

Известия РАН. Серия биологическая, 2022, № 1, стр. 70-81
Метагеномика – новое направление в экологии
М. В. Вечерский 1, *, М. В. Семенов 2, А. А. Лисенкова 3, 4, А. А. Степаньков 1
1 Институт проблем экологии и эволюции им А.Н. Северцова РАН
119071 Москва, Ленинский проспект, 33, Россия
2 Почвенный институт им. В.В. Докучаева
119017 Москва, Пыжевский пер., 7, Россия
3 Институт общей генетики им. Н.И. Вавилова РАН
117971 Москва, ул. Губкина, 3, Россия
4 Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, стр. 12, Россия
* E-mail: vecherskomy@mail.ru
Поступила в редакцию 19.03.2020
После доработки 24.07.2020
Принята к публикации 24.07.2020
- EDN: LZKBTH
- DOI: 10.31857/S1026347022010152
Аннотация
В работе обсуждаются перспективы применения метагеномных технологий в экологических исследованиях. Описаны успехи в изучении таксономического состава сообществ водных и сухопутных биотопов. Приводятся примеры выявления трофических и форических связей в экосистемах. Подробно показаны возможности метагеномики в исследовании сообществ, образованных прокариотами, и таких трудных для изучения субстратов, как почва и кишечник животных. Описана роль реликтовой ДНК в метагеноме и возможности изучения сообществ древних организмов. Особое внимание уделено критике метагеномных технологий, связанной с низкой воспроизводимостью результатов. Приводятся типичные методические ошибки в сфере биоинформатики, которые приводят к грубому искажению получаемых данных.
Развитие технологии секвенирования ДНК позволило перейти от изучения генома конкретного организма к изучению метагенома – всей совокупности геномов сообщества. Метагеном представляет собой набор геномов всех организмов, находящихся в образце среды, а также внеклеточную ДНК. В качестве объекта исследования могут выступать метагеномы почвы, водоемов, физиологического материала (содержимого кишечника, гноя, зубных бляшек), промышленных ферментеров (Borbón-García et al., 2017; Chai et al., 2018; Wilson et al., 2019). Существует два подхода к определению метагеномики. В строгом значении под метагеномикой подразумевается анализ совокупности геномов сообщества организмов (Riesenfeld et al., 2004). При более широкой трактовке термина объектом изучения метагеномики становятся не только геномы, но и отдельные гены в составе геномов. При анализе отдельных генов наибольшее значение приобретают филогенетически значимые участки, использующиеся для таксономической идентификации членов сообщества (Ranjan et al., 2016). Например, для получения таксономического состава прокариот исследуют совокупность генов 16S рРНК, грибов – последовательности ITS, животных – 18S рРНК или фрагмент митохондриального гена COI. Этот метод называется метабаркодингом (по аналогии с баркодингом для отдельных видов). Реже изучают разнообразие функциональных генов, например: целлюлаз, нитрогеназ, цитохромов, факторов антибиотикорезистентности (Ngara, Zhang, 2018). Данные, полученные в результате метагеномного анализа, носят преимущественно качественный характер, однако при правильно поставленном эксперименте дают возможность делать и осторожные количественные выводы. Метагеномные исследования позволяют получать информацию о таксономическом составе, трофической структуре и даже форических отношениях в сообществе. Во всех случаях исследователю не требуется наблюдать и идентифицировать отдельных представителей сообщества. Более того, в образце даже не обязательно должны быть представлены живые организмы. Достаточно наличия их фрагментов или внеклеточной ДНК.
Методически метагеномные технологии делятся на две большие группы – ампликонные и “shot-gun” (Ma et al., 2014). В первом случае метагеном образца является лишь матрицей для амплификации одного из генов путем полимеразной цепной реакции (ПЦР). Само секвенирование осуществляется на полученных продуктах ПЦР, а не на изначальной метагеномной ДНК-матрице. Для амплификации требуется использование праймеров, фланкирующих целевой участок нуклеотидной последовательности. Несовершенство существующих праймеров приводит к неполному анализу сообщества, а ПЦР формирует большое количество артефактов. Однако этот метод относительно недорогой и сравнительно быстрый. Напротив, при “shot-gun” секвенировании анализируется весь метагеном. В этом случае не происходит ошибок, связанных с выбором праймеров и несовершенством ПЦР. Однако это метод более трудоемок и дорог. Кроме того, если объект характеризуется очень высоким генетическим разнообразием (например, ДНК почвы), то “shot-gun” секвенирование сталкивается с проблемой полноты прочтения метагенома.
Метагеномный анализ при изучении экологии сообществ. Одной из важных задач экологии сообществ является изучение трофических взаимодействий между видами. При невозможности прямого наблюдения за процессом питания приходится опираться на содержимое пищеварительной системы. Однако сохранность останков не всегда позволяет определить жертву по морфологическим признакам. При этом генетические маркеры в содержимом пищеварительного тракта сохраняются достаточно хорошо (Pompanon et al., 2012). Например, было показано (Yoon et al., 2017), что в рацион крупнейшей южнополярной рыбы – антарктического клыкача (Dissostichus mawsoni) – входят минимум 16 видов жертв из класса головоногих. Кроме того, у него в желудке присутствуют 4 вида многоклеточных паразитов, относящихся к разным типам (рис. 1).
Рис. 1.
Метагеномный анализ содержимого желудка антарктического клыкача, Dissostichus mawsoni (по Yoon et al., 2017).

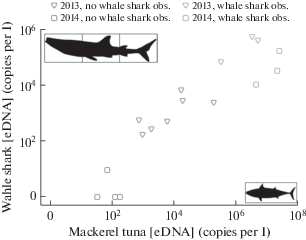
Анализ экзогенной ДНК позволяет проводить количественную оценку состава сообществ в водной среде. Из животного организма может выделяться экзогенная ДНК в составе слущивающихся покровов, эпителиальных слизей и кала. Экзогенная ДНК деградирует во внешней среде, но при достаточной численности организма ее количество может поддерживаться на технически определяемом уровне. В метагеноме образцов морской воды удалось установить количество копий митохондриальной ДНК китовой акулы и сопоставить его с численностью китовой акулы (Sigsgaard et al., 2016). В тех же образцах определяли количество копий митохондриальной ДНК малого восточного тунца, икрой которого питается китовая акула. Оказалось, что “копийность” мтДНК (число копий в образце) хорошо отражает реальное соотношение численностей соответствующих рыб (рис. 2).
Рис. 2.
Количество экзогенной ДНК китовой акулы и малого восточного тунца в пробах морской воды (по Sigsgaard et al., 2016).
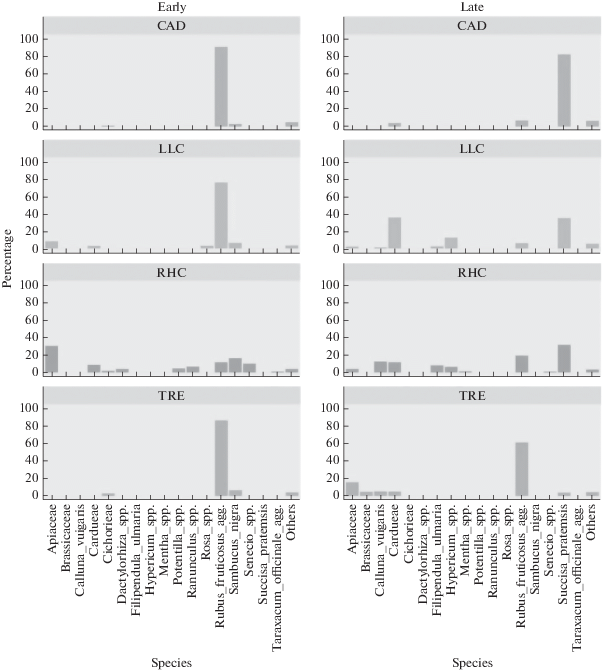
Метагеномные методики применяются в отношении не только животных, но и растений. Стало возможным изучение состава опыляемых растений по метагеному пыльцы на покровах насекомого-опылителя. Так, было показано (Lucas et al., 2018), что муха-журчалка опыляет цветки семнадцати видов растений (рис. 3). Были установлены ее пищевые предпочтения для разных биотопов в разные сезоны года.
Рис. 3.
Таксономический состав пыльцы на покровах мухи-журчалки в начале и конце лета в разных биотопах на западе Гренландии (по Lucas et al., 2018).
Метагеномные исследования также позволяют более точно установить состав симбиотических ассоциаций, в том числе хорошо изученных лишайников. Оказалось, что значительная часть биомассы аскомицетовых лишайников представлена, помимо мицелиального и бактериального компонентов, дрожжевым компонентом (Spribille et al., 2016). Пока не ясно, какую функцию выполняют эти базидиомицетовые дрожжи в составе лишайника, но очевидно, что их клетки составляют значительную долю этого организма.
Таким образом, анализ рибосомальных генов позволяет получать информацию о таксономическом составе и разнообразии изучаемого сообщества. Однако эти гены составляют менее десятой доли процента от размера метагенома. Оставшаяся часть также может быть высокоинформативной. Наиболее интересны в этом отношении работы по изучению функциональных генов, вовлеченных в различные биохимические пути. Примером такой работы может служить изучение биохимических путей синтеза бутирата кишечным микробиомом. Известно, что бутират является ключевым питательным веществом для клеток толстого кишечника (Vital et al., 2017). Дефицит бутирата приводит к деградации толстого кишечника, нарушению водно-электролитного баланса и повышению риска развития рака. Поэтому активно проводится изучение изменений в микробиоме, вызывающих дефицит бутирата. Однако только по таксономическому составу микробиома практически невозможно предсказать фактический выход бутирата. Более информативна в этом отношении количественная оценка встречаемости функциональных генов, вовлеченных в этот биосинтез. Анализ результатов полногеномного секвенирования позволил установить ключевые пути метаболизма бутирата у кишечных бактерий, а также связать отдельные метаболические пути с конкретными таксонами микроорганизмов (Vital et al., 2017).
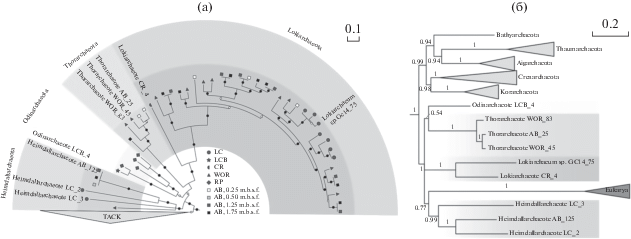
Одно из ключевых общебиологических открытий последних лет было сделано именно с помощью полногеномного “shot-gun” секвенирования метагенома прокариотных сообществ глубоководных гидротермальных источников (Zaremba-Niedzwiedzka et al., 2017). Метагеномный анализ выявил наличие генов, специфичных для эукариот и не встречающихся у прокариот. В частности, были обнаружены специфические варианты белков цитоскелета. Тем не менее, оказалось, что эти гены принадлежат не эукариотам, а эндемичным археям, образующим отдельный филум – надтип Asgard, асгардархеи. Последующий биоинформационный анализ показал, что Asgard археи демонстрируют признаки как прокариот, так и эукариот. Весьма вероятно, что именно представители Asgard осуществили успешный эндосимбиоз с Proteobacteria (будущими митохондриями) и сформировали первый эукариотический организм порядка двух миллиардов лет назад. Из ныне живущих A sgard архей ближе всего к эукариотам находится таксон Heimdallarchaeota (рис. 4). Столь позднее открытие этих уникальных организмов связано с невозможностью культивирования Asgard архей и труднодоступностью их местообитаний. Это блестящее подтверждение теории симбиогенеза смогло произойти только благодаря метагеномике. Однако нельзя утверждать, что теперь подобные работы будут выходить регулярно. Изучение Asgard архей обошлось в астрономическую сумму, значительно превосходящую годовой бюджет рядового российского НИИ. Кроме того, как и любое метагеномное исследование, данная работа оперирует не целыми организмами, а только характеризует набор генов в среде. Поэтому неизбежно возникает критика, связанная с принадлежностью анализируемых генов к конкретным видам. Есть вероятность существования тесных ассоциаций видов, разделяющих некую общую совокупность генов. Нельзя недооценивать и вклад внеклеточной ДНК, в том числе эукариотической, а также деградированной.
Рис. 4.
Филогенетическое дерево современных прокариот Asgard архей с указанием предположительного места отделения ветви эукариот (по Zaremba-Niedzwiedzka et al., 2017).
Метагеномный анализ почв. Одним из сложнейших объектов для экологических исследований является почва (Lombard et al., 2011). С точки зрения метагеномики можно выделить три большие группы особенностей почвы как объекта изучения и возникающих при этом методических проблем: 1) неоднородность почвенного покрова, неравномерное размещение микроорганизмов в почвенных агрегатах и возникающая в связи с этим проблема отбора почвенных образцов; 2) адсорбция клеток на почвенных частицах, ингибирование амплификации присутствующими в почве гуминовыми веществами и проблема экстракции и очистки ДНК; 3) очень высокое разнообразие сообществ, их разный физиологический статус и наличие внеклеточной ДНК.
Большая часть почвенного метагенома представлена прокариотической ДНК. Поэтому почвенная метагеномика чаще всего направлена на изучение таксономической и функционально-генетической структуры почвенных микробных сообществ. Результатом высокопроизводительного секвенирования почвенной ДНК является список таксонов с их относительной представленностью в составе сообщества, с помощью которого можно получить множество показателей биологического состояния почв: 1) таксономическая структура сообщества; 2) количественное соотношение отдельных таксонов и альфа-разнообразие; 3) степень различий между сообществами разных почв (бета-разнообразие); 4) состав и доля индикаторных таксонов в составе сообщества; 5) сложность и характер межвидовых сетей (networks); 6) состав сообществ, связанных с определенным процессом (анализ функциональных генов). Также по результатам анализа функциональных генов можно выявлять корреляции отдельных таксонов или сообществ в целом с отдельными почвенно-экологическими условиями.
Метагеномике принадлежит значительная роль в изучении экологии и распространения малоисследованных таксонов микроорганизмов в почвах. Наиболее яркими примерами могут служить филумы Thaumarchaeota и Verrucomicrobia, представители которых очень трудно культивируются в лаборатории. До сих пор всего три вида Thaumarchaeota выделены в чистую культуру. С помощью РТ-ПЦР и метабаркодинга было показано, что Thaumarchaeota являются самыми распространенными археями в почве и вообще на Земле, и, по всей видимости, являются основными окислителями почвенного аммония (нитрификаторами) (Pester et al., 2011). По сравнению с нитрифицирующими бактериями Thaumarchaeota хорошо адаптированы к низким концентрациям аммония, что дает им преимущество в олиготрофных условиях среды, характерных для почв (Valentine, 2007). Доля Thaumarchaeota составляет, в среднем, 5–15% от всего прокариотного сообщества.
Подобно археям Thaumarchaeota, подавляющее число видов веррукомикробий в настоящее время также не удалось выделить в чистую культуру. Тем не менее, с помощью метабаркодинга было показало, что наряду с бактериями Proteobacteria, Actinobacteria и Acidobacteria, данный филум является одним из основных доминантов в составе прокариотных сообществ, доля которого варьирует в диапазоне от 5 до 15%, а в черноземах – до 25% и выше (Semenov et al., 2018). Вопрос о факторах, ответственных за распределение Verrucomicrobia в почвах до сих пор остается открытым. Долгое время считалось, что Verrucomicrobia – олиготрофы, способные расти в условиях низкой доступности углерода (Rocha et al., 2010; Senechkin et al., 2010; Eilers et al., 2012). Тем не менее, с помощью метабаркодинга было показано, что основная численность Verrucomicrobia приурочена к верхним горизонтам почв, более обеспеченным органическим веществом (Semenov et al., 2018). Метагеномный анализ выявил связь между пространственным распределением Verrucomicrobia и потоком углерода (Fierer et al., 2012), что ставит под сомнение гипотезу относительно их олиготрофности. Кроме того, численность Verrucomicrobia падает при распашке, а также остро реагирует на снижение содержания органического вещества почвы (Navarrete et al., 2015; Semenov et al., 2018).
Помимо таксономического разнообразия, с помощью метагеномного анализа можно получать информацию о функциональных возможностях микробиома почвы. Для этого формируется иерархическая структура, в которой идентифицируемые гены обитающих в почве организмов собираются в функциональные субсистемы по принципу единства выполняемой функции. В состав такого метагенома входят функциональные субсистемы генов, ответственные за метаболизм белков, жиров и углеводов, вирулентность, дыхание, отклик на стресс и т.д. На более низком иерархическом уровне возможен анализ доли генов, ответственных за процессы цикла углерода и азота (например, нитрификацию), синтез или разложение определенных соединений.
Сравнение функциональных профилей метагеномов почв арктических и песчаных пустынь показало, что по соотношению субсистем генов разница между почвами столь контрастных экосистем незначительна. Сообщества песчаных пустынь характеризуются более высокой долей генов, связанных с осморегуляцией и переходом в покоящееся состояние, а также метаболизмом углеводов и ароматических соединений. В метагеноме арктических пустынь, по сравнению с песчаными, выявлено большее количество генов, ассоциированных с круговоротом питательных веществ и катаболизмом соединений, которые связаны с растениями (Fierer et al., 2012). Вместе с тем, метагеномный анализ успешно выявляет различия, возникающие в почвах разных типов землепользования. По сравнению с естественными экосистемами, чернозем под пашней демонстрирует более низкое разнообразие архей и грибов, а также функциональных показателей (Gorbacheva et al., 2018). В ряду “тропический лес – экосистема после вырубки леса – пашня и пастбище” показано, что сельскохозяйственные и пастбищные почвы характеризуются более низкой численностью микроорганизмов, при этом обладая самым высоким таксономическим и функциональным разнообразием, что является важным атрибутом для поддержания функционирования экосистемы после вырубки леса (Mendes et al., 2015). С другой стороны, экосистемное равновесие в нативной лесной экосистеме поддерживается на основе более низкого разнообразия, но более высокой численности микроорганизмов. Еще в одном исследовании показано, что обработка почвы и севооборот существенно сказываются на субсистемах функциональных генов в составе полного метагенома (Souza et al., 2015). В почве при вспашке с оборачиванием пласта оказалось больше микроорганизмов, связанных с разложением растительных остатков и циклами углерода и азота, а также эукариот. При минимальной обработке почвы более высокой численностью характеризуются азотфиксирующие ризобии и археи. Стоит отметить, что в данной работе не удалось аннотировать почти половину метагеномных последовательностей, а на бактерии приходилось основная часть всех прочтений. Суммарный вклад архей и эукариот в аннотированную часть изученного метагенома составил лишь 0.5% (Souza et al., 2015).
Также микробные сообщества почв сильно реагируют на другие агрогенные воздействия, например, внесение удобрений. Использование азотных удобрений увеличивает численность копиотрофов – Proteobacteria и Firmicutes, и уменьшает долю олиготрофов Acidobacteria, Nitrospirae и Chloroflexi. Особенно сильно негативное влияние сказывается на порядок Rhizobiales, включающих в себя множество ассоциативных азотфиксаторов. Внесение фосфорных удобрений повышает численность Armatimonadetes и Chlorobi (Ling et al., 2017).
Метагеномный анализ древней ДНК. Метагеномика может изучать генетический материал не только живых организмов, но и давно умерших. Древнюю ДНК экстрагируют из образцов, возраст которых исчисляется столетиями и тысячелетиями. С биохимической точки зрения древняя ДНК характеризуется крайне малыми размерами фрагментов, длина которых в большинстве случаев не превышает 100–150 пар оснований, хотя, конечно, известны случаи лучшей сохранности данного типа ДНК. Помимо этого, древняя ДНК имеет большое количество специфических постмортальных мутаций. Древняя ДНК сохраняется во многих органах и тканях, способных противостоять времени и воздействию внешней среды. Самыми популярными объектами исследования являются кости, зубы и волосы, так как они лучше всего сохраняются при большом диапазоне условий. Защитой для зубов от грубых механических воздействий с течением времени служит эмаль сверху и кости челюсти снизу. Различные типы ДНК (ядерная, митохондриальная, в случае растений – хлоропластная) имеют также различные свойства и сохраняются в разной степени в тех или иных частях органов и тканей. В случае зубов ядерная ДНК сохраняется наилучшим образом в клетках цементного слоя, инкапсулированная в его минеральной матрице, тогда как в пульпе и дентине источником ядерной ДНК являются мягкие ткани, наиболее подверженные разложению в начале постмортального периода. Многокопийная мтДНК, напротив, наилучшим образом сохраняется в дентине, в особенности в области корней зуба (Higgins et al., 2015). Волосы, благодаря гидрофобной структуре кератина, обладают значительной устойчивостью как к контаминации экзогенной ДНК, так и к воздействию воды (Gilbert et al., 2006). Правда, из-за особенностей развития волос, основным сохраняющимся типом ДНК является мтДНК, которая в большом количестве (имеется в виду копийность) может быть найдена в стержне волоса на всем его протяжении. Ядерная же ДНК сохраняется хорошо только в корне живого волоса и в ближайших к нему нескольких сантиметрах стержня, однако уже в выпавших волосах зачастую ядерная ДНК вообще не детектируется (Andréasson et al., 2006).
Мягкие ткани также могут сохраняться, если для этого имеются благоприятные условия – к примеру, вечная мерзлота. Благодаря низким температурам, приводящим как к лучшей сохранности клеточных компонентов и ДНК, так и к низкой активности микроорганизмов и клеточным ферментов, именно биологический материал из вечной мерзлоты характеризуется самой высокой устойчивостью по сравнению с другими видами палеоДНК. Помимо этого, вечная мерзлота открыла науке путь и к другим объектам: грибам (Bellemain et al., 2013), бактериям (Willerslev et al., 2004) и древним человеческим популяциям (Green et al., 2006; Noonan et al., 2006; Fu et al., 2015). Древняя ДНК находит свое применение и в областях биологии, связанных с медициной. Чтобы проследить эволюцию и распространение чумной палочки, были отсеквенированы геномы штаммов чумной палочки Yersinia pestis возрастом от 2800 до 4000 лет и построены филогенетические деревья. В ходе исследования выяснилось, что ~3000 лет назад в геноме Y. pestis появился ген Yersinia murin toxin (ymt), который кодирует фосфолипазу Д, защищающую чумную палочку в кишечнике артропод (в данном случае блох), и таким образом авторы смогли довольно точно предсказать время, когда блохи стали вектором распространения чумы. Другие авторы исследовали образцы плейстоценовых и голоценовых отложений из вечной мерзлоты Сибири, а также образцы пещерных и береговых отложений Новой Зеландии (Willerslev et al., 2003). ДНК экстрагировали из образцов весом ~2 г и затем проводили амплификацию с использованием праймеров на гены хлоропластов растений и мтДНК животных, в результате чего удалось выделить ДНК растений возрастом 300 000–400 000 лет и позвоночных животных 20 000–30 000 лет. Благодаря этому удалось оценить видовое разнообразие в каждой местности, а также проследить его динамику для растений на протяжении обширных временных периодов.
Проблема воспроизводимости и интерпретации метагеномных данных. Эффективность и высокая производительность метагеномного анализа обусловила резкий рост числа работ в этой области. Однако неаккуратность в проработке метагеномных методик приводит к невоспроизводимости результатов, от которой не застрахованы работы самого высокого уровня. Наибольшее количество противоречий в полученных результатах приходится на долю исследований микробиома кишечника человека (Poussin et al., 2018). Причем в подавляющем числе случаев под микробиомом кишечника человека авторы понимают микробиом кала человека. Интерес к этому объекту объясняется публикацией серии сообщений об исключительном влиянии микробиома кишечника на физиологию организма в целом. В разное время указывалось, что микробиота человека влияет на развитие сахарного диабета и ожирения, онкологических и аутоиммунных заболеваний, психологию человека посредством синтеза нейромедиаторов и, конечно, “иммунитет” (Yan, Charles, 2017; Malan-Muller et al., 2018; Dicks et al., 2018). Логика исследователей основывалась на корреляции между параметрами метагенома и частотой развития патологии. Хотя очевидно, что никакая корреляция не гарантирует прямой взаимосвязи. Например, есть хорошая корреляция между возрастом респондента и его микробиотой. Но до сих пор не появилась статья, где бы старение человека объяснялось активностью его бактерий. Кроме того, наблюдаемые различия вполне могут объясняться высокой микробиологической гетерогенностью образцов кала. Эта гетерогенность, а также многочисленные методические допущения приводят к невоспроизводимости метагеномных результатов. Так, в журнале “Cell” был поставлен вопрос о наследуемости микробиома человека, как органа, предположительно имеющего широчайшие функции (Goodrich et al., 2014). После проведения близнецового эксперимента с помощью метагеномных технологий оказалось, что состав микробиома наследуется генетически. Однако совсем скоро в журнале “Nаture” были опубликованы прямо противоположные данные (Rothschild et al., 2018). Подобное расхождение в результатах, опубликованных в журналах с импакт-фактором свыше 30, может объясняться как раз невозможностью буквального трактования метагеномных данных. Причины низкой воспроизводимости метагеномных данных кроются на всех этапах постановки эксперимента (Hoopen et al., 2017).
Этап выбора биологического образца. Образцы для метагеномных исследования характеризуются высокой гетерогенностью. Если речь идет о микробиоме, то его состав в образце зависит от микроструктуры объекта, наличия анаэробных зон, присутствия микроскопических слепых полостей, дистанции до активных зон, например: корневых волосков, стенки кишечника, копролитов дождевых червей. Очень сложно выполнить рандомизацию, учитывающую все эти факторы. Иногда рандомизацию выполнять просто нецелесообразно, поскольку различия микробиома в двух ближайших микролокусах (например: поверхность стенки кишечника и просвет кишечника) так велики, что их в принципе не следует рассматривать вместе. Также нельзя забывать о скорости изменений микробиома. Например, разница в отборе проб в один час может радикально изменить характер микробиома. Представление о микробиоме, как о неизменной и постоянной во времени системе совершенно не соответствует действительности. Ненамного проще ситуация с метагеномом сообществ животных. Разные части тела животного содержат разное количество ДНК. Кроме того, ДНК в разных частях животного деградирует с разной скоростью. Оптимальная ее сохранность достигается в пульпе зуба, наихудшая – в слущенном эпителии. Поэтому различия в представленности генов двух животных в среде, могут быть связаны как с различной численностью животных, так и с различиями в механизмах попадания и депонирования их ДНК в среде. К сожалению, многие исследователи, находящиеся под впечатлением от технической новизны метода, слишком формально выполняют методические протоколы, которые подкупают своей простотой. Но протоколы разрабатывают специалисты, незнакомые со спецификой каждого конкретного типа образцов. А ошибка в выборе биологического образца для метагеномного анализа заранее вызывает искажение результатов, которое нельзя скомпенсировать на более поздних этапах эксперимента.
Этап выделения суммарной ДНК. Данный этап работы высоко формализован. Ошибка может возникнуть только при сравнении результатов, полученных с помощью разных методов выделения ДНК. Внутри одного метода достигается достаточно хорошая воспроизводимость.
Этап амплификации. В метагеномных работах, выполненных по “shot-gun” методике, этот этап и связанные с ним ошибки отсутствуют. В ампликонных работах возникает необходимость множественной амплификации целевого фрагмента ДНК с помощью ПЦР. На этом этапе помимо копирования целевых фрагментов происходит появление фрагментов-артефактов (например, химер), которые изначально отсутствовали в образце. При неудачном ПЦР доля артефактов может превышать 50%. Кроме того, для ПЦР необходимо подобрать праймеры – специальные молекулы ДНК, которые отмечают начало и конец целевого фрагмента. У разных организмов эти участки немного отличаются. Даже наиболее универсальные праймеры хорошо подходят для амплификации требуемых участков ДНК одних организмов и не подходят для других. Поэтому на практике используется “коктейль” из десятков праймеров, которые потенциально должны подойти ко всем организмам. Но чем больше параллельно используется праймеров, тем выше количество артефактов ПЦР. Объективно отличить артефакт от целевого фрагмента невозможно. Контроль над появлением артефактов осуществляется на этапе биоинформатической обработки исключительно косвенными методами.
Этап секвенирования. Секвенирование может осуществляться на одной из нескольких аппаратных платформ. Наиболее популярные: Illumina, Pacific Bioscience, IonTorrent. Сам процесс секвенирования предусматривает ошибки. Наиболее высокий уровень аппаратных ошибок у платформы IonTorrent, но она является наиболее привлекательной по стоимости. Результаты, полученные на разных платформах, характеризуется невысокой воспроизводимостью по отношению друг к другу (Allali et al., 2017). Ключевым способом повышения воспроизводимости на аппаратном уровне является увеличение глубины секвенирования, измеряемой в количестве прочтений на образец (Zaheer et al., 2018). Если для работы по микробиому в начале 2010-х гг. можно было ограничиться 10 000 прочтений, то теперь требования выросли до 50 000 и даже 120 000 прочтений на образец. При постановке работы необходимо стремиться к тому, чтобы все сиквенсы эксперимента были получены на одной и той же платформе. Причем это актуально и при сравнении собственных результатов с литературными данными.
Этап биоинформатической обработки данных. Результат работы секвенатора представляет собой перечень нуклеотидных последовательностей, которые находились в образце и были фланкированы праймерами, а также набор всех артефактов секвенирования. Как правило, каждый реально существующий целевой фрагмент длиной 200–400 п.н. в результате ошибок отображается как группа фрагментов, все члены которой отличаются друг от друга на несколько нуклеотидов. Поэтому неверно воспринимать каждый вариант фрагмента, как фактически существующий в среде. Это приведет к ложному многократному увеличению биоразнообразия сообщества. Для борьбы с этим (а также для учета внутривидового полиморфизма) применяется кластеризация сиквенсов в операционные таксономические единицы – ОТЕ. Можно выделить три основные группы кластеризующих алгоритмов:
1. Динамические de novo алгоритмы. Подходят для кластеризации сиквенсов при анализе новых, ранее не изучавшихся сообществ, включающих редкие и отсутствующие в базах данных таксоны. Не могут быть реализованы на бытовых компьютерах, требуют наличия высокопроизводительных процессоров.
2. “Жадные” de novo алгоритмы. Подходят для кластеризации сиквенсов при анализе новых, ранее не изучавшихся сообществ, включающих редкие и отсутствующие в базах данных виды. Могут быть реализованы на любом компьютере. Но дают ложное завышение разнообразия, искажают количественную представленность кластеров.
3. Референсные алгоритмы. Подходят для кластеризации сиквенсов при анализе хорошо известных сообществ, все члены которых были ранее проанализированы по отдельности, и сиквенсы депонированы в базу данных. Могут быть реализованы на любом компьютере. Дают минимальное искажение результатов. Плохо работают при низком пороге идентичности (см. ниже).
4. Без кластеризации. Метод, привлекающий все большее внимание благодаря наиболее высокой разрешающей способности. Требует большого личного участия оператора и поэтому не может быть реализован на поточной основе.
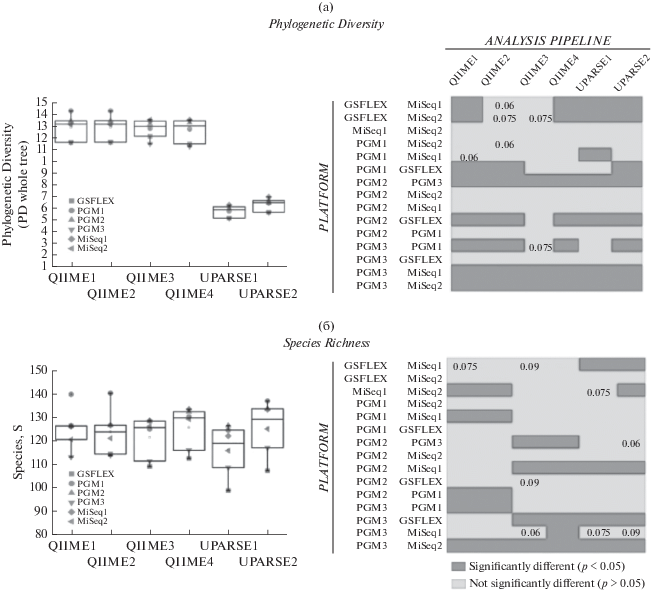
Очевидно, что кластеризация серьезно снижает разрешающую способность метода, поэтому исследователь должен задать порог идентичности (в % отличий нуклеотидной последовательности) в пределах которого будет формироваться ОТЕ. При большом количестве артефактов порог идентичности увеличивают, при высоком качестве сиквенсов – уменьшают. Стандартный порог для баркодирования по рибосомальным генам – 97%, что соответствует уровню род/вид. Очень важно, что при работе с неуниверсальными узкоспециализированными праймерами необходимо поднять порог идентичности, иначе это обесценит высокую разрешающую способность праймера. В редких случаях его могут поднимать до 100%, т.е. отказываться от кластеризации. При работе с очень разнообразными или низкокачественными метагеномными данными при использовании высокоуниверсальных праймеров порог уменьшают вплоть до 93% (уровень отряда–семейства). При работе с функциональными генами (целлюлазы, токсины и т.д.) порог обычно также уменьшают до 93–95% (Ngara, Zhang, 2018). Неудачный выбор алгоритма кластеризации и порога идентичности может сильно навредить работе. Принципиально важно, чтобы все сравнения проводили только между результатами, полученными с помощью одного и того же метода кластеризации (Clooney et al., 2016). Это актуально и при сравнении с литературными данными. В работе Allali et al., 2017 было наглядно продемонстрировано, что анализ одного и того же образца с помощью разных математических пакетов и/или разных платформ секвенирования ведет к невоспроизводимости данных (рис. 5). Стоит только отметить, что основным “инструментом” контроля является исключение всех нуклеотидных последовательностей, встречающихся в единственном экземпляре (синглтонов). Еще одним подходом является перенос внимания на таксоны более высокого уровня. Небольшие ошибки в нуклеотидной последовательности фрагмента могут “перевести” его в другой род организмов, но маловероятно – в другое семейство или отряд (порядок).
Рис. 5.
Воспроизводимость результатов метагеномного анализа образца с помощью разных аппаратных платформ для секвенирования (по вертикали), и различных математических пакетов обработки данных (по горизонтали) (по Allali et al., 2017). (a) Филогенетическое разнообразие. (б) Видовое богатство.
Ошибки, возникающие на перечисленных этапах метагеномного исследования, могут неоправданно увеличивать биоразнообразие анализируемого сообщества организмов и обуславливать ложные корреляции. Наиболее эффективный способ борьбы с подобными ошибками – контрольное исследование образцов без использования технологий метагеномики. Как правило, высокорейтинговые журналы требуют, чтобы вывод любой работы был подтвержден как минимум двумя экспериментами, основанными на разных методических принципах. Например, увеличение экспрессии гена (показанное методом секвенирования тотальной РНК) должно быть подтверждено увеличением количества целевого метаболита (показанным методом ГХ-МС). Этот же принцип должен соблюдаться и для метагеномных исследований, однако в подавляющем большинстве метагеномных работ этого не происходит. Поэтому любые рассуждения о составе и функциях микробиома, вытекающие только из метагеномных данных, являются в значительной мере спекулятивными. Вероятно, дальнейшее развитие метагеномных исследований будет заключаться в контроле параметров фенотипа, поскольку любые геномные данные лишь косвенно связаны с фактическим фенотипом. Более детальную информацию дают: метатранскриптом – совокупность всей матричной РНК в пробе, метапротеом – совокупность всех белков и, наконец, мета-метаболом, характеризующий все органические вещества, включая малые молекулы. Данные подходы иногда называются “омиксными” технологиями – от общего английского окончания для всех четырех терминов: metagenomics, metatranscriptomics, metaproteomics, meta-metabolomics. Сочетание омиксных технологий будет служить внутренним контролем для любого исследования. Например, наличие характерных изоформ белков будет подтверждать наличие конкретного вида при метабаркодинге. Наличие определенного метаболита будет подтверждать обнаружение соответствующего функционального гена. Без этого, метагеномика будет оставаться многообещающим, но пока недостаточно надежным методом.
Финансирование. Раздел статьи “Метагеномный анализ почв” подготовлен при финансовой поддержке РФФИ (проект 19-04-00315); раздел статьи “Метагеномный анализ древней ДНК” подготовлен при поддержке РФФИ (проект 18-34-00895).
Благодарности. Авторы выражают благодарность акад. РАН В.В. Рожнову и О.Л. Макаровой за помощь при подготовке статьи.
Список литературы
Allali I., Arnold J.W., Roach J., Cadenas M.B., Butz N., Hassan H.M., Koci M., Ballou A., Mendoza M., Ali R., Azcarate-Peril M.A. A comparison of sequencing platforms and bioinformatics pipelines for compositional analysis of the gut microbiome // BMC Microbiol. 2017. V. 17(1). P. 194.
Andréasson H., Nilsson M., Budowle B., Lundberg H., Allen M. Nuclear and mitochondrial DNA quantification of various forensic materials // Forensic Sci. Int. 2006. V. 164(1). P. 56–64.
Bellemain E., Davey M.L., Kauserud H., Epp L.S., Boessenkool S. Fungal palaeodiversity revealed using high-throughput metabarcoding of ancient DNA from arctic permafrost // Environ. Microbiol. 2013. V. 15(4). P. 1176–1189.
Borbón-García A., Reyes A., Vives-Flórez M., Caballero S. Captivity shapes the gut microbiota of Andean Bears: insights into health surveillance // Front. Microbiol. 2017. V. 8. P. 1316.
Chai Z.Y., He Z.L., Deng Y.Y., Yang Y.F., Tang Y.Z. Cultivation of seaweed Gracilaria lemaneiformis enhanced biodiversity in a eukaryotic plankton community as revealed via metagenomic analyses // Mol. Ecol. 2018. V. 27(4). P. 1081–1093.
Clooney A.G., Fouhy F., Sleator R.D., O’Driscoll A., Stanton C., Cotter P.D., Claesson M.J. Comparing apples and oranges?: next generation sequencing and its impact on microbiome analysis. PLOS ONE. 2016. V. 11(2). e0148028.
da Rocha U.N., Andreote F.D., de Azevedo J.L., van Elsas J.D., van Overbeek L.S. Cultivation of hitherto-uncultured bacteria belonging to the Verrucomicrobia subdivision 1 from the potato (Solanum tuberosum L.) rhizosphere // J. Soils Sediments. 2010. V. 10. P. 326–339.
Dicks L.M.T., Geldenhuys J., Mikkelsen L.S., Brandsborg E., Marcotte H. Our gut microbiota: a long walk to homeostasis // Benef. Microbes. 2018. V. 9(1). P. 3–20.
Eilers K.G., Debenport S., Anderson S., Fierer N. Digging deeper to find unique microbial communities: the strong effect of depth on the structure of bacterial and archaeal communities in soil // Soil Biol. Biochem. 2012. V. 50. P. 58–65.
Fierer N., Leff J.W., Adams B.J., Nielsen U.N., Bates S.T., Lauber C.L., Owens S., Gilbert J., Wall D., Caporaso J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes // PNAS. 2012. V. 109(52). P. 21390–21395.
Fu Q., Hajdinjak M., Moldovan O.T., Constantin S., Mallick S. et al. An early modern human from Romania with a recent Neanderthal ancestor // Nature. 2015. V. 524(7564). P. 216–219.
Gilbert M.T.P., Menez L., Janaway R.C., Tobin D.J., Cooper A., Wilson A.S. Resistance of degraded hair shafts to contaminant DNA // Forensic Sci. Int. 2006. V. 156(2–3). P. 208–212.
Goodrich J.K., Waters J.L., Poole A.C., Sutter J.L., Koren O., Blekhman R., Beaumont M., Van Treuren W., Knight R., Bell J.T., Spector T.D., Clark A.G., Ley R.E. Human genetics shape the gut microbiome // Cell. 2014. V. 159(4). P. 789–799.
Gorbacheva M.A., Melnikova N.V., Chechetkin V.R., Kravatsky Y.V., Tchurikov N.A. DNA sequencing and metagenomics of cultivated and uncultivated chernozems in Russia // Geoderma Regional. 2018. V. 14. e00180.
Green R.E., Krause J., Ptak S.E., Briggs A.W., Ronan M.T., Simons J.F. et al. Analysis of one million base pairs of Neanderthal DNA // Nature. 2006. V. 444. P. 330.
Higgins D., Rohrlach A.B., Kaidonis J., Townsend G., Austin J.J. Differential nuclear and mitochondrial DNA preservation in post-mortem teeth with implications for forensic and ancient DNA studies // PLoS One. 2015. V. 10(5). e0126935.
Hoopen P., Finn R.D., Bongo L.A., Corre E., Fosso F., Meyer F., Mitchell A., Pelletier E., Pesole G., Santamaria M., Willassen N.P. The metagenomic data life-cycle: standards and best practices // Gigascience. 2017. V. 6(8). P. 1–11.
Ling N., Chen D., Guo H., Wei J., Bai Y., Shen Q., Hu S. Differential responses of soil bacterial communities to long-term N and P inputs in a semi-arid steppe // Geoderma. 2017. V. 292. P. 25–33.
Lombard N., Prestat E., van Elsas J.D., Simonet P. Soil-specific limitations for access and analysis of soil microbial communities by metagenomics // FEMS Microbiol. Ecol. 2011. V. 78(1). P. 31–49.
Lucas A., Bodger O., Brosi B.J., Ford C.R., Forman D.W., Greig C., Hegarty M., Neyland P.J., de Vere N. Generalisation and specialisation in hoverfly (Syrphidae) grassland pollen transport networks revealed by DNA metabarcoding // J. Anim. Ecol. 2018. V. 87(4). P. 1008–1021.
Ma J., Prince A., Aagaard K.M. Use of whole genome shotgun metagenomics: a practical guide for the microbiome-minded physician scientist // Semin. Reprod. Med. 2014. V. 32. P. 5–13.
Malan-Muller S., Valles-Colomer M., Raes J., Lowry C.A., Seedat S., Hemmings S.M.J. The gut microbiome and mental health: implications for anxiety- and trauma-related disorders // OMICS. 2018. V. 22(2). P. 90–107.
Mendes L.W., Tsai S.M., Navarrete A.A., De Hollander M., van Veen J.A., Kuramae E.E. Soil-borne microbiome: linking diversity to function // Microb. Ecol. 2015. V. 70. P. 255–265.
Navarrete A.A., Soares T., Rossetto R., van Veen J. A., Tsai S.M., Kuramae E.E. Verrucomicrobial community structure and abundance as indicators for changes in chemical factors linked to soil fertility // Antonie van Leeuwenhoek. 2015. V. 108(3). P. 741–752.
Ngara T.R., Zhang H. Recent advances in function-based metagenomic screening // Genom. Prot. Bioinform. 2018. V. 16(6). P. 405–415.
Noonan J.P., Coop G., Kudaravalli S., Smith D., Krause J. et al. Sequencing and Analysis of Neanderthal Genomic DNA // Science. 2006. V. 314(5802). P. 1113–1118.
Pester M., Schleper C., Wagner M. The Thaumarchaeota: an emerging view of their phylogeny and ecophysiology // Curr. Opin. Microbiol. 2011. V. 14(3). P. 300–306.
Pompanon F., Deagle B.E., Symondson W.O., Brown D.S., Jarman S.N., Taberlet P. Who is eating what: diet assessment using next generation sequencing // Mol. Ecol. 2012. V. 21(8). P. 1931–1950.
Poussin C., Sierro N., Boué S., Battey J. Scotti E., Belcastro V., Peitsch M.C., Ivanov N.V., Hoeng J. Interrogating the microbiome: experimental and computational considerations in support of study reproducibility // Drug Discov. Today. 2018. V. 23(9). P. 1644–1657.
Ranjan R., Rani A., Metwally A., McGee H.S., Perkins D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing // Biochem. Biophys. Res. Commun. 2016. V. 469(4). P. 967–77.
Rothschild D., Weissbrod O., Barkan E., Kurilshikov A., Korem T., Zeevi D., Costea P.I., Godneva A., Kalka I.N., Bar N., Shilo S., Lador D., Vila A.V., Zmora N., Pevsner-Fischer M., Israeli D., Kosower N., Malka G., Wolf B.C., Avnit-Sagi T., Lotan-Pompan M., Weinberger A., Halpern Z., Carmi S., Fu J., Wijmenga C., Zhernakova A., Elinav E., Segal E. Environment dominates over host genetics in shaping human gut microbiota // Nature. 2018. V. 555(7695). P. 210–215.
Riesenfeld C.S., Schloss P.D., Handelsman J. Metagenomics: genomic analysis of microbial communities // Annu. Rev. Genet. 2004. V. 38. P. 525–552.
Semenov M., Blagodatskaya E., Stepanov A., Kuzyakov Y. DNA-based determination of soil microbial biomass in alkaline and carbonaceous soils of semi-arid climate // J. Arid Environ. 2018. V. 150. P. 54–61.
Senechkin I.V., Speksnijder A.G.C.L., Semenov A.M., van Bruggen A.H.C., van Overbeek L.S. Isolation and partial characterization of bacterial strains on low organic carbon medium from soils fertilized with different organic amendments // Microb. Ecol. 2010. V. 60. P. 829–839.
Sigsgaard E.E., Nielsen I.B., Bach S.S., Lorenzen E.D., Robinson D.P., Knudsen S.W., Pedersen M.W., Jaidah M.A., Orlando L., Willerslev E., Møller P.R., Thomsen P.F. Population characteristics of a large whale shark aggregation inferred from seawater environmental DNA // Nat. Ecol. Evol. 2016. V. 1(1). P. 4.
Souza R.C., Hungria M., Cantao M.E., Vasconcelos A.T.R., Nogueira M.A., Vicente V.A. Metagenomic analysis reveals microbial functional redundancies and specificities in a soil under different tillage and crop-management regimes // Appl. Soil Ecol. 2015. V. 86. P. 106–112.
Spribille T., Tuovinen V., Resl P., Vanderpool D., Wolinski H., Aime M.C., Schneider K., Stabentheiner E., Toome-Heller M., Thor G., Mayrhofer H., Johannesson H., McCutcheon J.P. Basidiomycete yeasts in the cortex of ascomycete macrolichens // Science. 2016. V. 353. P. 488–492.
Valentine D.L. Adaptations to energy stress dictate the ecology and evolution of the archaea // Nat. Rev. Microbiol. 2007. V. 5(4). P. 316–323.
Vital M., Karch A., Pieper D.H. Colonic butyrate-producing communities in humans: an overview using omics data // Systems. 2017. V. 2(6).
Willerslev E., Hansen A.J., Binladen J., Brand T.B., Gilbert M.T. et al. Diverse plant and animal genetic records from holocene and pleistocene Sediments // Science. 2003. V. 300(5620). P. 791–795.
Willerslev E., Hansen A.J., Rønn R., Brand T.B., Barnes I. et al. Long-term persistence of bacterial DNA // Curr. Biol. 2004. V. 14(1). P. 9–10.
Wilson J.J., Brandon-Mong G.J., Gan H.M., Sing K.W. High-throughput terrestrial biodiversity assessments: mitochondrial metabarcoding, metagenomics or metatranscriptomics? // Mitochondrial DNA A DNA Mapp. Seq. Anal. 2019. V. 30(1). P. 60–67.
Yan J., Charles J.F. Gut microbiome and bone: to build, destroy, or both? // Curr. Osteoporos Rep. 2017. V. 15(4). P. 376–384.
Yoon T.H., Kang H.E., Lee S.R., Lee J.B., Baeck G.W., Park H., Kim H.W. Metabarcoding analysis of the stomach contents of the Antarctic Toothfish (Dissostichus mawsoni) collected in the Antarctic Ocean // Peer J. 2017. V. 5. P. e3977.
Zaheer R., Noyes N., Polo R.O., Cook S.R., Marinier E., Van Domselaar G., Belk K.E., Morley P.S., McAllister T.A. Impact of sequencing depth on the characterization of the microbiome and resistome // Sci. Rep. 2018. V. 8(1). P. 5890.
Zaremba-Niedzwiedzka K., Caceres E.F., Saw J.H., Bäckström D., Juzokaite L., Vancaester E., Seitz K.W., Anantharaman K., Starnawski P., Kjeldsen K.U., Stott M.B., Nunoura T., Banfield J.F., Schramm A., Baker B.J., Spang A., Ettema T.J.. Asgard archaea illuminate the origin of eukaryotic cellular complexity // Nature. 2017. V. 541(7637). P. 353–358.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Серия биологическая