

Геология рудных месторождений, 2020, T. 62, № 5, стр. 391-413
Изотопное фракционирование серы при формировании сульфидов субмаринных гидротермальных систем на примере полей Логачев, Краснов и Рэйнбоу (САХ)
Е. О. Дубинина a, *, Н. С. Бортников a, О. О. Ставрова a, С. А. Коссова a
a Институт геологии рудных месторождений, петрографии, минералогии и геохимии РАН
119017 Москва, Старомонетный пер., 35, Россия
* E-mail: elenadelta@gmail.com
Поступила в редакцию 20.03.2020
После доработки 16.05.2020
Принята к публикации 17.05.2020
Аннотация
Изучен изотопный состав серы единичных зерен сульфидов с учетом последовательности их образования в образцах из построек трех гидротермальных полей (Логачев, Краснов и Рэйнбоу), расположенных в северной части САХ. Интервалы величин δ34S составили +3.9…+6, +7.1…+9.8 и +2.1…+8.4‰ для этих полей соответственно, причем сульфиды более поздних генераций в пределах изученных доменов (1–10 мм) образцов закономерно обогащены изотопом 34S относительно сульфидов ранних генераций, а изотопный состав серы не обнаруживает связи с составом сульфидных минералов. В единичных зернах гидротермальных баритов установлены величины δ34S, превышающие общепринятый диапазон для современного морского сульфата. Предложена модель изотопного фракционирования серы в гидротермальных системах, основанная на предположении о термогенном восстановлении сульфата при взаимодействии флюид-порода в системе, закрытой относительно флюида. В модели также учитывается одновременное извлечение серы из окружающих пород, доля которой зависит от протекания реакции термогенного восстановления сульфата. Применение модели объясняет широко известные противоречия, обнаруженные при изучении изотопного состава серы сульфидов других гидротермальных полей Мирового океана.
ВВЕДЕНИЕ
Изучение современных сульфидных гидротермальных отложений в океане является ключом к пониманию процессов формирования колчеданных месторождений, которые служат маркером геодинамических обстановок зон спрединга и задуговых бассейнов (Бортников, Викентьев, 2005; Гричук, 2000, Еремин и др., 2000; McDonald et al., 2018; Brueckner et al., 2015; Aggarwal, Nessbit, 1984; Hannington et al., 1998; Growe et al., 1992; Green, Vokes, 1990; Haymon et al., 1989; Neathery, Hollister, 1984 и многие другие работы). Колчеданные месторождения являются важными источниками меди, цинка, свинца, золота и серебра (Cu, Zn, Pb, Au и Ag), а также сопутствующих металлов Cd, Ga, In, Te, Se, широко используемых в высокотехнологичных производствах (Бортников и др., 2016). Эти месторождения уникальны среди других типов рудных месторождений. Процесс их формирования охватывает чуть ли не три четверти истории Земли, начавшись примерно 3.55 млрд лет назад и продолжаясь в настоящее время на дне Мирового океана (Бортников, Викентьев, 2005; Смирнов, 1968; Shanks, 2001; Hannington et al., 2005: Huston et al., 2010; Shanks, Thurston, 2012). Колчеданные месторождения являются одними из самых ранних источников металлов благодаря высокому качеству руд и относительно простому способу их извлечения. Тем не менее, дискуссии об условиях их образования, природе минералообразующего флюида, источниках металлов и серы не прекращаются до сих пор. В 1960-е годы исследования, проведенные с привлечением методов изучения флюидных включений, стабильных и радиогенных изотопов, а также открытие современных субмаринных гидротермальных систем привели к тому, что на смену гипотезе о магматогенном эпигенетическом происхождении колчеданных руд (Lindgren, 1913) пришла идея о сингенетичном гидротермально-эксгаляционном отложении их на морском дне (Смирнов, 1968; Oftedahl, 1958). Колчеданные руды стали рассматривать как результат взаимодействия восходящего гидротермального флюида с окружающей морской водой, в результате чего происходит отложение сульфидов либо на поверхности морского дна, либо непосредственно под поверхностью (Ohmoto et al., 1983). Согласно этим представлениям, восходящий гидротермальный флюид мог представлять собой смесь флюида, отделившегося при кристаллизации магм, с нагретой морской водой, которая прошла стадию взаимодействия с нижележащими породами. Считалось, что в большинстве гидротермальных систем преобладала модифицированная нагретая морская вода (Franklin et al., 1981, 2005), а источниками металлов и серы могли являться как магмы, так и вмещающие породы, по которым эта вода фильтруется. Весомым аргументом в пользу данных представлений служит синхронное изменение величин δ34S в колчеданных рудах и сульфате морской воды в течение неопротерозоя и фанерозоя с сохранением постоянной разницы в 18‰ между сульфатом и сульфидами, что указывает на сульфат морской воды как на основной источник серы сульфидов колчеданных месторождений (Sangster, 1968) и примерно одинаковые условия формирования сульфидов.
Принципиальная схема циркуляции флюида в океанских гидротермальных системах была предложена около 30 лет назад и претерпела мало изменений (Woodruff, Shanks, 1988; Janecky, Shanks, 1988; Tivey, 2007; Fisher 1998; и другие работы). Основными источниками серы для образования гидротермальных сульфидных построек на дне океана являются сульфат океанской воды и сера, извлеченная из подстилающих базальтов или ультраосновных и основных пород (Бортников, Викентьев, 2005; Shanks, 2001; McDonald et al., 2018), в некоторых случаях привлекается вклад магматогенного источника серы (Herzig et al., 1998; Huston et al., 2011; Brueckner et al., 2015 и другие работы). Морской сульфат, вовлекаемый в процесс образования гидротермальных сульфидов, проходит стадию восстановления при взаимодействии с основными или ультраосновными породами океанской коры, содержащими двухвалентное железо. Этот процесс (термогенная сульфат-редукция, или TSR) протекает при повышенных температурах (200–400°С) и, поскольку реакция восстановления сульфата характеризуется большими сдвигами в изотопной системе серы, следы TSR должны присутствовать в изотопных характеристиках серы гидротермальных сульфидов (Canfield, 2001).
Исходя из хорошо развитых представлений о деятельности океанских гидротермальных систем, можно ожидать, что и поведение изотопов серы в гидротермальном флюиде и сульфидах гидротермальных построек легко предсказуемо. Однако анализ накопленного к настоящему времени материала показывает, что в поведении изотопов серы прослеживаются закономерности, которые до сих пор не находят согласованных объяснений. В основном, эти закономерности можно суммировать в следующих пунктах:
1. Величины δ34S гидротермальных океанских сульфидов (в том числе, массивных сульфидов некоторых колчеданных месторождений) заметно превышают таковые в породах океанской коры и магматогенных флюидах (Zheng et al., 2016; Shanks 2001; Seal et al., 2006; McDonald et al., 2018). За редким исключением (в основном, Красноморского рифта и некоторых островодужных вулканов и задуговых бассейнов) сера океанских гидротермальных сульфидов характеризуется величинами δ34S > 0. Частично эти повышенные величины δ34S можно объяснить протеканием TSR, однако расчеты показывают, что равновесное восстановление современного морского сульфата при 300–350°С может привести к генерации сероводорода с величинами δ34S от +1 до +4‰ (согласно Ohmoto, Lasaga, 1982). Эти величины могут опускаться до отрицательных значений при более низких температурах (например, δ34S(Н2S) = –3‰ при 250°С) или повышаться до +6‰ с повышением температуры до 400°С. Поскольку процесс термогенного восстановления сульфата протекает эффективно при Т > 250°С (Ohmoto, Lasaga, 1982), небольшие положительные величины δ34S в сульфидах – на уровне первых промилле – можно считать атрибутом TSR, при которой восстанавливается сера с изотопным составом, отвечающим современному морскому сульфату. Действительно, в сульфидах полей, расположенных в зонах быстрого спрединга, величины δ34S в целом близки к диапазону равновесия с морским сульфатом (ВТП, 21 и 9–11 градусы СШ, хребет Хуан-де-Фука, Shanks, 2001, Woodruff, Shanks, 1988; Rouxel et a., 2008). Однако для сульфидов гидротермальных полей, приуроченных к зонам медленного спрединга (например, в районе САХ), характерны более высокие величины δ34S сульфидов. Например, для TAG верхний предел величин δ34S сульфидов составляет +10.3‰ (Zeng et al., 2016), для SMAR – почти +10‰ (Peters et al., 2009), для Рэйнбоу +12.5‰ (Lein et al., 2001) и для поля Логачев +13.8‰ (Богданов и др., 1997). Эти величины не могут отвечать равновесному фракционированию серы в процессе TSR при характерных температурах генерации и разгрузки гидротермальных флюидов.
2. Немногочисленные прямые измерения изотопного состава серы сероводорода в гидротермальных растворах показывают, что величины δ34S(H2S)фл не отвечают равновесию с серой сульфата морской воды при наблюдаемой температуре разгрузки флюидов (Shanks, 1995, 2001). Это установлено как для полей, расположенных в зонах быстрого спрединга океанской коры (ВТП, 21 и 9–11 градусы СШ, хребет Хуан-де-Фука, Shanks, 2001; Woodruff, Shanks, 1988; Rouxel et al., 2008), так и для полей зон медленного спрединга (SMAR, Логачев, MARK, Peters et al. 2009). Более того, для большинства гидротермальных полей установлено и отсутствие изотопного равновесия серы между сосуществующими сульфидными минералами в пределах одного и того же образца. Это наблюдается как в современных, так и в древних сульфидных гидротермальных отложениях (Brueckner et al., 2015). Возможно, это результат проявления кинетических эффектов, сопровождающих быструю кристаллизацию сульфидов (аналогично кристаллизации карбонатов из раствора, например, Dietzel et al., 2009; Gabitov et al., 2012; Kim, O’Neil, 1997; и другие работы). Однако к такому же результату приводит и разновременное осаждение сульфидов из гидротермального флюида, если изотопный состав серы во флюиде варьирует.
3. Наблюдаются закономерные вариации изотопного состава серы в сульфидах в зависимости от последовательности отложения минералов. Эти вариации имеют разный масштаб. Например, обнаруженные пространственные вариации величин δ34S в пределах отдельно взятых гидротермальных построек на 11–13° с.ш. ВТП были объяснены тем, что при формировании внешних и внутренних зон построек участвовали более ранние и более поздние порции гидротермального флюида с величинами δ34S(Н2S) 1.7 и 5.0‰ соответственно (Bluth, Ohmoto, 1988). Различие в величинах δ34S более ранних и более поздних генераций сульфидов отмечалось также для пермских массивных руд месторождения Ice, ЮВ Юкон, Канада (McDonald et al., 2018), для которых очередность генераций устанавливалась по типам минерального парагенезиса сульфидов. Наконец, наши результаты (Дубинина и др., 2018), в том числе и приведенные в данной работе, показывают наличие временных вариаций величин δ34S в локальном масштабе. На данный момент специальных исследований, посвященных связи изотопных параметров сульфидов со временем или последовательностью их отложения, немного, но имеющиеся результаты показывают, что временные вариации в изотопном составе серы в гидротермальном флюиде, по-видимому, наследуются изотопными характеристиками сульфидных отложений.
4. Наименее освещенная в литературе, но очень важная, на наш взгляд, проблема касается изотопного состава серы гидротермальных сульфатов, которые в крайне малом количестве обычно присутствуют среди сульфидов гидротермальных построек. Изотопных данных для серы гидротермальных океанских сульфатов (ангидрита, барита) опубликовано на порядок меньше, чем для сульфидов гидротермальных полей Мирового океана. Тем не менее, во многих случаях величины δ34S гидротермальных сульфатов либо соответствуют верхней планке диапазона, принимаемого для современного сульфата морской воды, либо находятся выше него на 1–2‰. Например, для полей TAG-1 и TAG-2 величины δ34S гидротермальных сульфатов достигают +23‰ (Chiba et al., 1998), в то время как изотопный состав серы современного морского сульфата находится в узком интервале +21.0 ± 0.2‰ (Rees et al., 1978). На первый взгляд, это превышение не кажется критичным, но для современных методов изотопного анализа получаемые различия существенно превышают аналитическую погрешность. Появление избыточного количества изотопа 34S в гидротермальном сульфате заслуживает особого внимания, поскольку кристаллизация сульфатов происходит в окружении огромного изотопного резервуара сульфатной серы морской воды, который обладает максимально высокими величинами δ34S по сравнению остальными геохимическими источниками серы в океане. Следовательно, повышенные величины δ34S в гидротермальных сульфатах необходимо объяснять с привлечением механизмов, подразумевающих обогащение тяжелым (или обеднение легким) изотопом серы сульфата в гидротермальном флюиде.
Перечисленные проблемы инициировали исследования, которые были нами проведены для гидротермальных полей, расположенных в зонах низких скоростей спрединга САХ (Логачев, Краснов и Рэйнбоу), поскольку именно в этих зонах наблюдается больше всего аномалий в изотопных характеристиках серы (Chiba et al., 1998; Gemmell, Sharpe, 1998; Peters et al., 2009; Shanks et al., 1995; и другие работы). Чтобы оценить роль вариаций изотопных параметров флюида во времени, мы применили дифференцированный анализ изотопного состава серы в единичных зернах сульфидных минералов. Отбор зерен проводился после детального минералогического изучения последовательности их формирования в пределах 1-сантиметровых доменов образцов сульфидных построек. В результате была получена классификация сульфидных зерен по времени их отложения для большого количества доменов. Специальное внимание мы уделили поиску гидротермальных сульфатов в образцах и изучению их изотопного состава серы. Изотопный анализ серы проводился прецизионным методом классической масс-спектрометрии в единичных зернах сульфидных минералов.
Для описания собственных, а также опубликованных данных нами была разработана модель, некоторые аспекты которой были освещены ранее (Дубинина, Бортников, 2019). Модель адекватно описывает приведенные выше особенности поведения серы в гидротермальных океанских системах, и центральная роль в ней отведена эффекту закрытой системы на этапе взаимодействия флюид–порода.
МАТЕРИАЛЫ И МЕТОДЫ
Образцы были отобраны в рейсах 35 и 41 НИС “Академик Мстислав Келдыш” и “Serpentine” RV “Pourquoi Pas?”. Были изучены минералы сульфидных построек “Ирина”, “Ирина-2”, “Анна-Луиза” и фрагменты неизвестных построек гидротермального поля Логачев, из массивных сульфидных руд и рудных брекчий поля Краснов, активных и реликтовых построек поля Рэйнбоу. Все три поля расположены в северной части САХ (N MAR), их основные характеристики суммированы в табл. 1, там же приведены ссылки на ключевые работы, в которых описано геологическое строение, минералогия и геохимические особенности этих полей. Детальную информацию об этих и других полях можно найти на специальном интернет-ресурсе (InterRidge Vents Database). Изучение структурных взаимоотношений и последовательности отложения сульфидов в анализируемых образцах, а также отбор единичных зерен пирита, халькопирита и сфалерита проводился вручную под микроскопом с высоким разрешением (Olympus). Зерна ранних и поздних генераций, отобранные в пределах одного домена, располагались друг от друга на расстоянии от 1 до 10 мм. Размер зерен не превышал 1–2 мм.
Таблица 1.
Характеристики гидротермальных полей, материалы которых изучены в данной работе
| Поле, координаты (СШ, ЗД), активность | Тmax, °С | Глубины, м | Скорость спрединга, мм/г | Вмещающие породы | Источники |
|---|---|---|---|---|---|
| Логачев (14.7520, 44.9785), активное |
370 | 2925–3050 | 25.5 | Cерпентиниты, габбро, MORB, | Petersen et al., 2009; Schmidt 2007; Rouxel et al., 2004; Bogdanov et al., 1997 |
| Краснов (16.6400 46.4750), неактивное |
3700–3900 | 25.2 | MORB | Cherkashov et al., 2008; Ockert 2008 | |
| Рэйнбоу (36.2300, 33.9020), активное |
362 | 2270–2320 | 20.6 | Cерпентиниты, габбро, MORB, | Edmonds, German 2004; Rouxel et al., 2004; Douville et al., 2002; Charlou et al., 2002; Andreani et al., 2014 и другие работы |
Поле “Логачев” представлено образцами сульфидных построек “Ирина”, “Ирина-2”, “Анна-Луиза” и несколькими фрагментами неизвестных построек (образцы с порядковым номером 1–10 в табл. 2, фиг. 1а, б). Среди сульфидов резко преобладают халькопирит и изокубанит, в незначительном количестве присутствует сфалерит. В наиболее поздних ассоциациях присутствуют борнит и халькозин, в небольших количествах – ковеллин и атакомит. В участках, сложенных массивным халькопиритом, по трещинам и порам развит борнит. Общая последовательность формирования минералов состоит в смене наиболее раннего пирротина, представленного удлиненными зернами призматической формы, более поздним халькопиритом, изокубанитом и сфалеритом. Гидротермальный сульфат представлен баритом (обр. 6, табл. 2).
Таблица 2.
Изотопный состав серы в гидротермальных отложениях полей Логачев, Краснов и Рэйнбоу (САХ)
| Номер пп | Номер образца | Локализация образца | Описание минеральной фазы | δ34S, ‰ |
|---|---|---|---|---|
| Поле Логачев | ||||
| 1 | 3868-4-2 | Постройка “Ирина-2”, восточный склон, вершина курильщика | Пластинчатый сфалерит ранней генерации (Sph-1) | 4.9 |
| Мелкозернистый халькопирит ранней генерации (Cpy-1) | 4.5 | |||
| Дендритовидный сфалерит поздней генерации (Sph-2) | 5.3 | |||
| Массивный халькопирит поздней генерации (Сру-2) | 5.1 | |||
| 2 | 3863-10-7 | Постройка “Ирина-2”, западный склон, вершина курильщика | Дендрит сфалерита поздней генерации (Sph-2) | 5.9 |
| 3 | 3868-10-1 | Постройка “Ирина”, западный склон, неактивная зона курильщика | Копьевидный халькопирит поздней генерации (Сру-2) | 5.6 |
| Дендрит сфалерита поздней генерации (Sph-2). | 5.6 | |||
| 4 | 3863-9-6 | Там же | Мелкокристаллический сфалерит поздней генерации (Sph-2) | 5.4 |
| Массивный халькопирит поздней генерации (Сру-2) | 5.8 | |||
| 5 | 3863-6-4 | Неактивный курильщик, восточный борт кратера “Анна-Луиза” | Крупнозернистый халькопирит ранней генерации (Сру-1) | 4.7 |
| Мелкозернистый халькопирит поздней генерации (Сру-2) | 4.5 | |||
| Поздний сфалерит из окисленной зоны (Sph-2) | 5.4 | |||
| 6 | DV6-1 | Фрагмент неизвестной сульфидной постройки | Халькопирит поздней генерации (Сру-2) | 5.7 |
| Халькопирит поздней генерации (Сру-2) | 5.7 | |||
| Поздний борнит из зоны окисления (Bn-2) | 6.0 | |||
| Гидротермальный барит | 21.3 | |||
| 7 | DV6-6 | То же | Халькопирит дендритовый поздней генерации (Сру-2) | 5.0 |
| Сфалерит ранней генерации (Sph-1) | 4.5 | |||
| 8 | DV7-18 | То же | Сфалерит ранней генерации, массивный (Sph-1) | 4.0 |
| Халькопирит ранней генерации (Сру-1) | 4.3 | |||
| 9 | DV7-17 | То же | Сфалерит ранней генерации (Sph-1) | 3.9 |
| Халькопирит ранней генерации (Сру-1) | 4.5 | |||
| 10 | DV7-11 | То же | Халькопирит ранней генерации (Сру-1) | 4.5 |
| Сфалерит ранней генерации (Sph-1) | 4.2 | |||
| Поле Краснов | ||||
| 11 | DV8-13 | Массивные руды | Пирит поздней генерации | 9.6 |
| Халькопирит поздней генерации | 9.6 | |||
| 12 | DV-8-17 | Массивные руды | Пирит поздней генерации | 9.8 |
| 13 | DR-16 | Рудная брекчия | Пирит ранней генерации | 8.0 |
| Барит | 21.7 | |||
| 14 | DR-9 | Рудная брекчия | Пирит ранней генерации | 7.7 |
| Пирит ранней генерации | 7.1 | |||
| 15 | DR-11 | Рудная брекчия | Пирит ранней генерации | 7.4 |
| Пирит ранней генерации | 8.1 | |||
| 16 | DR-15 | Рудная брекчия | Пирит ранней генерации | 7.5 |
| Пирит ранней генерации | 8.1 | |||
| Поле Рэйнбоу | ||||
| 17 | 4816-6 | Вершина активной сульфидной постройки | Сфалерит поздней генерации | 6.6 |
| Халькопирит поздней генерации | 6.6 | |||
| 18 | 4816-3 | Склон активной сульфидной постройки | Изокубанит поздней генерации | 5.6 |
| Сфалерит ранней генерации | 4.9 | |||
| Почковидный сфалерит поздней генерации | 5.0 | |||
| 19 | 4820-6 | Реликтовая сульфидная постройка | Массивный сфалерит ранней генерации | 2.9 |
| Массивный сфалерит ранней генерации | 2.9 | |||
| Почковидный сфалерит поздней генерации | 4.4 | |||
| 20 | 4815-8 | Реликтовая сульфидная постройка | Изокубанит поздней генерации | 3.0 |
| Халькопирит ранней генерации | 2.8 | |||
| Сфалерит поздней генерации из зоны окисления | 3.3 | |||
| 21 | 4815-3 | Реликтовая сульфидная постройка | Сфалерит поздней генерации | 4.4 |
| Сфалерит поздней генерации | 4.8 | |||
| 22 | 4816-4 | Неактивная сульфидная постройка | Сфалерит ранней генерации | 5.9 |
| Сфалерит поздней генерации | 6.1 | |||
| Халькопирит поздней генерации | 6.3 | |||
| Сфалерит поздней генерации | 6.1 | |||
| 23 | 4817-9 | Фрагмент массивных руд | Халькопирит ранней генерации | 3.8 |
| 24 | 4819-8 | Фрагмент гидротермальной постройки (метка Г) | Дендриты сфалерита | 5.4 |
| Сфалерит, ассоциирующий с халькопиритом | 6.0 | |||
| Халькопирит, ассоциирующий со сфалеритом | 6.1 | |||
| 25 | 4820-5 | Гидротермальная постройка | Ранний почковидный сфалерит | 4.3 |
| Ранний сфалерит | 4.1 | |||
| Ранний халькопирит | 3.9 | |||
| 26 | 4816-5 | Активная гидротермальная постройка | Халькопирит длиннопризматический | 5.1 |
| Халькопирит длиннопризматический | 5.2 | |||
| Пирит-марказит | 5.8 | |||
| Почковидный сфалерит | 5.2 | |||
| Крупнокристаллический сфалерит | 5.6 | |||
| 27 | 4819-16 | Активная гидротермальная постройка | Барит гидротермальный, в пустотах между кристаллическими агрегатами сульфидов | 21.9 |
| То же | 21.1 | |||
| То же | 21.5 | |||
| Пирит-марказит ранней генерации, ассоциирующий с баритом | 2.5 | |||
| Дендриты сфалерита поздней генерации | 4.3 | |||
| Халькопирит ранней генерации | 2.1 | |||
| 28 | 4817-13 | Неактивная часть гидротермальной постройки | Массивный сфалерит ранней генерации | 4.3 |
| Мелкокристаллический сфалерит ранней генерации | 4.2 | |||
| 29 | 4812-1 | Склон гидротермальной постройки | Почковидный марказит ранней генерации | 3.9 |
| Сфалерит ранней генерации | 4.9 | |||
| 30 | 4815-2 | Фрагмент сечения постройки | Сфалерит ранней генерации | 3.4 |
| Борнит ранней генерации | 3.3 | |||
| Ковеллин ранней генерации | 3.5 | |||
| 31 | 4812-2 | Фрагмент массивных руд | Почковидный поздний марказит | 7.7 |
| Сфалерит ранней генерации | 6.0 | |||
| Пирит поздней генерации | 8.4 | |||
Фиг. 1.
Микрофотографии характерных участков гидротермальных сульфидных руд, служивших доменами для отбора единичных зерен минералов. Цифры на фото – измеренные величины δ34S. Сульфиды поля Логачев: а – наиболее ранние тонкозернистые агрегаты сфалерита и халькопирита, б – поздние дендриты халькопирита (изокубанита) в порах. Сульфиды поля Краснов: в – ранний тонкозернистый массивный пирит, частично в срастаниях с марказитом, г – кубические зерна позднего пирита в порах и трещинах. Сульфиды поля Рэйнбоу: д – ранний тонкокристаллический халькопирит (изокубанит), пересеченный трещинами, выполненными поздним баритом, е – поздний сфалерит среди кристаллических агрегатов барита, выполняющего одну из пустот. Сульфаты: пластинчатые агрегаты барита в пирит-марказитовой руде поля Краснов (ж) и тонкоигольчатые агрегаты барита в порах и пустотах сульфидных руд поля Рэйнбоу.

Поле Краснов представлено образцами массивных сульфидных руд (обр. 11-16 в табл. 2, фиг. 1в, г) и рудных брекчий. В массивных рудах стадия формирования массивных пиритовых и пирит-марказитовых руд сменяется отложением халькопирита с редкими включениями сфалерита и развитием по трещинам в халькопирите халькозина и окислов железа. Образцы рудных брекчий сложены обломками подстилающих базальтов, сцементированных сульфидным цементом, который состоит из массивного марказита и пирита. В рудных брекчиях отложение массивного мелкокристаллического пирита сменяется развитием марказита и пирит-марказитовых агрегатов, после чего происходит отложение пирита второй генерации. Гидротермальный сульфат представлен баритом (обр. 13, табл. 2, фиг. 1ж).
Поле Рэйнбоу представлено образцами активных и реликтовых построек и двумя образцами (обр. 17–31 в табл. 2, фиг. 1д, е) массивных руд. Для этого поля характерно развитие существенно цинковой и существенно медной минерализации. В образцах с преобладанием цинковых минералов проявлено последовательное выделение генераций сфалерита от плотных массивных разновидностей до дендритов и микрокристаллических разновидностей. В образцах с существенно медной минерализацией к более ранним минералам относится массивный халькопирит (изокубанит) с последующими выделениями его дендритовых форм. Более поздней является борнит-халькозин-ковеллиновая ассоциация, которая сменяется ассоциацией пирита и марказита, представленных кубическими и натечными формами соответственно. Гидротермальный сульфат представлен баритом (обр. 27, табл. 2, фиг. 1з).
Изотопный анализ серы проведен в лаборатории изотопной геохимии и геохронологии ИГЕМ РАН. Перед анализом каждое зерно истиралось до тонкой пудры и от гомогенизированной пробы отбиралась необходимая навеска от 100 до 400 мг для сульфидов и 360 мг для сульфатов (эквивалентно 50 мг в пересчете на чистую серу). При анализе сульфатов в навеску образца добавлялся высокочистый реактив V2O5 в соотношении 1 : 1 по массе. Конверсия сульфидной серы в SO2 проводилась с помощью элементного анализатора FlashHT 1112 при 1020°С в окислительно-восстановительном реакторе, заполненном Cu0 и WO3. Изотопный состав серы в газе SO2 измерялся on-line методом масс-спектрометрии в постоянном потоке гелия (CF-IRMS) на масс-спектрометре DELTA V + (Finnigan, Germany). В конце и начале измерительных серий анализировались международные стандартные образцы сульфида серебра IAEA-S-1 и IAEA-S-2 при анализе сульфидов и сульфат бария NBS 127 – при анализе сульфатов. Для представления результатов в международной шкале V-CDT использовались референтные значения δ34S для вышеуказанных стандартов, равные –0.3‰, +22.67 и +21.1‰ соответственно. Изотопный состав серы в данной работе выражен в относительных величинах δ (Coplen, 2011):
Воспроизводимость полученных данных находилась на уровне ±0.25‰ (1σ).
Результаты измерений представлены в табл. 2, и на фиг. 1–4. Усредненные изотопные характеристики для сульфидов изученных полей суммированы в табл. 3.
ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ
Сульфиды
Величины δ34S, полученные для сульфидов всех трех полей, существенно превышают значения, характерные для мантии (δ34S ≈ 0 ± 2‰, Thode et al., 1961) и магматических пород океанской коры (δ34SMORB= –0.3 ± 2.3‰, δ34SOIB = 1.0 ± 1.9‰, Sakai et al., 1984; Chaussidon et al., 1989). Величины δ34S в сульфидах поля Логачев составляют интервал +3.9…+6‰, аналогичный несколько более широкому интервалу, установленному в сульфидах поля Рэйнбоу: +2.1…+8.4‰ (фиг. 2). Согласно ранее опубликованным данным (Lein et al., 2001), на поле Рэйнбоу отмечаются еще более широкие вариации изотопного состава серы сульфидов – от +5 до +12‰. Однако наши данные лежат в более компактном диапазоне и близки к опубликованным данным Rouxel et al., 2004. Изотопный состав серы сульфидов поля Логачев соответствует опубликованным данным Peters et al., 2009 и Rouxel et al., 2004 и далек от величин δ34S, опубликованных для массивных сульфидов этого поля (Zheng et al., 2016). Для поля Краснов, в отличие от полей Логачев и Рэйнбоу, характерны более высокие величины δ34S (+7.1…+9.8‰), слабо варьирующие, с хорошо проявленной разницей между сульфидами ранней и поздней генераций. Имеющиеся в литературе данные для этого поля немногочисленны (Ockert, 2008; Peters et al., 2009) и показывают широкий разброс (от +5 до +10‰), в который попадают все наши определения, проведенные как для ранних, так и для поздних генераций сульфидов.
Фиг. 2.
Сравнение наших и опубликованных данных по полям Логачев, Краснов и Рэйнбоу. Литературные данные: O 08 – (Ockert, 2008; Peters et al., 2009), L 01 – (Lein et al., 2001), R 04 – (Rouxel et al., 2004), P 09 – (Peters et al., 2009), Z 16 – (Zheng et al., 2016). “Ранние” и “Поздние” – обозначения сульфидов более ранних и более поздних генераций, зерна которых отбирались для изотопного анализа.
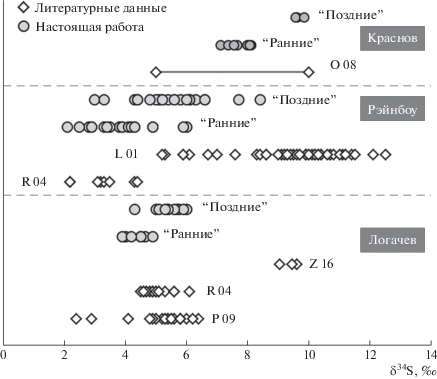
Связь величин δ34S сульфидов с последовательностью их генерации, установленной по структурным соотношениям отдельных зерен в образце, оказалась устойчивой. Сульфиды ранних генераций имеют более низкие значения величины δ34S, чем сульфиды поздних генераций (фиг. 2). Это проявлено для всех трех полей, и статистическая обработка данных показывает, что полученные различия в 1–2 промилле между сульфидами ранних и поздних генераций являются значимыми (табл. 3). Подобные различия в изотопном составе серы ранних и поздних генераций сульфидов были получены для массивных руд на Юконе, Канада (McDonald et al., 2018). Этот результат согласуется с наблюдаемой зональностью величин δ34S в пределах отдельных построек на 11о–13о (Bluth, Ohmoto, 1988) и на 21о с.ш. ВТП (Woodruff, Shanks, 1988), выраженной в изотопном утяжелении серы от внешних к внутренним зонам трубообразных построек. Наши данные, в дополнение к предыдущим, показывают, что закономерное изменение изотопного состава серы сульфидов может быть проявлено и в локальном масштабе, в пределах доменов, размером 1–10 мм.
Интервалы измеренных величин δ34S пирита, халькопирита и сфалерита из построек одних и тех же полей полностью перекрываются (фиг. 3), из чего следует, что ни состав, ни структура минералов не являются контролирующим фактором в формировании изотопных отношений серы при осаждении сульфидов из гидротермальных растворов. Независимость изотопного состава серы от минерального состава и отчетливая связь величин δ34S с последовательностью отложения приводят к выводу о том, что изотопный состав серы сульфидов определяется исключительно изотопными характеристиками серы в гидротермальном флюиде, которые закономерно эволюционируют во времени.
Фиг. 3.
Систематика изотопного состава серы сульфатных минералов в зонах подводной гидротермальной деятельности. Вертикальные линии – интервал изотопного состава серы современного сульфата морской воды (21.0 ± 0.2‰, Rees et al., 1978). Литературные данные: С 98 – Chiba et al., 1998, dR 05 – de Ronde et al., 2005, GS 98 – Gemmell, Sharpe, 1998, H 98 – Herzig et al., 1998, K 90 – Kusakabe et al., 1990, M 99 – Marumo et al., 1999, P 09 – Peters et al., 2009, SN 98 – Shanks, Niemitz, 1982, Z 84 – Zierenberg et al., 1984.

Сульфаты
Нам удалось обнаружить несколько редко встречающихся кристаллических выделений барита в материале изученных сульфидных образцов. По морфологическим признакам и положению в образце отобранные для анализа единичные кристаллы барита могут быть отнесены к гидротермальным образованиям (фиг. 1). Согласно фиг. 3, величины δ34S гидротермального барита, установленные для полей Краснов (+21.7‰) и Рэйнбоу (+21.1, +21.9‰), слабо, но устойчиво превышают величины, характерные для сульфата морской воды (21.0 ± 0.2‰, Rees et al., 1978). Образец игольчатого барита с поля Логачев имеет величину δ34S = +21.3‰, что соответствует верхнему пределу интервала, характерного для современного сульфата морской воды. Установленные нами сдвиги уверенно превышают аналитическую погрешность, что не позволяет считать их случайными. На фиг. 4 результаты данной работы приведены в сравнении с опубликованными данными для сульфатов других зон гидротермальной активности в океане (Chiba et al., 1998; de Ronde et al., 2003; Gemmell, Sharpe, 1998; Herzig et al., 1998; Kusakabe et al., 1990; Marumo et al., 1999; Peters et al., 2009; Shanks, Niemitz, 1982; Zierenberg et al., 1984). Видно, что высокие величины δ34S в гидротермальных сульфатах возникают повсеместно, вне зависимости от тектонической обстановки, скорости спрединга и строения участков коры, на которых расположены гидротермальные поля. Например, отчетливое утяжеление изотопного состава серы сульфатов установлено как в зонах медленного (SMAR, Peters et al., 2009; TAG-1, Chiba et al., 1998), так и быстрого спрединга (21-й градус ВТП, Zierenberg et al., 1984), на перекрытом осадками поле Гуаймас (Shanks, Niemitz, 1982) и в гидротермах островодужных вулканов Brothers (de Ronde et al., 2005).

ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Полученные нами данные для трех гидротермальных полей САХ подтверждают, что величины сульфидов δ34S характеризуются высокими положительными значениями, практически не зависят от типа сульфидного минерала и имеют устойчивую связь с последовательностью генерации сульфидов, устанавливаемой по структурно-текстурной позиции отдельных зерен в образце. Эти черты исключают равновесные соотношения изотопов серы между сульфидными минералами, что может быть отнесено к атрибутам сульфидов океанских гидротермальных полей (например, Shanks, 1995, 2001; Herzig et al., 1998; Seal et al., 2000, 2006; Zheng et al., 2016; и другие обзоры). Закономерное изменение величин δ34S сульфидов в зависимости от времени и отсутствие связи с типом минерала приводит к выводу, что изотопные параметры серы осаждающихся сульфидов контролируются исключительно величиной δ34S(H2S) флюида, которая изменяется во времени. Изотопное утяжеление серы гидротермальных сульфатов, формирующихся на выходе гидротермальных растворов вместе с сульфидами, позволяет предположить, что в процессе TSR, который протекает в зоне генерации сульфидов с участием железистых минералов (Woodruff, Shanks, 1988; Seal et al., 2006), происходят закономерные сдвиги в изотопном составе серы флюида. Это означает, что система должна быть закрыта относительно флюида, чтобы реализовалось частичное исчерпание изотопа 32S в растворенном сульфате флюида.
Как и в других циркуляционных моделях, для океанских гидротерм полагается, что система открыта по отношению к флюиду, поскольку конвективная ячейка находится в окружении бесконечного резервуара океанской воды. Однако в зоне генерации сульфидов, где происходит основная стадия взаимодействия флюид-порода, имеет значение соотношение характерных скоростей протекания TSR и скорости фильтрации флюида. Если время пребывания флюида в зоне взаимодействия с породой существенно выше, чем время установления изотопного равновесия серы между ее формами в этом флюиде, по отношению к изотопной системе серы система может рассматриваться как закрытая относительно флюида. Время восстановления сульфата с участием базальтов и других пород океанской коры составляет от первых часов при Т = 350°С, до первых суток при 250°С (Shanks et al., 1981). Установление изотопного равновесия серы между сероводородом и сульфатом в гидротермальном растворе требует больше времени. При рН = 4…7 и Т = 300оС реакция изотопного обмена между сульфидной и сульфатной формами серы протекает на 90% за 140 дней, при повышении температуры до 350°С – за 17 дней, а снижение рН до величины менее 4 сократит это время до первых дней или часов (Ohmoto, Lasaga, 1982). Время пребывания флюида в субмаринных гидротермальных системах оценить трудно, поскольку оно зависит от многих факторов: размеров и глубины залегания конвективной ячейки, строения порово-трещинного пространства пород океанского дна, скорости спрединга и наличия осадочного чехла. Одна из немногочисленных прямых оценок времени пребывания флюида в гидротермальной системе на хребте Хуан-де- Фука, проведенная по 210Pb, составила 22.3 года (Kadko et al., 1985/86). Можно ожидать, что в гидротермальных ячейках полей медленно-спрединговых зон, например САХ, время пребывания флюида является существенно более длительным. Сопоставление времени фильтрации флюида в гидротермальной ячейке (годы – десятки лет) с характерным временем восстановления морского сульфата и установлением изотопного равновесия между сульфатом и сульфидом во флюиде (часы – десятки суток) показывает, что условия закрытой относительно флюида системы при восстановлении сульфата должны реализовываться во многих океанских гидротермах. Таким образом, может наблюдаться релеевское исчерпание сульфата флюида относительно изотопа 32S, который концентрируется в сульфидной фазе в процессе TSR.
1. Схема и основные допущения
В предлагаемой модели значительная роль отводится так называемому эффекту закрытой системы в процессе TSR, учитывается дополнительный вклад серы, извлекаемой флюидом из пород океанского дна, а также процессы смешения флюида с океанской водой в зоне подводной разгрузки. Последовательность этих процессов приведена на фиг. 5, где разделы модели соответствуют трем стадиям, выделяемым в традиционных циркуляционных схемах (например, Woodruff, Shanks, 1988; Janecky, Shanks, 1988; Бортников, Викентьев, 2005; Tivey, 2007; McDonald et al., 2018). В модели принято допущение о том, что при осаждении сульфидов и сульфатов из раствора имеет место локальное изотопное равновесие между минералом и соответствующей формой серы в растворе. Численные параметры, которые использованы для модельных расчетов, суммированы в табл. 4.
Фиг. 5.
Схема основных стадий деятельности подводной гидротермальной системы по Woodruff, Shanks, 1988 (а) и схема модели для описания поведения изотопной системы серы на данных стадиях (б). Серыми полями обозначены процессы: (ТSR) – термогенное восстановление сульфата при участии магматических пород, (М-1) – смешение восстановленной серы флюида с серой, извлеченной из магматических пород, (М-2) – смешение сульфата флюида с сульфатом морской воды в зоне осаждения сульфидов и сульфатов. Пунктир – уровень изотопного состава серы сульфата морской воды (SW).

Таблица 4.
Численные параметры, принятые для расчетов
| Название | Величина | Единицы измерения | Ссылка на источник |
|---|---|---|---|
| Содержание сульфата в морской воде | 27 | ммоль,кг | Shanks, 2001 |
| Содержание FeO в базальтах | 7.3–8.2 | вес. % | Le Maitre, 1976 |
| Содержание FeO в перидотитах | 7.0 | вес. % | Le Maitre, 1976 |
| Содержание S в базальтах | 800 | г/т | Принято в данной работе |
| Содержание S в перидотитах | 250 | г/т | То же |
| Величина δ34S морского сульфата | +21.0 ± 0.2 | ‰ | Rees et al., 1978 |
| Величина δ34S серы в магматических породах (базальтах) | –0.3 ± 2.3 | ‰ | Sakai et al., 1984 |
| Уравнение изотопного фракционирования серы при восстановлении сульфата | 103Lnα(TSR) = 6.463 × 106T–2 + 0.56 | Ohmoto, Lasaga, 1982 | |
| Уравнение изотопного фракционирования серы при осаждении сульфидов из флюида | 103Lnα (Py- H2S) = 0.4 × 106T–2 103Lnα (Sl- H2S) = 0.1 × 106T–2 |
Ohmoto, Rye, 1979 | |
Стадия осаждения ангидрита. Этот процесс протекает при нисходящей фильтрации океанской воды в зону нагрева. В силу ретроградной растворимости ангидрита, сульфатная сера почти полностью выводится из раствора при Т > 150°C (Bishoff, Seyfried, 1978). При осаждении ангидрита происходит вывод подавляющей доли морского сульфата из разогретой морской воды, однако часть (SO4)2– остается во флюиде в соответствии с равновесием по отношению к ангидриту (Bishoff, Seyfried, 1978; Seyfried, Bishoff, 1981). Изотопное фракционирование серы при осаждении сульфатов невелико (менее 1‰, Shanks, 2001), и можно считать, что изотопный состав серы сульфата, оставшегося во флюиде после осаждения ангидрита, не отличается от изотопного состава серы сульфата морской воды. Таким образом, в модели для этой стадии можно допустить частичный вывод морского сульфата из флюида без изменения изотопного состава серы в оставшемся сульфате.
Стадия взаимодействия флюид-порода. Согласно схеме циркуляции (фиг. 5), эта стадия приурочена к зоне генерации сульфидов (Woodruff, Shanks, 1988). Здесь протекают два процесса, каждый из которых является ключевым в формировании изотопных характеристик серы в гидротермальном флюиде. Первый из них – TSR: восстановление сульфата в условиях повышенных температур и при участии Fe(II)-содержащих минералов основных пород – прежде всего, оливина (Seal et al., 2006):
(1)
$\begin{gathered} 4{\text{F}}{{{\text{e}}}_{{\text{2}}}}{\text{Si}}{{{\text{O}}}_{4}} + 3{\text{M}}{{{\text{g}}}_{{\text{2}}}}{\text{Si}}{{{\text{O}}}_{4}} + {\text{CaS}}{{{\text{O}}}_{4}} + \\ + \,\,{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to 5{\text{M}}{{{\text{g}}}_{{\text{2}}}}{\text{Si}}{{{\text{O}}}_{3}} + 4{\text{F}}{{{\text{e}}}_{{\text{2}}}}{{{\text{O}}}_{3}} + \\ + \,\,{\text{CaMgS}}{{{\text{i}}}_{{\text{2}}}}{{{\text{O}}}_{6}} + {{{\text{H}}}_{{\text{2}}}}{\text{S}}{\text{.}} \\ \end{gathered} $Во время этого процесса эффект закрытой системы должен играть ключевую роль, в результате чего величины δ34S гидротермального флюида должны возрастать, что приводит к существенным вариациям в изотопном составе серы осаждающихся сульфидов. Второй процесс, протекающий на данной стадии – экстракция серы из пород. Медленная фильтрация флюида, высокая Т и наличие свежих деформаций способствуют эффективному выщелачиванию серы из пород. В процессе выщелачивания во флюид поступает только сульфидная сера, которая смешивается с восстановленной серой, генерируемой при TSR, и влияет на величину δ34S(H2S) во флюиде.
Стадия подводной разгрузки. На этой стадии происходит смешение гидротермальных растворов с океанской водой и осаждение основной части гидротермальных сульфидов и сульфатов. В модели принимается, что процесс частичного окисления сероводорода при смешении флюида с океанской водой не играет заметной роли и не влияет на изотопный состав серы сероводорода и осаждающихся сульфидов. В результате смешение гидротермального флюида с морской водой не влияет на изотопные характеристики серы сульфидов. Однако на изотопный состав серы сульфатов смешение с океанской водой влияет резко, поскольку концентрация сульфатной серы в океанской воде намного выше, чем в гидротермальном растворе, из которого сульфат был извлечен сначала при осаждении ангидрита, а потом за счет процесса TSR. Осаждение ангидрита понижает концентрацию сульфата морской воды примерно втрое, а после взаимодействия с базальтами она может снизиться еще на порядок. По оценкам (Seyfried, Bishoff, 1981; Shanks et al., 1995), при 300°С взаимодействие морской воды с базальтами приводит к падению концентрации сульфата в ней до 0.2–0.5 ммоль/кг. При подводной разгрузке гидротермальный раствор смешивается с морской водой, содержащей 27 ммоль/кг сульфата (Shanks, 2001), имеющего постоянный изотопный состав серы. На этой стадии гидротермальный сульфат быстро теряет свою изотопную метку даже при участии очень небольшой примеси свежей морской воды.
2. Описание модели
Алгоритм, который воспроизведен в математическом описании модели (фиг. 5б), подразумевает, что основная часть сульфата морской воды выведена на стадии осаждения ангидрита без изменения величины δ34S оставшегося во флюиде сульфата. В процессе TSR на стадии взаимодействия флюид–порода восстановлению подвергается некая доля сульфата гидротермального раствора, так что в любой момент величина f, которую можно определить как степень протекания реакции восстановления сульфата, равна :
(2)
$f = \frac{{{{{[{{{\text{H}}}_{{\text{2}}}}{\text{S}}]}}_{{SR}}}}}{{{{{[{\text{S}}{{{\text{O}}}_{{\text{4}}}}{\text{]}}}}^{0}}}},$(3)
$\delta {{\left( {{\text{S}}{{{\text{O}}}_{4}}} \right)}^{0}} = ~\delta {{\left( {{\text{S}}{{{\text{O}}}_{4}}} \right)}_{{SR}}}\left( {1 - f} \right) + \bar {\delta }{{\left( {{{{\text{H}}}_{{\text{2}}}}{\text{S}}} \right)}_{{SR}}}f.$где δ(SO4)0 и δ(SO4)SR – изотопный состав серы сульфата до начала восстановления и к данному моменту, а δ(H2S)SR – усредненный изотопный состав серы сероводорода, образованного к моменту f. Состав последнего можно выразить интегралом:
(4)
$\bar {\delta }{{\left( {{{{\text{H}}}_{{\text{2}}}}{\text{S}}} \right)}_{{SR}}} = \left( {\frac{1}{f}} \right)\mathop \smallint \limits_0^f \left( {\delta {{{\left( {{\text{S}}{{{\text{O}}}_{4}}} \right)}}_{{SR}}} - \Delta {\text{ТSR}}} \right)df,$(5)
$\frac{{d\delta {{{\left( {{\text{S}}{{{\text{O}}}_{4}}} \right)}}_{{{\text{TSR}}}}}}}{{df}} = \frac{{\Delta {\text{TSR}}}}{{1 - f}}.$Решением уравнения (5) при граничном условии: если f = 0, $\delta \left( {{\text{S}}{{{\text{O}}}_{4}}} \right)$ = $\delta {{\left( {{\text{S}}{{{\text{O}}}_{4}}} \right)}^{0}}$ будет выражение, описывающее изменение изотопного состава серы остающегося в растворе сульфата по мере его исчерпания за счет восстановления:
(6)
$\delta {{\left( {{\text{S}}{{{\text{O}}}_{4}}} \right)}_{{{\text{TSR}}}}} = \delta {{\left( {{\text{S}}{{{\text{O}}}_{4}}} \right)}^{0}} - \Delta {\text{TSRLn}}\left( {1 - f} \right).$В любой момент состояния системы в равновесии с сульфатной серой флюида будет генерироваться сероводород с изотопным составом:
(7)
$\delta {{\left( {{{{\text{H}}}_{2}}{\text{S}}} \right)}_{{{\text{TSR}}}}} = \delta {{\left( {{\text{S}}{{{\text{O}}}_{4}}} \right)}_{{{\text{TSR}}}}} - \Delta {\text{TSR}}.$Одновременно с восстановлением сульфата взаимодействие флюид–порода приводит к выщелачиванию сульфидной серы из пород. Нами в модель был введен коэффициент, который задает долю серы, извлеченной из пород, во всей восстановленной сере, находящейся во флюиде:
(8)
$k = ~\frac{{{{{\left[ {{{{\text{H}}}_{2}}{\text{S}}} \right]}}_{R}}}}{{{{{\left[ {{{{\text{H}}}_{2}}{\text{S}}} \right]}}_{{{\text{TSR}}}}} + {{{\left[ {{{{\text{H}}}_{2}}{\text{S}}} \right]}}_{R}}}}.$Задавая пропорцию смешения через этот коэффициент, можно записать изотопный состав сероводорода флюида как результат смешения H2S, поступившего из двух источников – при восстановлении сульфата флюида и при выщелачивании пород:
(9)
$\delta {{\left( {{{{\text{H}}}_{2}}{\text{S}}} \right)}_{{{\text{Fl}}}}} = k\delta {{\left( {{{{\text{H}}}_{2}}{\text{S}}} \right)}_{R}} + \left( {1 - k} \right)\delta {{\left( {{{{\text{H}}}_{2}}{\text{S}}} \right)}_{{{\text{TSR}}}}}.$Изотопный состав серы сульфида, равновесного с флюидом в момент осаждения, можно записать:
(10)
$\delta \left( {{\text{Sfd}}} \right) = \delta {{\left( {{{{\text{H}}}_{2}}{\text{S}}} \right)}_{{{\text{Fl}}}}} + {{\Delta }_{{{\text{Sfd}} - {{{\text{H}}}_{2}}{\text{S}}}}},$(11)
$\bar {\delta }\left( {{\text{Sfd}}} \right) = \left( {\frac{1}{f}} \right)\mathop \smallint \limits_0^f \left( {\delta {{{\left( {{{{\text{H}}}_{2}}{\text{S}}} \right)}}_{{{\text{Fl}}}}} + {{\Delta }_{{{\text{Sfd}} - {{{\text{H}}}_{2}}{\text{S}}}}}} \right)df.$В уравнениях (6–11) фигурируют два параметра – переменная f (степень восстановления сульфата в процессе TSR) и коэффициент k – доля серы, извлеченной из пород в общем количестве сульфидной серы флюида. В предыдущих моделях эти параметры задаются независимо друг от друга (например, Brueckner et al., 2015; McDonald et al., 2018). Однако, если извлечение серы из пород сопровождает процесс взаимодействия флюид-порода, при котором также протекает и TSR, неизбежен вывод о связи переменной f и коэффициента k через стехиометрию реакции TSR. Зная стехиометрию конкретной реакции TSR и содержания Fe(II) и S в породе, можно оценить величину коэффициента k, которая дает представление о доле извлеченной из пород серы в общем содержании ее восстановленной формы во флюиде. Существующие оценки этой доли различны – от преобладания сульфидной серы, полученной в результате TSR (Brueckner et al., 2015), до преобладания серы, извлеченной из пород (Zheng, 2016). Последняя оценка получена прямым балансовым расчетом смешения двух компонентов с величинами δ34S, равными 0 (породы) и 21‰ (морской сульфат), без учета каких-либо процессов фракционирования серы. В результате было подсчитано, что в океанские гидротермальные сульфиды из сульфата морской воды поступает не более 36% серы, а остальное (не менее 64%) извлекается из пород океанской коры, т.е. величина k должна быть выше 0.64. Если принять во внимание мольные соотношения в реакции восстановления сульфата за счет двухвалентного железа, содержащегося в породе, расчет приводит к оценке, которая почти на порядок ниже. Например, если по реакции (1) восстановить 1 моль сульфатной серы, должно окислиться 8 молей двухвалентного железа. При содержании FeО в базальтах на уровне 7–9 вес. % (Le Maitre, 1976) и S ≈ 800 г/т, доля сульфидной серы, конгруэнтно извлекаемой из породы, составит не более 0.15. При более низких содержаниях серы (например, общепринятая величина 250 г/т для мантии, Alt et al., 1989; Sakai et al., 1984) величина k может опуститься до значения ≈0.06. Исходя из интервалов концентраций двухвалентного железа и серы в породах океанской коры (например, Marini, 2011; Alt et al., 2007), коэффициент k для большинства океанских гидротерм должен находиться примерно в этих пределах. На фиг. 6 приведен расчет величин δ34S сульфата и сероводорода во флюиде, а также интегральных составов пирита и сфалерита, проведенный для k = 0.1 и протекания реакции TSR при 300°С. Эффект закрытой системы приводит к резкому возрастанию величин δ34S в остаточном сульфате и сероводороде флюида, что связано с большим изотопным сдвигом, при термогенном восстановлении сульфата (Ohmoto, Rye, 1979; Ohmoto, Lasaga, 1982). Интегральный изотопный состав серы сульфидов достигает высоких величин, например, при f > 0.7 можно получить сульфиды с величинами δ34S > > +10…+15‰ (фиг. 7), но даже при f → 1 невозможно получить гидротермальные сульфиды с величинами δ34S, равными или выше состава сульфата морской воды. Данные, представленные в многочисленных обзорах (например, Zheng et al., 2016; Shanks, 2001; Seal et al., 2006), подтверждают этот вывод – величины δ34S гидротермальных сульфидов в океане никогда не достигают состава морского сульфата.
Фиг. 6.
Изменение изотопного состава форм серы (${\text{SO}}_{4}^{{2 - }}$ и H2S) во флюиде и серы гидротермальных сульфидов – пирита и сфалерита при взаимодействии флюид–порода в закрытой относительно флюида системе (частичное восстановление сульфата и выщелачивание серы из магматических пород при k = 0.1, Т = 300°С).

Фиг. 7.
Интегральный изотопный состав серы сульфидов, сформированных в равновесии с флюидом, который прошел взаимодействие с породами при разной температуре. Звездочки – формирование “ранних” и “поздних” минералов из одного и того же флюида, закономерно различающихся по изотопному составу серы.

При пониженных температурах (ниже 250°С) и небольших величинах f (менее 0.1) гидротермальные сульфиды могут иметь небольшие отрицательные величины δ34S, на уровне первых промилле (фиг. 7). На данном рисунке иллюстрируется еще одно наблюдение – соотношение величин δ34S в ранних и поздних сульфидах, формирующихся при участии одной и той же флюидной системы или порции флюида. Как следует из приведенных расчетов, по мере развития процесса TSR интегральный изотопный состав серы сульфидов возрастает монотонно, следовательно, более ранние минералы должны иметь более низкие величины δ34S, чем более поздние. Данные, полученные нами для ранних и поздних сульфидов в настоящей работе, прекрасно иллюстрируют этот вывод.
В отличие от сульфидов, изотопный состав серы гидротермальных сульфатов почти не сохраняет информацию о составе сульфата в гидротермальном флюиде из-за контаминации серой сульфата океанской воды. Считая, что в кристаллизующемся минерале (барите, ангидрите) находится сера, поступающая из двух источников, можно описать величины δ34S(MeSO4) балансовым уравнением:
(12)
$\delta \left( {Me{\text{S}}{{{\text{O}}}_{4}}} \right) = \delta {{\left( {{\text{S}}{{{\text{O}}}_{4}}} \right)}_{{{\text{TSR}}}}}X + \delta {{\left( {{\text{S}}{{{\text{O}}}_{4}}} \right)}_{{SW}}}\left( {1 - X} \right),$
3. Сопоставление модели с природными наблюдениями
Поскольку процессы осаждения гидротермальных сульфидов слабо влияют на их изотопный состав серы (Ohmoto, Rye, 1979), величины δ34S гидротермальных сульфидов могут служить источником информации об условиях протекания процесса TSR в зоне генерации сульфидов (зона 2 на схеме фиг. 5). Согласно модели, высокие величины δ34S в сульфидах могут быть признаком высоких, а низкие – низких степеней протекания TSR в закрытой системе. Величины δ34S сульфидов, слабо варьирующие и близкие к составу сульфидной серы магматических пород, указывают на реализацию условий открытой системы по отношению к флюиду.
В качестве примера можно провести реконструкцию условий взаимодействия флюид-порода по опубликованным данным для гидротермальных полей, в которых приводится информация не только по изотопному составу серы сульфидов, но и по величинам δ34S(H2S) в гидротермальных растворах (Peters et al., 2009; Woodruff, Shanks, 1988; Shanks, Seyfried, 1987). На фиг. 9 приведены два варианта расчетов в координатах δ34S(H2S)Fl – δ34S(Sfd) при зафиксированном параметре k = 0.1. Расчет с применением температуры в качестве переменной (200 < T°С < 400) при четырех фиксированных величинах f (0.05, 0.15, 0.25 и 0.35) показывает, что имеющиеся данные для полей ВТП, САХ и хребта Хуан-де-Фука могут формироваться при степени протекания TSR не более 0.35. Основная часть опубликованных данных попадает в диапазон значений 0.15 < f < 0.35 (фиг. 9а). Отсюда легко подсчитать эффективное (т.е. геохимическое) соотношение флюид-порода, при котором протекает TSR по реакции (1). Интервал величин f = (0.15–0.35) соответствует интервалу соотношений W/R от 1000 до 440. Это максимальная, и явно завышенная оценка, т.к. предполагается, что все двухвалентное железо породы участвует в реакции (1). Тем не менее, наш расчет находится в согласии с другими оценками геохимического отношения W/R, которое для океанских гидротерм варьирует, в зависимости от глубины, от 100 до 104 (Fisher, 1998).
Фиг. 9.
Модельный расчет и природные данные (обобщение результатов работ Peters et al., 2009; Woodruff, Shanks, 1988; Shanks, Seyfried, 1987) в координатах δ34S(H2S)Fl – δ34S(Sfd). В расчетах был зафиксирован параметр k = 0.1, варьировались степень восстановления сульфата (а) и температура (б). Обозначения: 1 – поля САХ (Логачев, SMAR), 2 – поля ВТП (9–10 и 21 градусы СШ), 3 – хребет Хуан-де-Фука.

Во втором варианте расчета в координатах δ34S(H2S)Fl – δ34S(Sfd) в качестве переменной использовалась величина f (0.05 < f < 0.35) при фиксированных температурах 200, 250, 300, 350 и 400°С. На этой диаграмме (фиг. 9б) все имеющиеся данные попадают в узкий интервал температуры 250–350°С (редко до 400°С). Таким образом, можно утверждать, что TSR протекает в разных гидротермальных ячейках примерно в одном узком диапазоне температуры. Этот расчет согласуется с результатами эксперимента по восстановлению сульфата морской воды с участием фаялита и магнетита, которые показали, что ниже 250°С процесс восстановления сульфата двухвалентным железом крайне затруднен (Shanks et al., 1981). Таким образом, модель действительно позволяет судить об условиях протекания TSR – температуре и степени протекания реакции, если имеются данные по изотопному составу серы не только сульфидов, но и сероводорода флюида.
Модель объясняет отсутствие изотопного равновесия серы H2S флюида с сульфатом океанской воды при наблюдаемых температурах выхода гидротерм. Литературные данные по изотопному составу серы сероводорода флюидов океанских гидротермальных полей мы сопоставили с расчетом в координатах δ34S(H2S) – Т, в котором в качестве переменной использовалась температура, а величины f задавались с шагом 0.1 в интервале от 0 до 0.5 (фиг. 10). Стоит оговориться, что данная диаграмма должна интерпретироваться с осторожностью, поскольку модельный расчет подразумевает температуру процесса взаимодействия флюид-порода, а природные данные охарактеризованы температурой выхода гидротерм. Диаграмма, приведенная на фиг. 10, имеет определенные топологические черты. Линия SW – изотопное равновесие серы сероводорода с морским сульфатом, и все точки, лежащие левее и выше нее, показывают эффект закрытой системы. Точки, лежащие ниже этой линии, представляют открытую систему, в которой с удалением от линии изотопного равновесия серы сероводорода с сульфатом морской воды возрастает степень извлечения серы из магматических пород. Обобщение имеющихся в литературе данных показывает, что почти для всех гидротермальных полей наблюдается эффект закрытой системы, и только отдельные проявления, в основном, гидротермы ВТП, попадают в область открытой системы с повышенным коэффициентом извлечения серы из пород океанского дна. Для полей САХ характерен вертикальный тренд (вертикальная стрелка на фиг. 10), который может быть интерпретирован как изотермическое смешение флюида с океанской водой в субповерхностной зоне. Часть точек полей ВТП тоже лежат вдоль вертикального тренда, но для этого района характерен и другой тренд – горизонтальный (горизонтальная стрелка на фиг. 10), который можно отнести к процессу кондуктивного остывания флюида. Интересно, что для хребта Хуан-де-Фука проявлены оба процесса. Как и в предыдущем расчете, ни для одного из полей не выявляется высоких степеней восстановления сульфата. За небольшим исключением, величины f не превышают 0.3, а имеющиеся данные для полей САХ соответствуют величинам f не выше 0.2.
Фиг. 10.
Величины δ34S(H2S) и температура разгрузки гидротермальных растворов на океанских гидротермальных полях: ВТП – поля Восточно-Тихоокеанского поднятия (по Woodruff, Shanks, 1988, Shanks et al., 1995, Rouxel et al., 2008); ХдФ – хребет Хуан-де-Фука (по Shanks et al., 1995); САХ – поля Срединно-Атлантического хребта (Логачев, MARK и SMAR, по Shanks et al., 1995; Peters et al., 2009). Результаты модельного расчета величин δ34S(H2S): сплошные кривые – расчет без учета серы, извлекаемой из пород, пунктирные линии – с учетом серы, выщелачиваемой из пород (k = 0.1). Расчет проведен для пяти фиксированных величин f (доли восстановленного сульфата во флюиде) от 0.1 до 0.5, жирная линия SW – изотопный состав серы H2S в равновесии с сульфатом морской воды. Пунктирные линии ниже линии SW – расчет величин δ34S(H2S) флюида, который проходит стадию восстановления сульфата и выщелачивания серы из пород в открытой системе (показано 4 варианта расчета, для k = 0.2, 0.4, 0.6 и 0.8). Стрелки – пояснения в тексте.

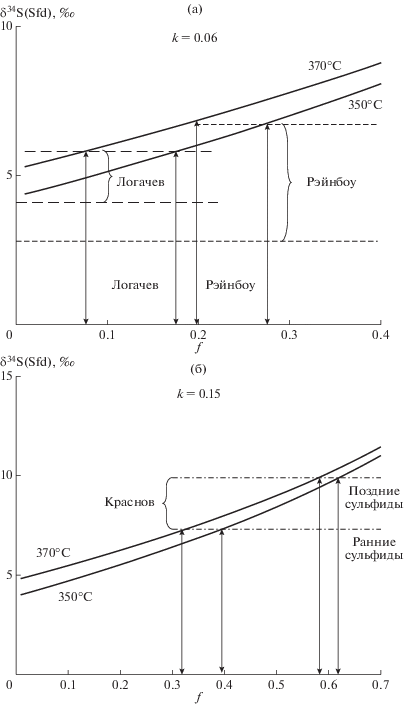
Данные по изотопному составу серы гидротермальных сульфидов тоже можно сопоставить с модельными расчетами, например, чтобы найти степень восстановления сульфата. Наши данные, полученные для полей САХ, показывают, что оценки величины f могут варьировать в пределах каждого конкретного поля (фиг. 11). В соответствии с известными данными о температуре выхода флюида на этих полях (350–370°С), а также содержании серы и Fe(II) в толеитовых базальтах и перидотитах (Douville et al., 2002; Shanks, 2001; Alt et al., 2007; Zhao, Zheng, 2003), был проведен расчет эволюции составов гидротермальных сульфидов по уравнениям (5–10). Для поля Краснов, приуроченного к базальтам, использован параметр k, равный 0.15, а для полей Логачев и Рэйнбоу, расположенных на перидотитовом основании, этот параметр составил 0.06. Оценка величин f составила интервалы 0.08–0.18 и 0.2–0.38 для сульфидов поздних генераций полей Логачев и Рэйнбоу. Для ранних сульфидов оценка оказалась близка к 0, для поля Рэйнбоу нижняя граница уходит за пределы расчета при указанных температурах (фиг. 11а). Для поля Краснов величины f в ранних и поздних сульфидах составили 0.32–0.40 и 0.58–0.62 соответственно (фиг. 11б).
Фиг. 11.
Расчет величин f для сульфидов (пирит + сфалерит) полей Логачев, Рэйнбоу (а) и Краснов (б). Расчет проведен для взаимодействия флюида с породами при 350 и 370°С, величины k были заданы 0.06 для полей, приуроченных к перидотитам, и 0.15 для поля Краснов, расположенного на базальтовом основании.
ЗАКЛЮЧЕНИЕ
Дифференцированный подход к изотопному анализу серы в единичных зернах сульфидов с одновременным анализом их временной позиции в образце позволил установить, что последовательность формирования минеральных зерен является одним из факторов, контролирующих величину δ34S гидротермальных сульфидов. На примере сульфидных руд гидротермальных полей Логачев, Краснов и Рэйнбоу удалось показать, что ранние минеральные зерна в пределах одного и того же образца имеют более низкие величины δ34S, чем минералы поздних генераций. При этом минеральный состав сульфидов практически не имеет значения для формирования их изотопного состава серы.
Полученные данные указывают на то, что основным источником серы сульфидов является морской сульфат, восстановленный в процессе TSR, протекающем в зонах взаимодействия океанской воды с нагретыми породами океанского дна, причем процесс восстановления протекает в системе, закрытой относительно флюида. Эффект закрытой системы приводит к появлению аномально высоких величин δ34S в сульфидах (до +15 ‰ и выше) и гидротермальных сульфатах, имеющих величины δ34S выше, чем в сульфате океанской воды.
Предложенная модель предсказывает появление аномальных изотопных характеристик серы в океанских гидротермах и объясняет неравновесные соотношения величин δ34S в сосуществующих сульфидных минералах, а также зависимость изотопных параметров от последовательности генерации сульфидов. Модель объясняет возникновение неравновесных соотношений величин δ34S(H2S) гидротермальных флюидов с сульфатом морской воды при температуре разгрузки гидротерм. Примеры верификации модели на собственных и обобщенных литературных данных показывают, что изотопные характеристики серы гидротермальных сульфидов отражают, прежде всего, условия протекания процесса TSR, т.е. дают представление об условиях взаимодействия флюид–порода на восходящей ветви конвективных ячеек.
Кроме реализации условий закрытой системы во время протекания TSR, в модели используется постулат о связи прогресса TSR со степенью извлечения серы из магматических пород, поскольку оба эти процесса протекают одновременно при взаимодействии флюид–порода. Такой подход позволяет описать закономерности, проявленные в разных океанских гидротермах, в том числе, приуроченных к участкам океанской коры с разной скоростью спрединга (фиг. 12). Численные оценки и сравнение модели с наблюдаемыми изотопными характеристиками серы для большого числа гидротермальных полей показывают, что основным источником сульфидной серы в гидротермальном флюиде является восстановленный сульфат морской воды, а доля серы, извлеченной из пород, не превышает 15%.
Фиг. 12.
Изотопный состав серы сульфидов некоторых гидротермальных полей и скорость спрединга участков коры, к которым эти поля приурочены. Залитые значки – данные настоящей работы, остальные – литературные данные (Blum, Pushelt, 1991; Bluth, Ohmoto, 1988; Chiba et al., 1998; Lein et al., 2001; Ono et al., 2007; Peters et al., 2009; Rouxel et al., 2004; Rouxel et al., 2008; Shanks, Seyfried, 1987; Woodruff, Shanks, 1988, Zheng et al., 2016; Zierenberg et al., 1984). Скорость спрединга взята из интернет-ресурса InterRidge Vents Database (https://vents-data.interridge.org/).

Из наших построений следует, что об условиях протекания TSR можно судить и просто по характеру поведения величин δ34S в сульфидах построек. В условиях, когда TSR протекает в открытой относительно флюида системе, величины δ34S сульфидов должны изменяться в узком диапазоне значений, которые соответствуют равновесному фракционированию серы при восстановлении морского сульфата плюс небольшая добавка серы, извлекаемой из магматических пород. При более высокой температуре протекания TSR величины δ34S должны быть ниже, чем при восстановлении сульфата при более низкой температуре. Поскольку оба источника (морской сульфат и породы океанского дна) гомогенны в отношении изотопного состава серы, предпосылок для возникновения значительных изотопных вариаций серы во флюид-доминирующей системе не возникает. В областях с низкими скоростями спрединга реализация закрытой системы в зоне протекания TSR обеспечивает широкие вариации величин δ34S во флюиде и в отлагаемых сульфидах. Возрастание разброса величин δ34S в сульфидах гидротермальных полей с возрастанием скорости спрединга отмечалось ранее (Zheng et al., 2016), но это объяснялось авторами вероятным присутствием дополнительных источников серы в гидротермах, приуроченных к зонам коры с низкой скоростью спрединга, например, серы, поступающей из серпентинизированных ультраосновных пород. Действительно, в серпентинизированных перидотитах может содержаться до 2000–8000 ppm сульфидной серы с варьирующим изотопным составом (Delacour et al., 2008). Однако, по нашему мнению, более вероятной причиной является то, что с уменьшением скорости возрастает вероятность проявления эффекта закрытой системы. С этой точки зрения находят объяснение другие особенности поведения изотопов серы в субмаринных гидротермах – широкие вариации и появление “аномально” высоких значений δ34S в сульфидах и сульфатах, что проявлено не только для полей Логачев, Краснов и Рэйнбу, но и для других гидротермальных полей Мирового океана.
Список литературы
Богданов Ю.А., Бортников Н.С., Викентьев И.В., Гурвич Е.Г., Сагалевич А.М. Новый тип современных минерал-формирующих систем: черные курильщики гидротермального поля на 14°45′ с.ш., Срединно-Атлантический хребет // Геология руд. месторождений. 1997. Т. 39. № 1. С. 68–90.
Бортников Н.С., Викентьев И.В. Современное сульфидное полиметаллическое минералообразование в Мировом океане // Геология руд. месторождений. 2005. № 1. С. 16–50.
Бортников Н.С., Волков А.В., Галямов А.Л., Викентьев И.В., Аристов В.В., Лаломов А.В., Мурашов К.Ю. Минеральные ресурсы стратегических металлов в России: состояние и перспективы развития // Геология руд. месторождений. 2016. Т. 58. № 2. С. 97–119.
Гричук Д.В. Термодинамические модели субмаринных гидротермальных систем. 2000, М: Научный мир. 304 с.
Дубинина Е.О., Бортников Н.С. Новая модель поведения изотопов серы в современных субмаринных гидротермальных системах // ДАН. 2019. Т. 486. № 5. С. 73–77.
Дубинина Е.О., Ставрова О.О., Бортников Н.С. Аномально высокие величины δ34S в постройках Срединно-Атлантического хребта: эффект закрытой системы в зоне генерации сульфидов // ДАН. 2018. Т. 483. № 6. С. 650–653.
Еремин Н.И., Дергачев А.Л., Сергеева Н.Е., Позднякова Н.В. Типы колчеданных месторождений вулканической ассоциации // Геология руд. месторождений. 2000. № 2. С. 177–190.
Смирнов В.И. Генезис эндогенных рудных месторождений. М.: Недра, 1968. 719 с.
Aggarwal P.K., Nessbit B.E. Geology and geochemistry of the Chu Chua massive sulfide deposit, British Columbia // Econ. Geol. 1984. V. 79. P. 815–825.
Alt J. C., Anderson T. F., Bonnell L. The geochemistry of sulfur in a 1.3 km section of hydrothermally altered oceanic crust, DSDP Hole 504B // Geochim. Cosmochim. Acta. 1989. V. 53. P. 1011–1023.
Alt J.C., Shanks III W.C., Bach W., Paulick H., Garrido C.J., Beaudoin G. Hydrothermal alteration and microbial sulfate reduction in peridotite and gabbro exposed by detachment faulting at the Mid-Atlantic Ridge, 15-20oN (ODP Leg 209): A sulfur and oxygen isotope study // Geochemistry, Geophysics, Geosystems. 2007. V. 8. Q08002. https://doi.org/10.1029/2007GC001617
Andreani M., Escartin J., Delacour A., Ildefonse B., Godard M., Dyment J., Fallick A.E, Fouquet Y. Tectonic structure, lithology, and hydrothermal signature of the Rainbow massif (Mid-Atlantic Ridge 36°, 14′ N) // Geochemistry, Geophysics, Geosystems. 2014. V. 15. P. 3543–3571. https://doi.org/10.1002/2014GC005269
Bischoff J.L., Seyfried W.E. Hydrothermal chemistry of seawater from 25° to 350°. Am. J. Sci. 1978. V. 278, June. P. 838–860.
Blum N., Puchelt H. Sedimentary-hosted polymetallic massive sulfide deposits of the Kerbit and Shaban Deeps, Red Sea // Miner. Deposita. 1991. V. 26. P. 217–227.
Bluth G.J., Ohmoto H. Sulfide-sulfate chimneys on the East Pacific Rise, 11° and 13°N latitude. Part II: Sulfur isotopes // Can. Mineral. 1988. V. 26. P. 505–515.
Bogdanov Yu.A., Bortnikov N.S., Vikent’ev I.V., Gurvich E.G., Sagalevich A.M. A new type of modern mineral-forming system: black smokers of the hydrothermal field at 14°45′ N latitude, mid-atlantic ridge // Geology of Ore Deposits. 1997. V. 39. № 1. P. 58–78.
Brueckner S.M., Piercey S.J., Layne G.D., Piercey G., Sylvester P.J. Variations of sulfur isotope signatures in sulfides from the metamorphosed Ming Cu(-Au) volcanogenic massive sulfide deposit, Newfoundland Appalachians, Canada // Mineralium Deposita. 2015. V. 50. P. 619–640.
Charlou J.L., Donval J.P., Fouquet Y., Jean-Baptiste P., Holm N. Geochemistry of high H2 and CH4 vent fluids issuing from ultramafic rocks at the Rainbow hydrothermal field (36°14′ N, MAR). Chem. Geol. 2002. V. 191. P. 345–359.
Chaussidon M., Albarède F., Sheppard S.M.F. Sulphur isotope variations in the mantle from ion microprobe analyses of micro-sulphide inclusions // Earth Planet. Sci. Lett. 1989. V. 92. P. 144–156.
Cherkashov G., Bel’tenev V., Ivanov V., Lazareva L., Samovarov M., Shilov V., Stepanova T., Glasby G.P., Kuznetsov V. Two new hydrothermal fields at the Mid-Atlantic Ridge // Marine Georesources & Geotechnology. 2008. V. 26. № 4. P. 308–316.
Chiba H., Uchiyama N., Teagle D.A.H. Stable isotope study of anhydrite and sulfide minerals at the TAG Hydrothermal Mound, Mid-Atlantic ridge, 26°N // Proceedings of the Ocean Drilling Program, Scientific Results. 1998. V. 158. P. 85–90.
Coplen T. Guidelines and recommended terms for expression of stable-isotope-ratio and gas-ratio measurement results // Rapid Communications in Mass Spectrometry. 2011. V. 25. P. 2538–2560.
de Ronde C., Faure K., Bray C.M., Chappell D.A., Wright I.C. Hydrothermal fluids associated with seafloor mineralization at two southern Kermadec arc volcanoes, off-shore New Zealand // Miner. Deposita. 2003. V. 38. P. 217–233.
Delacour A., Früh-Green G.L., Bernasconi S.M., Kelley D.S. Sulfur in peridotites and gabbros at Lost City (30°N, MAR): implications for hydrothermal alteration and microbial activity during serpentinization // Geochim. Cosmochim. Acta. 2008. V. 72. P. 5090–5110.
Dietzel M., Jianwu T., Leis A., Köhler S.J. Oxygen isotopic fractionation during inorganic calcite precipitation — effects of temperature, precipitation rate and pH // Chem. Geol. 2009. V. 268. № 1–2. P. 107–115.
Douville E, Charlou J.L., Oelkers E.H., Bienvenu P., Jove Colon C.F., Donval J.P., Fouquet Y., Prieur D., Appriou P. The Rainbow Vent fluids (36 degrees 14′ N, MAR); the influence of ultramafic rocks and phase separation on trace metal content in Mid-Atlantic Ridge hydrothermal fluids // Chem. Geol. 2002. V. 184 (1–2). P. 37–48.
Edmonds H.N., German C.R. Particle geochemistry in the Rainbow hydrothermal plume, Mid-Atlantic Ridge // Geochim. Cosmochim. Acta. 2004. V. 68. № 4. P. 759–772.
Fisher A.T. Permeability within basaltic oceanic crust // Reviews of Geophysics. 1998. V. 36. P. 143–182.
Franklin J.M., Gibson H.L., Jonasson I.R., Galley A.G. Volcanogenic massive sulfide deposits, in Hedenquist. J.W., Thompson, J.F.H., Goldfarb, R.J., Richards, J.P. (Eds.), Economic Geology 100th anniversary volume, 1905–2005: Littleton, Colo., Society of Economic Geologists. 2005. P. 523–560.
Franklin J.M., Lydon J.W., Sangster D.M. Volcanic-associated massive sulfide deposits // Economic Geology 75th Anniversary Volume. 1981. P. 485−627.
Gabitov R.I., Watson E.B., Sadekov A. Oxygen isotope fractionation between calcite and fluid as a function of growth rate and temperature: An in situ study // Chem. Geol. 2012. V. 306–307. P. 92–102.
Gemmell J.B., Sharpe R. Detailed sulfur-isotope investigation of the TAG hydrothermal mound and stockwork zone // Proc. Ocean Drill. Program Sci. Results. 1998. V. 158. P. 71–84.
Green T., Vokes F.M. Sea-floor sulfides at the Hoydal volcanogenic deposit, Central Norwegian Caledonides // Econ. Geol. 1990. V. 85. P. 344–359.
Growe D.E., Nelson S.W., Brown P.E., Shanks W.C., Valley J.W. Geology and geochemistry of volcanogenic massive sulfide deposits and related igneous rocks, Prince William Sound, South-Central Alaska // Econ. Geol. 1992. V. 87. P. 1722–1746.
Hannington M.D., Galley A.G., Herzig P.M., Petersen S. Comparison of the TAG mound and stockwork complex with Cyprus-type massive sulfide deposits // Proceed. of the Ocean Drilling Program. Scientific results. 1998. V. 158. P. 389–415.
Hannington M.D., de Ronde C.E.J., Petersen S. Sea-floor tectonics and submarine hydrothermal systems // Economic Geology 100th Anniversary Volume 1905–2005. Hedenquist, J.W., Thompson, J.F.H., Goldfarb, R.J., Richards, J.P. (Eds.). Littleton, Colo., Society of Economic Geologists, 2005. P. 111–141.
Haymon R.M., Koski R.A., Abrams M.J. Hydrothermal discharge zones beneath massive sulfide deposits mapped in the Oman ophiolites // Geology. 1989. V. 17. P. 531–535.
Herzig P.M., Petersen S., Hannington M.D. Geochemistry and sulfur isotopic composition of the TAG hydrothermal mound, Mid-Atlantic Ridge, 26°N. Proc.Ocean Drill. Program Sci. Results. 1998. V. 158. P. 47–70.
Huston D.L., Pehrsson S., Eglington B.M., Zaw K. The geology and metallogeny of volcanic-hosted massive sulfide deposits: Variations through geologic time and with tectonic setting // Econ. Geol. 2010. V. 105. № 3. P. 571–591.
Huston D.L., Relvas J.M., Gemmell J.B. & Drieberg S. The role of granites in volcanic-hosted massive sulphide ore-forming systems: an assessment of magmatic–hydrothermal contributions // Miner. Deposita. 2011. V. 46. № 5–6. P. 473–507.
InterRidge Vents Database ver.3.4. https://vents-data.interridge.org/
Janecky D.R., Shanks W.C. Computational modeling of chemical and sulfur isotopic reaction processes in seafloor hydrothermal systems: chimneys, massive sulfides, and subjacent alteration zones // Can. Mineral. 1988. V. 26. P. 805–825.
Kadko D., Koski R., Tatsumoto M., Bouse R. An estimate of hydrothermal fluid residence times and vent chimney growth rates based on 210Pb/Pb ratios and mineralogic studies of sulfides dredged from the Juan de Fuca Ridge // Earth and Planetary Science Letters. 1985/86. V. 76. P. 35–44.
Kim S.T., O’Neil J.R. Equilibrium and nonequilibrium oxygen isotope effects in synthetic carbonates // Geochimica et Cosmochimica Acta. 1997. V. 61. № 16. P. 3461–3475.
Kusakabe M., Mayeda S., Nakamura E. S, O and Sr isotope systematics of active vent materials from the Mariana backarc basin spreading axis at 18°N // Earth Planet. Sci. Lett. 1990. V.100. P. 275–282.
Le Maitre R.W. The chemical variability of some common igneous rocks // J. Petrol. 1976. V. 17. P. 589–637.
Lein A.Y., Ulyanova N.V., Ulyanov A.A., Cherkashev G.A., Stepanova T.V. Mineralogy and geochemistry of sulfide ores in ocean-floor hydrothermal fields associated with serpentinite protrusions // Russian J. of Earth Sciences. 2001. V. 3. P. 371–393.
Lindgren W. Mineral deposits. New York: McGrawHill Book Company, 1913. 883 p.
Marini L. Sulfur isotopes in magmatic-hydrothermal systems, melts, and magmas // Rev. Mineral. Geochem. 2011. V. 73. P. 423–492.
Marumo K., Hattori K.H. Seafloor hydrothermal clay alteration at Jade in the back-arc Okinawa Trough: mineralogy, geochemistry and isotope characteristics // Geochim. Cosmochim. Acta. 1999. V. 63. P. 2785–2804.
McDonald M.J., Piercey S.J., Layne G.D., Pigage L.C., Piercey G. Mineral assemblages, textures and in situ sulphur isotope geochemistry of sulphide mineralization from the cyprus-type ice volcanogenic massive sulphide (VMS) Deposit, Yukon, Canada // Minerals. 2018. V. 8. P. 501. https://doi.org/10.3390/min8110501
Neathery T.L., Hollister V.F. Volcanogenic sulfide deposits in the Southernmost Appalachians // Econ. Geol. 1984. V. 79. P. 1540–1560.
Ocker C. Variability of sulfur isotopes of hydrothermal sulfides and sulfates of slow and intermediate spreading ridges. Master thesis, Univ. of Münster, 2008.
Oftedahl C. A theory of exhalative-sedimentary ores: Geologiska Föreningens I Stockholm Förhandlingar. 1958. V. 80. № 492. P. 1–19.
Ohmoto H., Rye R.O. Isotope of sulfur and carbon // Geochemistry of Hydrothermal deposits. Barnes H.L. (Eds.). John Wiley & Sons, 1979. P. 509–567
Ohmoto H., Lasaga A.C. Kinetics of reactions between aqueous sulfates and sulfides in hydrothermal systems // Geochim. Cosmochim. Acta. 1982. V. 46. P. 1727–1745.
Ohmoto H., Mizukami M., Drummond S.E., Eldridge C.S., Pisutha-Arnond V., Lenaugh T.C. Chemical processes in Kuroko formation // Econ. Geol. Monograph 5. 1983. P. 570−604.
Ono S., Shanks III W.S., Rouxel O.R., Rumble D. S-33 constraints on the seawater sulfate contribution in modern seafloor hydrothermal vent sulfides // Geochim. Cosmochim. Acta. 2007. V. 71. P. 1170–1182.
Peters M., Strauss H., Farquhar J., Ockert C., Eikmann B., Jost C.L. Sulfur cycling at the Mid-Atlantic Ridge: A multiple sulfur isotope approach // Chem. Geology. 2009. https://doi.org/10.1016/j.chemgeo.2009.09.016
Petersen S., Kuhn K., Kuhn T., Augustin N., Hékinian R., Franz L., Borowski C. The geological setting of the ultramafic-hosted Logatchev hydrothermal field (14°45′ N, Mid-Atlantic Ridge) and its influence on massive sulfide formation // Lithos. 2009. V. 112. P. 40–56.
Rees C.E., Jenkins W.J., Monster J. The sulphur isotope geochemistry of ocean water sulphate // Geochim. Cosmochim. Acta. 1978. V. 42. P. 377–382.
Rouxel O., Shanks W.C.I.I.I., Bach W., Edwards K.J. Integrated Fe- and S-isotope study of seafloor hydrothermal vents at East Pacific Rise 9°–10°N // Chem. Geol. 2008. V. 252. P. 214–227.
Rouxel O., Fouquet Y., Ludden J.N. Copper isotope systematics of the Lucky Strike, Rainbow and Logatchev seafloor hydrothermal fields on the Mid Atlantic Ridge // Econ. Geol. 2004. V. 99. P. 585–600.
Sakai H., Des Marais D.J., Ueda A, Moore J.G. Concentrations and isotope ratios of carbon, nitrogen, and sulfur in ocean-floor basalts // Geochim. Cosmochim. Acta. 1984. V. 48. P. 2433–2442.
Sangster D.F. Relative sulphur isotope abundances of ancient seas and strata-bound sulphide deposits // Proceedings of the the Geological Association of Canada. 1968. V. 19. P. 79−91.
Schmidt K., Koschinsky A., Garbe-Schönberg D., de Carvalho L.M., Seifert R. Geochemistry of hydrothermal fluids from the ultramafic-hosted Logatchev hydrothermal field, 15°N on the Mid-Atlantic Ridge: Temporal and spatial investigation // Chem. Geol. 2007. V. 242. № 1–2. P. 1–21.
Seal II R.R. Sulfur isotope geochemistry of sulfide minerals // Rev. Mineral. Geochem. 2006. V. 61. P. 633–677.
Seal R.R. II, Rye R.O., Alpers C.N. Stable isotope systematics of sulfate minerals // Rev. Mineral. Geochem. 2000. V. 40. P. 541–602.
Seyfried W.E. Jr., Bischoff J.L. Experimental seawater-basalt interaction at 300°C, 500 bars, chemical exchange, secondary mineral formation and implications for the transport of heavy metals // Geochim. Cosmochim. Acta. 1981. V. 45. P. 135–147.
Shanks W.C. III. Stable isotopes in seafloor hydrothermal systems: vent fluids, hydrothermal deposits, hydrothermal alteration, and microbial processes. In: Valley, J.W., Cole, D.R. (Eds.), Stable Isotope Geochemistry // Rev. Mineral. Mineralogical Society of America, Washington, D.C. 2001. V. 43. P. 469–525.
Shanks W.C. III, Niemitz J. Sulfur isotope studies of hydrothermal anhydrite and pyrite, Deep Sea Drilling Project Leg 64, Guaymas Basin, Gulf of California. Initial Rep. Deep Sea Drill. Proj. 1982. V. 64. P. 1137–1142.
Shanks W.C. III., Seyfried W.E. Jr. Stable isotope studies of vent fluids and chimney minerals, Southern Juan de Fuca Ridge’ sodium metasomatism and seawater sulfate reduction // J. Geophys. Res. 1987. V. 92. № 11. P. 387–399.
Shanks W.C. III, Bischoff J.L., Rosenbauer R.J. Seawater sulfate reduction and sulfur isotope fractionation in basaltic systems: interaction of seawater with fayalite and magnetite at 200–350°C // Geochim. Cosmochim. Acta. 1981. V. 45. P. 1977–1995.
Shanks W.C. III, Böhlke J.K., Seal R.R.II. Stable isotopes in mid-ocean ridge hydrothermal systems: interactions between fluids, minerals, and organisms. In: Seafloor Hydrothermal Systems: Physical, Chemical, Biological, and Geological Interactions. Geophysical Monograph 91. 1995. Amer. Geophys. Union (Eds.: S.E. Humphris, R.A. Zierenberg, L.S. Mullineaux, R.E. Thomson).https://doi.org/10.1029/GM091p0194
Shanks W.C. Pat III, Thurston R. Volcanogenic massive sulfide occurrence model // U.S. Geological Survey Scientific Investigations Report 2010–5070–C. 2012. 345 p.
Thode H.G., Monster J., Dunford H.B. Sulfur isotope geochemistry // Geochim. Cosmochim. Acta. 1961. V. 25. P. 159–174.
Tivey M.K. Generation of seafloor hydrothermal vent fluids and associated mineral deposits // Oceanography. 2007. V. 20. № 150. P. 50–65.
Woodruff L.G., Shanks W.C. III Sulfur isotope study of chimney minerals and vent fluids from 21°N, East Pacific Rise: Hydrothermal sulfur sources and disequilibrium sulfate reduction // J. Geophys. Res. 1988. V. 93. P. 4562–4572.
Zeng Zh., Maa Y., Chen S., Selby D., Wang X., Yin X. Sulfur and lead isotopic compositions of massive sulfides from deep-sea hydrothermal systems: Implications for ore genesis and fluid circulation // Ore Geol. Rev. 2016. V. 87. P. 155–171.
Zhao Z.F., Zheng Y.F. Calculation of oxygen isotope fractionation in magmatic rocks // Chem. Geol. 2003. V. 193. P. 59–80.
Zierenberg R.A., Shanks W.C., Bischoff J.L. Massive sulfide deposits at 21°, East Pacific Rise: chemical composition, stable isotopes, and phase equilibria // Bulletin of the Geological Society of America. 1984. V. 95. P. 922–929.
Дополнительные материалы отсутствуют.
Инструменты
Геология рудных месторождений