракции насыщенных углеводородов элюент – гексан, ароматических соединений – толуол. Полнота разделения подтверждена данными газохроматомасс-спектрометрии (ГХ/МС) с ионизацией электронами.
ГХ/МС анализ проводили на газохроматомасс-спектрометре Thermo Focus DSQ II. Использована капиллярная колонка HP-5, длина 15 м, внутренний диаметр 0.25 мм, толщина фазы 0.25 мкм, газ-носитель-гелий. Режим работы: температура инжектора 300°С, начальная температура печи хроматографа – 70°С, нагрев со скоростью 2°С/мин до 310°С, после чего – изотерма в течении 20 мин; режим работы масс-спектрометра: ионизация электронами (энергия ионизации 70 эВ), температура источника 250°С, сканирование в диапазоне 10–650 Да со скоростью 1.0 скан/с, разрешение единичное по всему диапазону масс.
Количественные характеристики отдельных соединений и их групп определяли с использованием площадей хроматографических пиков на хроматограммах, построенных по соответствующим характеристичным ионам: для н-алкилбензолов – ионы с m/z = 91 и m/z = 92, н-алкилнафталинов – m/z = = 141 и m/z = 142, н-алкилтолуолов – m/z = 105 и m/z = 106, метил-н-алкилнафталинов – m/z = 155 и m/z = 156.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
При анализе состава фракций ароматических соединений ряда нефтей было обнаружено нестандартное молекулярно-массовое распределение н-алкилбензолов (I) и α-н-алкилнафталинов (II), а именно – существенно повышенные концентрации С21 и С23 гомологов (рис. 1). Для количественной характеристики превышения содержания этих гомологов использованы коэффициенты КX, равные отношению площадей суммы соответствующих ионов данного гомолога к среднему геометрическому площадей сумм тех же ионов с числом С‑атомов, на 1 большим и на 1 меньшим. Например, для С21н-алкилбензола КБ, 21 = S21(91 + + 92)/[S20(91 + 92) × S22(91 + 92)]0.5, где S – площади пиков на масс-фрагментограммах, построенных по указанным в скобках ионам, нижние индексы указывают на число С-атомов в гомологе, индекс “Б” обозначает, что коэффициент вычислен для н-алкилбензолов, “Н” – н-алкилнафталинов, “Т” – н-алкилтолуолов. Наибольшие значения коэффициентов составили: КБ, 21 = 5.5, КБ, 23 = 6.5, Кα-Н, 21 = 8.6, Кα-Н, 23 = 7.2. В ряду β-н-алкилнафталинов К21 и К23 = 1.0 с точностью до ошибки измерения. Детальный анализ распределения этих классов соединений и следствия из него применительно к дифференциации нефтей Татарстана приведены в (Смирнов и др., 2018б).
Рис. 1.
Типичное молекулярно-массовое распределение н-алкилбензолов (верхний) и н-алкилнафталинов (нижний) в образцах с высоким содержанием С21 и С23 гомологов. Числа над пиками здесь и далее соответствуют числу С-атомов в соединении.
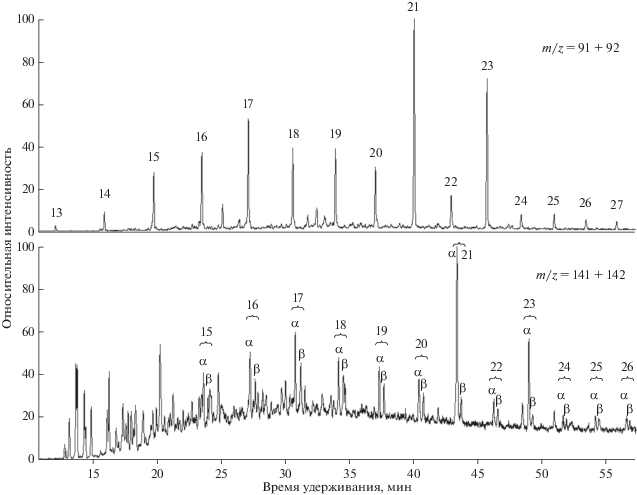
Повышенная концентрация С21-н-алкилбензолов и н-алкилнафталинов, а также некоторых других рядов н-алкилароматических соединений наблюдали в нефтях разных регионов и ранее (Williams et al., 1988; Connan et al., 1995; Гончаров и др., 2000, 2003; Остроухов, 2009; Иванова, Каширцев, 2010; Бушнев, Валяева, 2015; Бушнев и др., 2017). Наиболее убедительной возможной схемой их образования представляется предложенная в (Остроухов, 2009), где исходным соединением является полиеновый пигмент V и образование наблюдаемых в нефтях соединений происходит в результате его циклизации, восстановления функциональных групп и гидрирования остающихся после циклизации двойных связей (рис. 2). Отмечены также повышенные концентрации гомологов С23 (Williams et al., 1988; Connan et al., 1995; 26, Иванова, Каширцев, 2010; Бушнев, Валяева, 2015, Бушнев и др., 2017, Гончаров и др., 2003), (в (Connan et al., 1995) это следует из приведенной масс-фрагментограммы), хотя содержание компонентов С23 везде по крайней мере вдвое меньше, чем гомологов С21. Количественные оценки имеются только для н-алкилбензолов в нефти Талаканского месторождения (Восточная Сибирь) (Иванова, Каширцев, 2010) и нефтей Западной Сибири (Гончаров и др., 2003). Следует отметить, что наблюдаемое в (Иванова, Каширцев, 2010) распределение н-алкилбензолов отличалось от прочих явно проявляющейся нечетностью во всем диапазоне от С17 до С27 (например, КБ, 25 = 2.0 при КБ, 21 = 6,.5).
Из цитированных работ только в (Williams et al., 1988; Connan et al., 1995) наряду с н-алкилбензолами качественно рассматривали ряды н-алкилтолуолов (III) и диметил-н-алкилбензолов (последние – суммарно). Все три изученные в (Connan et al., 1995) пробы характеризовались повышенными концентрациями С22н-алкилтолуолов и С23 диметил-н-алкилбензолов. Объяснения этому факту предложено не было. В (Williams et al., 1988) установлено также повышенное содержание двух изомеров монометилзамещенных н-алкилнафталинов С22 и С24.
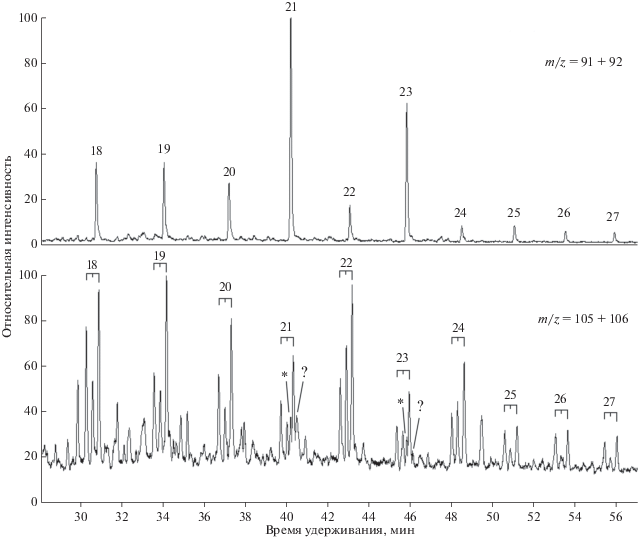
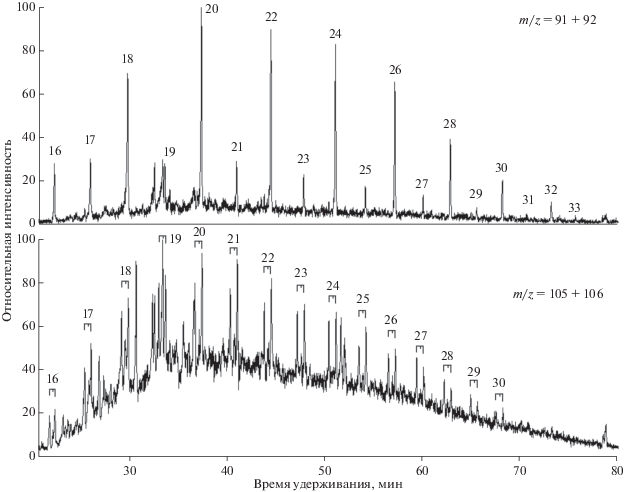
Во всех изученных в данной работе пробах, для которых КБ, 21 > 2.5, наблюдалась повышенная концентрация всех трех изомеров н-алкилтолуолов С22 и С24 (рис. 3). При этом выявлена следующая закономерность: во всех пробах Кп-Т, 22 > Ко-Т, 22 > Км-Т, 22 (п-, о-, м- – пара-, орто- и мета-изомеры; индекс “Т” указывает, что рассматриваются соответствующие изомеры н-алкилтолуолов) и то же – для гомологов С24. Наибольшая величина Кп-Т, 22 = 3.9, Ко-Т, 22 = 2.7, Км-Т, 22 = 1.9. То, что наибольшее превышение наблюдается для пара-, а наименьшее – для мета-изомеров свидетельствует в пользу того, что “дополнительные по сравнению с фоном” С22 и С24н-алкилтолуолы являются продуктами электрофильного замещения метильной группой бензольного цикла. Действительно, пусть исходными для этих алкилтолуолов являются иные соединения с соответствующим числом С-атомов. Согласно существующим представлениям, на первом этапе основным среди образующихся н-алкилтолуолов является орто-изомер, который в процессе созревания изомеризуется, причем при достижении равновесия преобладающим становится мета-изомер, а пара-изомеры в течение всего процесса изомеризации остаются минорной компонентой (см (Бушнев, Валяева, 2015) и цитируемую там литературу). При такой схеме получить наблюдаемую картину невозможно. Второй теоретически возможный вариант – наличие исходного соединения, непосредственным продуктом превращения которого является С22 или С24пара-н-алкилтолуол. Тогда поскольку изомеризация должна идти в последовательности пара- → мета- → орто- и мета-изомер имеет меньшую свободную энергию Гиббса, чем орто-, а среди “фоновых концентраций” везде орто-компонентов больше, чем мета-, должно получиться Км-Т, 22 > Ко-Т, 22, тогда как наблюдается обратное соотношение: Ко-Т, 22 > Км-Т, 22. При нуклеофильном замещении н-алкилбензолов среди продуктов преобладает мета-изомер, при радикальной реакции особой селективности ожидать не приходится.
Рис. 3.
Типичное молекулярно-массовое распределение н-алкилтолуолов (нижний) в образцах с высоким содержанием С21 и С23н-алкилбензолов (верхний) при наличии электрофильного метилирования. Порядок выхода н-алкилтолуолов: мета-, пара-, орто-изомеры (Остроухов и др., 1983; Остроухов, 2015). На масс-фрагментограмме по пикам m/z = 105 + 106 знаком “*” отмечены пики н-алкилбензолов С21 и С23, “?” – неидентифицированные пики, частично перекрывающиеся с пиками н-алкилтолуолов.
Величины К зависят не только от соотношения масс образовавшихся продуктов, но и от “фоновых” концентраций изомеров. Для более корректного анализа вопроса перейдем от величин К к рассмотрению концентраций компонентов, превышающих “фоновые”. Массовое содержание (в %) компоненты X (CX), у которой площадь на хроматограмме, построенной по некоторому иону (группе ионов) j составляет SX,j % от полного ионного тока образца может быть вычислено при известных величинах доли площади данного иона (или их группы) от полного ионного тока компоненты X $k_{{X,j}}^{t}$, отнесенной к единице массы относительной величины сечения ионизации компоненты X $k_{{X,}}^{i}$ и отнесенной к единице массы средней относительной величины сечения ионизации для образца $k_{{ev}}^{i}{\text{:}}$
(1)
${{C}_{X}} = {{{{S}_{{X,j}}}k_{{ev}}^{i}} \mathord{\left/ {\vphantom {{{{S}_{{X,j}}}k_{{ev}}^{i}} {\left( {k_{{X,j}}^{t}k_{X}^{i}} \right)}}} \right. \kern-0em} {\left( {k_{{X,j}}^{t}k_{X}^{i}} \right)}}.$По данным (Остроухов и др., 1983; Остроухов, 2015), масс-спектральной базы данных NIST 14 (NIST/EPA/NIH) и собственным измерениям величины $k_{{X,j}}^{t}$ в пределах ряда (н-алкилбензолы, н‑алкилтолуолы и т.д.) с высокой точностью не зависят от молекулярной массы при длине цепи по крайней мере начиная с С8. Так, для н-алкилбензолов измеренные разницы этого коэффициента лежат в пределах ±4 отн. % от среднего значения $k_{{X,j}}^{{t,ev}},$ равного 0.50, что соответствует ошибке измерения. Из указанных данных определено, что для н-алкилнафталинов $k_{{X,j}}^{t}$ = 0.55, для н-алкилтолуолов – 0.53. Сечение ионизации, отнесенное к единице массы в пределах одного такого ряда согласно (Бродский, 1985; Хмельницкий и др., 1980) можно считать не зависящим от молекулярной массы. То есть величина ${{{{S}_{{X,j}}}} \mathord{\left/ {\vphantom {{{{S}_{{X,j}}}} {k_{{X,j}}^{{t,ev}}}}} \right. \kern-0em} {k_{{X,j}}^{{t,ev}}}}$ в пределах соединений одного ряда пропорциональна CX с коэффициенттом пропорциональности aX = ${{k_{{ev}}^{i}} \mathord{\left/ {\vphantom {{k_{{ev}}^{i}} {k_{X}^{i}}}} \right. \kern-0em} {k_{X}^{i}}}{\text{:}}$
(2)
${{C}_{X}} = {{{{S}_{{X,j}}}{{a}_{X}}} \mathord{\left/ {\vphantom {{{{S}_{{X,j}}}{{a}_{X}}} {k_{{X,j}}^{{t,ev}}}}} \right. \kern-0em} {k_{{X,j}}^{{t,ev}}}}.$При этом величина aX во фракциях нефтей в основном зависит от числа ароматических колец, так что для н-алкилбензолов и н-алкилтолуолов можно принимать одинаковые значения этого коэффициента (Бродский, 1985; Хмельницкий и др., 1980). Обозначим его a1, а аналогичный коэффициент для н-алкилнафталинов – a2. Кроме того, поскольку анализируемые фракции представляли собой совокупность ароматических соединений широкого диапазона полярности С12+, исходя из известных закономерностей состава нефтей (например, не бывает, чтобы при наличии моно- и тетрациклической ароматики отсутствовала би- и трициклическая ароматика), из приведенных в (Бродский, 1985; Хмельницкий и др., 1980) данных следует, что для алкилбензолов и алкилнафталинов коэффициенты a относительно мало отличаются от 1 (заведомо меньше, чем в 1.3 раза).
Для анализа глубины протекания реакций и соотношений между массами образовавшихся параллельно продуктов достаточно оперировать концентрациями, выраженными в любых, в том числе – в относительных единицах. Последние задаются величиной ${{{{S}_{X}}_{{,j}}} \mathord{\left/ {\vphantom {{{{S}_{X}}_{{,j}}} {k_{{X,j}}^{{t,ev}}}}} \right. \kern-0em} {k_{{X,j}}^{{t,ev}}}},$ если рассматриваются только н-алкилбензолы и н-алкилтолуолы и величиной $\left( {{{{{S}_{{X,j}}}} \mathord{\left/ {\vphantom {{{{S}_{{X,j}}}} {k_{{X,j}}^{{t,ev}}}}} \right. \kern-0em} {k_{{X,j}}^{{t,ev}}}}} \right)$ × (a1/a2) при включение в рассмотрение алкилнафталинов. Из приведенных в (NIST/EPA/NIH) данных получено, что a1/a2 ≈ 1.14.
В настоящей работе для компонентов с повышенным содержанием принимали величины “фоновых” концентраций (CX,b) равными среднему геометрическому концентраций двух ближайших по молекулярной массе гомологов. Например, для С21-н-алкилбензола C21-B,b = (S20-B, 91+92 × × S22-B,91+92)0.5/0.5, для С22пара-н-алкилтолуола C22-p-T,b = (S21-p-T,105+106 × S23-p-T,105+106)0.5/0.53 (если рассматривают н-алкилбензолы и н-алкилтолуолы) и т.д. (нижний индекс “b” указывает, что речь идет о фоновой концентрации). Во всех образцах, в которых точность измерений была достаточно высока, разности между измеренными и “фоновыми” концентрациями CX,h изомеров н-алкилтолуолов дали следующую картину: C22-p-T,h ≈ ≈ C22-o-T,h ≈ 2–4 × C22-m-T,h, что типично именно для электрофильного метилирования н-алкилбензолов. Для н-алкилтолуолов С24 точность измерения намного ниже вследствие их существенно меньшего содержания, так что достоверно можно говорить только о том, что C24-m-T,h в несколько раз меньше, чем содержание двух других изомеров.
Очевидно, если имеет место электрофильное метилирование н-алкилбензолов, аналогичные реакции должны идти с участием других ароматических соединений. Однако выявить продукты этой реакции в большинстве случаев крайне сложно из-за особенностей состава нефтей. Например, во всех изученных образцах содержание нафталина было много меньше содержания метилнафталинов, концентрация которых, в свою очередь, составляла не более 15 мас. % суммарной концентрации диметилнафталинов. Поэтому вычленить на общем фоне продукты реакции метилирования если и возможно, то только при специальной постановке работы, ориентированной на прецезионное измерение концентраций в большом ряду образцов с последующим статистическим анализом результатов. При этом гарантировать, что точность измерения окажется достаточной для выявления эффекта метилирования, нельзя. Поэтому в настоящей работе рассмотрены только возможные продукты метилирования выделяющихся на общем фоне С21 и С23 α-н-алкилнафталинов. Анализ упрощался тем, что пики метилалкилнафталинов на масс-фрагментограмме по ионам m/z = 155 и суммарным интенсивностям ионов m/z = 155 и m/z = 156 за исключением гомологов С22 и С24 малы (рис. 4). Правда, электрофильное метилирование алкилнафталинов не изучено, так что установить, какое именно метилирование имеет место на основании изомерного состава продуктов, на данный момент невозможно. Поэтому в настоящей работе идентификацию изомеров метилалкилнафталинов не проводили.
Рис. 4.
Типичный вид масс-фрагментограмм по ионам m/z = 155 + 156 (пики метилалкилнафталинов, нижний) при высоком содержании С21 и С23 α-н-алкилнафталинов (верхний).
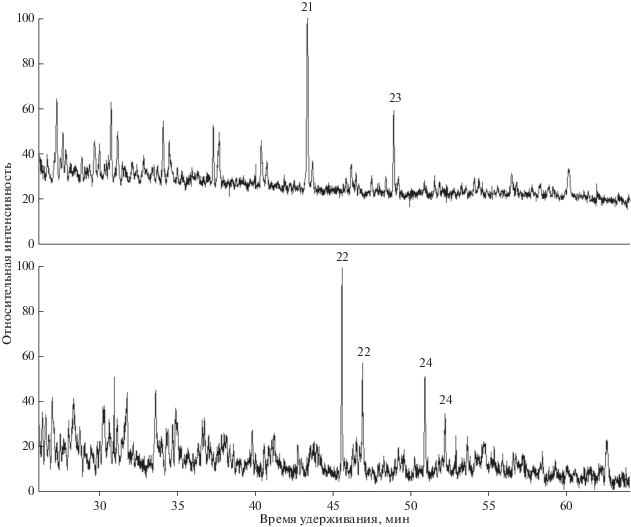
Во всех образцах нефтей с повышенной концентрацией С21 и С23 α-н-алкилнафталинов (КН > 3) наблюдались пики двух изомеров метил-н-алкилнафталинов С22 и С24 (рис. 4). Их строение подтверждено во-первых, по масс-спектрам (молекулярные ионы– m/z 296 и m/z 324 соответственно; наиболее интенсивные пики фрагментных ионов с m/z 155 и m/z 156 соответствуют характеристическому для алкилзамещенных ароматических соединений α-разрыву; интенсивные пики других фрагментных ионов, появление которых можно было бы связать с разветвлениями в алкильной цепи, отсутствуют). Во-вторых – по времени удерживания: линейные индексы удерживания α-н-алкилнафталинов С21 – 2305, С22 – 2412, С23 – 2520, С24 – 2627, наблюдаемых метил-н-алкилнафталинов С22 – 2379 и 2432, С24 – 2592 и 2647, при наличия заместителя в цепи у метилалкилнафталинов индексы удерживания должны быть меньше. Отношение интенсивностей пиков пар изомеров одинаково в пределах ошибки измерения во всех образцах, где оно могло быть вычислено с приемлемой точностью. Полученная картина аналогична наблюдавшейся в (Williams et al., 1988).
Для оценки значимости метилирования вычислены отношения концентраций описанных продуктов этой реакции к их сумме с исходными моно-алкилзамещенными соединениями YX. Для алкилнафталинов C21 → C22
(3)
${{Y}_{{N - 21/22}}} = {{C}_{{\Sigma 22\,{\text{ - }}m{\text{ - }}N}}}/({{C}_{{21 - ,{\text{ }}h}}} + {{C}_{{\Sigma 22 - m - N,}}}),$Для алкилбензолов точность оценки величин YX хуже, чем для алкилнафталинов по следующим причинам. Во-первых, пики С21 и С23н-алкилтолуолов частично перекрываются с пиками н-алкилбензолов и пиками иных компонентов (рис. 3), вследствие чего погрешность измерения их площадей и определения “фоновых” концентраций увеличивается примерно до 7–10 отн. %. Во-вторых, может не строго выполняться предположение о равенстве “фоновой” концентрации среднему геометрическому концентраций ближайших гомологов. Расчеты показали, что даже для образцов, в которых перекрывание пиков н-алкилтолуолов с пиками иных компонентов было наименьшим, а содержание их в образце выше среднего по изученной выборке, невозможно достоверно установить отсутствие таких отклонений на уровне менее 10 отн. % при использованной в работе аппаратуре и принятой методике анализа. В третьих, “фоновые” концентрации н-алкилтолуолов намного больше, чем метилалкилнафталинов, так что оценка содержания продуктов метилирования выполняется по разности. А это приводит к росту всех ошибок: в 2 раза, если концентрация продуктов равна “фоновой” концентрации, в 3 раза, если она составляет половину “фоновой” и т.д. В изученных же образцах содержание продуктов метилирования лишь в редких случаях слегка превышала “фоновое” содержание (максимум – в 1.3 раза). Дополнительная ошибка появляется и в случае, когда КБ < 3, поскольку начинает сказываться то, что концентрация оставшегося непрометилированным соединения определяется также по разности. В итоге оценки YX для бензолов проводили только для образцов, у которых КТ > 1.5 и КБ > 3, но и для них ошибка составляла от ~25 до 35 отн. %.
Для превращения алкилбензола C21 в алкилтолуол C22:
(4)
${{Y}_{{B - 21/22}}} = {{C}_{{\Sigma 22 - T,h}}}/({{C}_{{\Sigma 22 - T,h}}} + {{C}_{{21 - B,h}}}),$Интерпретировать величины YX как выход продуктов метилирования или глубину протекания реакции при имеющемся уровне изученности можно лишь весьма условно. Во-первых, образовавшиеся монометилзамещенные продукты способны вступать в ту же реакцию, давая диметилалкилароматические соединения. То есть и н-алкилтолуолы, и метил-н-алкилнафталины, строго говоря, являются промежуточными продуктами реакции и для получения концентрации исходных соединений это следует учитывать. В изученных пробах однозначно определяемых диметилзамещенных продуктов (достоверное превышение С23 и/или С25 компонентов в ряду диметил-н-алкилбензолов) не наблюдалось, а относительно небольшое превышение невозможно достоверно зафиксировать из-за большого числа пиков в масс-фрагментограммах по ионам m/z = 119, 120, 169, 170 или их сумме. К тому же пики на масс-фрагментограммах m/z = 169, 170 малы настолько, что не наблюдаются соответствующие им молекулярные ионы. Поэтому относительно рассмотренных образцов можно лишь утверждать, что доля диметилзамещенных компонентов в них существенно меньше доли монометилзамещенных. В (Connan et al., 1995) четко зафиксировано повышенное содержание как С22н-алкилтолуолов, так и С23 диметилалкилбензолов. Но отсутствие количественных данных в (Connan et al., 1995) и не изученность соотношений между константами скоростей реакций электрофильного метилирования алкилбензолов и метилалкилбензолов не позволяет ввести необходимые поправки при определении концентрации “исходных” соединений. Во-вторых, не исключено, что н-алкилбензолы и/или н-алкилнафталины вступают еще в какие-то неизвестные реакции, что приводит к уменьшению их остаточной (т.е. фиксируемой при измерениях) концентрации. Так что, говоря о глубине протекания метилирования, следует учитывать, что полученные в рамках представленной на рис. 2 схемы оценки возможно придется существенно корректировать.
Что касается возможных метилирующих агентов, то в составе природных газов (исключая газы вулканов) таковые не известны. В реакциях электрофильного метилирования в органическом синтезе чаще всего, как известно, используют метилгалогениды, присутствие которых в природных обстановках крайне сомнительно. Известны работы (Vogt et al., 1988; Kazuo et al., 1986), в которых получение ксилолов из толуола проводили при реакции с метанолом, который в принципе может существовать в осадках. Кроме того, возможны и реакции переметилирования, когда донорами метильной группы выступают полиметилзамещенные ароматические соединения (Szczerba et al., 2010; Ellis et al., 1995).
Реакции электрофильного метилирования, как известно, протекают в кислой среде. В качестве катализаторов при метилировании метанолом использовали алюмосиликаты: как цеолиты, так и глины (Vogt et al., 1988). В принципе для этого пригодны любые кислоты Льюиса, в том числе и соединения металлов. Однако поскольку в органической химии внимание сосредоточено на представляющих промышленный интерес катализаторах (например, FeCl3, AlCl3 и т.д.), относительно распространенных в осадочном чехле соединений данные не найдены.
Соотношение между скоростями реакций циклизации с образованием нафталинов и гидрирования до стадии, исключающей дальнейшую циклизацию vc/vh|21 (для исходного V) с точностью до всего, сказанного относительно интерпретации параметров YX, можно оценить следующим образом:
(5)
${{{v}}_{c}}/{{{v}}_{h}}|{{}_{{21}}} = ({{C}_{{21 - N,{\text{ }}h}}} + {{C}_{{\Sigma 22 - m - N,}}})/({{C}_{{\Sigma 22 - T,{\text{ }}h}}} + {{C}_{{21 - B,{\text{ }}h}}}),$Во всех изученных нефтях отношение пристан/фитан ≤0.7 (в ряде проб – <0.5), то есть типичное для восстановительной среды осадконакопления и последующей генерации углеводородов. Измеренные по составу стеранов С27 отношения диа-/рег- в большей части проб менее 0.25 (в отдельных пробах, где имело место метилирование – менее 0.1), наибольшее значение – 0.33; величина Ts/Tm лишь в одном образце оказалась равной 0.65, в остальных не более 0.4 (наименьшие значения меньше 0.15) при том, что по стерановым показателям нефти зрелые. Такие характеристики ранее считались характерными для карбонатных материнских пород (Peters et al., 2005). Однако в (Смирнов, Фадеева, 2019) было показано, что по крайней мере для доманиковой формации северных и центральных районов Волго-Урала ни один из критериев, по которым согласно (Peters et al.,2005) выделяли нефти, генерированные в карбонатных породах, полностью не работоспособны. То есть при анализе полученных данных можно опираться только на известные закономерности протекания химических реакций восстановления полиенов, их циклизации и метилирования ароматических циклов.
Для протекания реакций гидрирования и циклизации полиенов должна быть благоприятна восстановительная среда. Однако реакция электрофильного метилирования протекает только в кислой среде. Это позволяет предположить, что формирование наблюдаемого состава ароматических соединений протекает в два разделенных во времени этапа. Первый включает все процессы генерации компонентов нефти в диагенезе и протекает, по-видимому, в восстановительной среде, унаследованной со стадии осадконакопления (низкие значения отношения Pr/Ph). На втором образовавшееся вещество попадает в условия, характеризующиеся повышенной концентрацией кислотных центров (например, в результате внедрения в материнскую породу кислых вод). И тогда состав ароматических соединений изменяется в результате электрофильного метилирования. То есть метилирование протекает, скорее всего, на стадии катагенеза, следующей после завершение генерации того флюида, из которого сформировалась изучаемая залежь нефти. Однако в карбонатных породах возникновение таких условий очевидно исключено. Следовательно, метилирование ароматических циклов является четким индикатором того, что нефтематеринскими породами являются либо глины, либо силициты с существенной долей глинистого материала. И в отличие от ранее предлагавшихся этот критерий имеет достаточно понятную и прочную основу, опираясь на хорошо известные в органической химии факты. К сожалению, органическое вещество из отложений, залегающих ниже доманиковой формации, в Волго-Уральском бассейне не изучено; мы соответствующими образцами не располагаем. Поэтому обсуждать подробнее этот вопрос на имеющемся материале не представляется возможным. Очевидно, требуется проведение соответствующих работ для РОВ из глинистых отложений. Можно лишь добавить, что в принципе, при иных условиях осадконакопления возможна и реализация другой кинетической схемы, когда генерация углеводородов совмещена по времени с их метилированием.
То, что метилирование ароматических циклов не идет в карбонатных породах, подтверждено при изучении состава ароматических соединений битумоидов из доманикитов (семилукский горизонт). Среди них найдены пробы с необычным распределением н-алкилбензолов, а именно, с большим преобладанием четных гомологов над нечетными в диапазоне С18–С32 (рис. 5). Однако в этих образцах не наблюдалось какого-либо преобладания нечетных н-алкилтолуолов над четными. Так, в пробе с наибольшими концентрациями н-алкилбензолов и н-алкилтолуолов, для которой точность измерений наибольшая, среднее значение КБ,j (j – четные от 20-ти до 30-ти) составило 4.7, а среднее значение КТ,j (j – нечетные от 21-го до 29-ти) – 1.02. То есть реакция метилирования не является общераспространенной даже в пределах Волго-Уральского бассейна.
Рис. 5.
Молекулярно-массовое распределение н-алкилбензолов (верхний) и н-алкилтолуолов (нижний) в образце, где отсутствует метилирование ароматических соединений. Порядок выхода н-алкилтолуолов – см. подпись к рис. 3.
Следует также отметить, что следствием такого метилирования может стать изменение содержания изомеров ароматических соединений, используемых при расчетах коэффициентов зрелости, что приведет к некорректной интерпретации получаемых результатов. В первую очередь это, по-видимому, касается фенантреновых индексов и дибензотиофенового индекса (Radke et al., 1984; Radke et al., 1986; Radke, 1988), поскольку электрофильное метилирование фенантренов и дибензотиофенов не изучено.
ЗАКЛЮЧЕНИЕ
Изучение состава алкилзамещенных бензолов и алкилзамещенных нафталинов позволил установить, что одним из процессов, формирующих состав ароматических углеводородов нефтей, в отдельных регионах является реакция электрофильного метилирования ароматических циклов. Вывод основан на сравнении содержания соединений в парах гомологических рядов н-алкилбензолы/н-алкилтолуолы и н-алкилнафталины/метил-н-алкилнафталины, а также на изомерном составе отдельных гомологов н-алкил. По предварительным данным в нефтях до ~40% от исходных соединений может оказаться преобразованными в продукты метилирования. Показано, что метилирование протекает, скорее всего, на стадии катагенеза, следующей после завершение генерации того флюида, из которого сформировалась изучаемая залежь нефти. Метилирование ароматических циклов является четким индикатором того, что нефтематеринскими породами являются либо глины, либо силициты с существенной долей глинистого материала. Установлено, что реакция метилирования не является общераспространенной даже в пределах одного бассейна. Следовательно, анализ на наличие продуктов электрофильного метилирования ароматических соединений может быть эффективно использован при геохимических реконструкциях. Отмечено, что следствием метилирования может стать изменение содержания изомеров ароматических соединений, используемых при расчетах коэффициентов зрелости, что приведет к некорректной интерпретации получаемых результатов.
Авторы благодарят Т.К. Баженову и Б.И. Песоцкого за помощь в проведении работы.
Работа выполнена в рамках Государственного задания ИНХС РАН.
Список литературы
Арефьев О.А., Забродина М.Н., Русинова Г.В., Петров Ал.А. (1994) Биомаркеры нефтей Волго-Уральской нефтегазоносной провинции. Нефтехимия 34(6), 483-502.
Бродский Е.С. (1985) Масс-спектрометрический анализ углеводородов и гетероатомных соединений нефти. В кн.: Методы исследования состава органических соединений нефти и битумоидов. М.: Наука, 57-118.
Бушнев А.Д., Валяева О.В. (2015) н-Алкилбензолы и н‑алкилнафталины в ряде нефтей позднедевонского возраста Тимано-Печорской провинции. Вестник ИГ Коми НЦ Уро РАН. (10), 17-20.
Бушнев А.Д., Бурдельная Н.С., Валяева О.В., Деревесникова А.А. (2017) Геохимия нефтей позднего девона Тимано-Печорского бассейна. Геология и геофизика 58(3–4), 410-422.
Гончаров И.В., Носова С.В., Вяткина Н.В. (2000) О механизме образования длиноцепочечных алкилбензолов и алкилнафталинов. Материалы IV Международной конференции “Химия нефти и газа”. Томск. 2000 г. 1, 166-169.
Гончаров И.В., Носова С.В., Самойленко В.В. (2003) Генетические типы нефтей Томской области // Материалы V Междунар. конф. “Химия нефти и газа” Томск: Изд-во Института оптики атмосферы СО РАН, 2003 г. 10-13.
Гордадзе Г.Н., Тихомиров В.И. (2005) Геохимическая характеристика нефтей и РОВ пород центральных районов Волго-Урала (по УВ-биомаркерам). Геохимия (11). С. 1208-1223.
Gordadze G.N., V.I. Tikhomirov. (2005) Geochemical Characteristics of Oils and Dispersed Organic Matter from the Rocks of the Central Volga–Ural Basin: Hydrocarbon Biomarker Data. Geochem. Int. 43(11), 1108-1123.
Гордадзе Г.Н., Тихомиров В.И. (2007) Об источниках нефтей на северо-востоке Татарстана. Нефтехимия 47(6), 422-431.
Иванова И.К., Каширцев В.А. (2010) Особенности распределения моноалкилбензолов состава С12Н18–С27Н48 в венд-кембрийских нефтях Сибирской платформы. Геология и геофизика 51(11), 1539-1544.
Каткова Г.П., Нигмедзянова Л.З., Романов Г.В., Шарипова Н.С., Смелков В.М., Дахнова М.В., Жеглова Т.П., Храмова Э.В. (2004) Закономерности состава и распределения углеводородов – биомаркеров в нефтях и битумоидах Бавлинской площади Южно-Татарского свода. Нефтехимия 44(6), 440-448.
Каюкова Г.П., Миннегалиева А.М., Романов А.Г., Киямова А.М., Шарипова Н.С., Смелков В.М., Дахнова М.В., Нечитайло Г.С. (2006) Дифференциация нефтей Ромашкинского месторождения по биомаркерным параметрам. Нефтехимия 46(5), 341-351.
Кендэл М. (1975) Ранговые корреляции. М.: Статистика, 216 с.
Остроухов С.Б., Арефьев О.А., Пустильникова С.Д., Забродина М.С., Петров Ал.А. (1983) н-Алкилбензолы состава С12–С30 в нефтях. Нефтехимия 23(1), 20-30.
Остроухов С.Б. (2000) К вопросу о происхождении нефтяных алкилбензолов состава С10 и выше. Материалы IV международной конференции “Химия нефти и газа”. Томск. 2000 г. 1, 349-354.
Остроухов С.Б. (2009) К вопросу о происхождении н‑алкилбензола состава С21 в нефтях. Химия нефти и газа: Материалы VII Международной конференции. Томск. 2009 г. Издательство Института оптики атмосферы СО РАН. 189-190.
Остроухов С.Б. (2015) Высокомолекулярные нефтяные алкилтолуолы: оценка термодинамической преобразованности. Нефтехимия 55(3), 206-212.
Петров Ал.А. (1984) Углеводороды нефти. М.: Наука, 263 с.
Смирнов М.Б., Ванюкова Н.А. (2015) Закономерности распределения основных структурно-групповых параметров состава нефтей Волго-Уральского нефтегазоносного бассейна по данным ЯМР. Нефтехимия 55(6), (473-486).
Смирнов М.Б., Полудеткина Е.Н. (2018) Характеристики источников нефтей Ромашкинского месторождения по результатам изучения состава насыщенных и ароматических биомаркеров. Геохимия (2), 175-184.
Smirnov M.B. and Poludetkina E.N.. (2018) Characteristics of Sources of Oils of Romashkino Field by Composition of Saturated and Aromatic Biomarkers. Geochem. Int. 56(2), 162-170.
Смирнов М.Б., Фадеева Н.П., Борисов Р.С., Полудеткина Е.Н. (2018а) Характеристика органического вещества доманикоидных отложений верхнего девона северных и центральных районов Волго-Урала по составу насыщенных биомаркеров. Геохимия (8), 774-790.
Smirnov M.B., Fadeeva N.P., Borisov R.S., Poludetkina E.N. (2018a) The Characteristics of the Organic Matter of the Upper Devonian Domanik-Type Deposits in the Northern and Central Regions of the Volga-Ural Basin According to Saturated Biomarkers Composition. Geochem. Int. 56(8), 812-827.
Смирнов М.Б., Фадеева Н.П., Полудеткина Е.Н. (2018б) Выявление генетической неоднородности нефтей Татарстана на основании данных о составе алкилзамещенных ароматических соединений. Геология и геофизика 59(9), 1418-1430.
Смирнов М.Б., Фадеева Н.П. (2019) К вопросу об информативность масс-спектральных диагностических отношений и критериев на их основе для проверки геохимических гипотез. Масс-спектрометрия 16(1), 73-78.
Тиссо Б., Вельте Д. (1981) Образование и распространение нефти. М.: Мир, 501 с.
Хмельницкий Р.А., Лукашенко И.Н., Бродский Е.С. (1980) Пиролитическая масс-спектрометрия высокомолекулярных соединений. М.: Химия, 111-112.
Aizenshtat Z., Feinshtein S., Miloslavski I., Yakubson Z., Yakubson C.I. (1998) Oil-oil correlation and potential source rocks in the Tatarian and Perm basin, Russia. Org. Geochem. 29(1–3), 701-712.
Burnham A.K., Gregg H.R., Ward R.L., Knauss K.G., Copenhaver S.A., Reynolds J.G., Sanborn R. (1997) Decomposition kinetics and mechanism of n-hexadecane-1,2-13C2 and dodec-1-ene-1,2-13C2 doped in petroleum and n-hexadecane. Geochim. Cosmochim. Acta 61, 3725-3737.
Burnham A.K., Gregg H.R., Sanborn R.H. (1998) Thermal dealkylation of dodecylbenzene and dodecylcyclohexane. Org. Geochem. 28(11), 755-758.
Burkle-Vitzthum V., Michels R., Scacchi G., Marquaire P.-M., Dessort D., Pradier B., Brevart O. (2004) Kinetic effect of alkylaromatics on the thermal stability of hydrocarbons under geological conditions. Org. Geochem. 35(1), 3-31.
Connan J, Dessot D, Poirier Y, Guzman-Vega M.A. (1995) Unusual bacterially-derived biomarker spectra in crude oils and source rocks from Southern Chiapas-Guatemala area. Organic geochemistry: Developments and applicatoins to energy, climate, environment and human history. 17-th International Meeting of Organic Geochemistry. 1995. 4th–8th September. Donostia-San. Sebastian, Spain. 198-201.
Domine F., Bounaceur R., Scacchi G., Marquaire P.-M., Dessort D., Pradier B., Brevart O. (2002) Up to what temperature is petroleum stable? New insights from a 5200 free radical reactions model. Org. Geochem. 33(12), 1487-1499.
Ellis L., Singh R.K., Alexander R., Kagi R.I. (1995) Geosynthesis of organic compounds: III. Formation of alkyltoluenes and alkylxylenes in sediments. Geochim. Cosmochim. Acta. 59(24), 5133-5140.
Kagi R.I., Alexander R., Ton E. (1990) Kinetics and mechanism of the cyclisation reaction of orto-methylbiphenyls. Advaces in Organic Geochemistry, 1989. Org. Geochem. 16(1–3), 161-166.
Kazuo U., Hiroaki S., Yusuke S. (1986) J. Chem. Soc., Chem, Commun. (14), 1074-1076.
Koopmans M.P., Koster J., van-Kaam-Peters H.M.E., Kenig F., Schouten S., Hartgers W.A., de Leeuw J.W., Damste J.S.S. (1996) Diagenetic and catagenetic products of isorenieratene: Molecular indicators for photic zone anoxia. Geochim. Cosmochim. Acta 60(22), 4467-4496.
NIST/EPA/NIH Mass Spectral Database (NIST 14). Standard Reference Data Program. National Institute of Standards and Technology. US Department of Commerce, Gaithersburg, MD, USA.
Peters K.E., Walters C.C., Moldovan J.M. (2005) The Biomarker Guide / Second edition. Biomarkers and Isotopes in Petroleum Systems and Earth History. Cambridge University Press, 1029 p.
Radke M., Leythaeuser D., Teichmuller M. (1984) Relationship between rank and composition of aromatic hydricarbons for goals of different origins. Org. Geochem. 6(1), 423-430.
Radke M., Willsch H., Welte D.H. (1986) Maturity parameters based on aromatic hydrocarbons: influence of the organic matter type. Org. Geochem. 10(1), 51-63.
Radke M. (1988) Application of aromatic compounds as maturity indicators in source rocks and crude oils. Marine and Petroleum Geology. 5(3), 224-236.
Rospondek M.J., Marynovski L., Chachaj A., Gora M. (2009) Novel aryl polycyclic aromatichydrocarbons; Phenylphenantrene and phenylantracene identification, accurence and distribution in sedimentary rocks. Org. Geochem. 40, 986-1004.
Szczerba M., Rospondek M.J. (2010) Controls of distribution of methylphenantrenes in sedimentary rock extracts: Critical evaluation of existing geochemical data from molecular modelling. Org. Geochem. 41, 1297-1311.
Vogt F., Wendlandt K.-P., Isakov Ja.S., Isakova T.A., Minachev Ch.M. (1988) Alkylierung und Umalkylierung von Aromaten an Alumosilicaten. J. f. prakt. Chemie. 330(5), 717-723.
Williams J.A., Dolcater D.L., Torkelson B.E., Winters J.S. (1988) Anomaleous concentrations of specific alkilaromatic and alkilcycloparaffin components in West Texas and Michigan crude oils. Org. Geochem. 13(1–3), 47-59.
Дополнительные материалы отсутствуют.