

Известия РАН. Серия биологическая, 2022, № 6, стр. 678-684
Анализ мтДНК верхнеенисейского (Thymallus svetovidovi) и монгольского (Thymallus brevirostris) хариусов, уточнение их ареалов
Е. В. Пономарева 1, *, А. А. Волков 2, М. В. Пономарева 1, Е. А. Шубина 3
1 Московский государственный университет им. М.В. Ломоносова
119991 Москва, Россия
2 ВНИИ рыбного хозяйства и океанографии
105187 Москва, Окружной проезд,19, Россия
3 НИИ физико-химической биологии им. А.Н. Белозерского МГУ
119991 Москва, Россия
* E-mail: kponom@mail.ru
Поступила в редакцию 04.03.2021
После доработки 12.04.2022
Принята к публикации 05.05.2022
- EDN: UNDVJU
- DOI: 10.31857/S1026347022060130
Аннотация
Проанализированы выборки хариусов из притоков Большого Енисея, Черного Иртыша и проведено их сравнение с монгольским хариусом T. brevirostris из реки Кобдо на основе двух фрагментов мтДНК (CO1 и CR). Показано, что гаплотипы хариусов из Черного Иртыша относятся к монгольскому хариусу. На основе проведенного анализа сделан вывод, что в притоках Большого Енисея обитает верхнеенисейский хариус T. svetovidovi, обладающий низким генетическим разнообразием по сравнению с монгольским хариусом.
Представители рода хариусов Thymallus (Cuvier, 1829) встречаются в большинстве водоемов Европейского Севера, Сибири и Дальнего Востока. Морфологические и систематические исследования хариусов имеют длительную историю, при этом систематика рода Thymallus неоднократно пересматривалась (Световидов, 1936; Берг, 1948; Froufe et al., 2005; Weiss et al., 2020 и др.). Наибольший теоретический интерес представляют хариусы в азиатской части ареала, что связано с большим разнообразием их форм. Несмотря на значительно увеличившееся количество исследований, из-за труднодоступности многих мест обитания хариусов еще недостаточно известно как о морфологических, биологических и генетических особенностях, так и о границах ареалов многих представителей этого рода.
Световидов в обзоре (1936) определял хариусов, обитающих на всем протяжении Енисея, включая и верховья, как один вид – сибирский хариус (Thymallus arcticus Pallas, 1776). В 1960–1970-х гг. Гундризер (1967, 1979) описал морфологические отличия хариусов из высокогорных озер Большого Енисея от сибирского хариуса и предложил выделить их в подвиды сибирского. В обзоре видов Монголии Котела (Kottelat, 2006) предположил, что в притоке Малого Енисея Шишкид Гол обитает вид, отличный от сибирского. Эти предположения были подтверждены генетическими исследованиями (Weiss et al., 2020). При дальнейшем изучении хариусов из этого притока Книжин и Вайс (2009) на основе морфологических и морфометрических признаков описали новый вид Thymallus svetovidovi, (верхнеенисейский хариус). В российской части ареала подтверждений существования верхнеенисейского хариуса до настоящего времени не было получено.
Световидов (1936) полагал, что на всем протяжении Оби, как и в Енисее, обитает только сибирский хариус. Сейчас ареал сибирского хариуса в Оби ограничивается нижним течением реки (Romanov, 2017; Dyldin et al., 2017). В китайской части верховьев Иртыша обитает монгольский хариус (Dyldin et al., 2017). Монгольский хариус (Thymallus brevirostris) обитает в реках Центрально-Азиатского бассейна (Световидов, 1936; Берг, 1948; Kottelat, 2006). Впервые монгольский хариус был описан во второй половине XIX в., с тех пор его видовой статус большинством исследователей не подвергался сомнению.
Целью настоящего исследования было определение с помощью генетических маркеров видовой принадлежности хариусов, обитающих в верховьях Иртыша на территории Казахстана и в верховьях Енисея и сравнение с монгольским хариусом из реки Кобдо. В работе были проанализированы два участка мтДНК: контрольный регион (Д-петля, CR) и участок, кодирующий ген цитохромоксидазы субъединицы I (COI). Контрольный регион – это наиболее часто применяющийся во внутривидовых и межвидовых исследованиях хариусов (Froufe et al., 2005; Weiss et al., 2020). Участок CO1 был выбран как основной фрагмент, который используется в работах по паспортизации видов.
МАТЕРИАЛЫ И МЕТОДЫ
Географическое расположение выборок показано на рис. 1, координаты и характеристики выборок – в табл. 1.
Рис. 1.
Локализация выборок. Обозначения на карте-схеме: 1–2 – озера из притоков Большого Енисея, 3 – река Черный Иртыш (Казахстан), 4 – река Кобдо (Монголия).
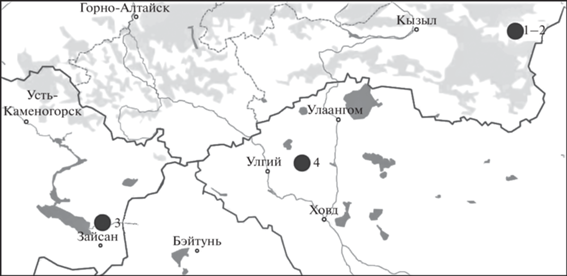
Таблица 1.
Характеристика выборок и количество выявленных гаплотипов, N – объем выборок
| Вид | Водоем, приток, бассейн | Год | Обозначение | Координаты | N | Количество гаплотипов с учетом инделов |
||
|---|---|---|---|---|---|---|---|---|
| CO1 | CR | CR + CO1 | ||||||
| T. svetovidovi | Озеро без названия, река Маймамыш, Большой Енисей | 2016 | En_1 | 51.67305, 97.459849 | 14 | 1 | 2 | 2 |
| T. svetovidovi | Озеро без названия, река Хан, Большой Енисей |
2016 | En_2 | 51.64000, 97.047222 | 14 | 1 | 1 | 1 |
| T. brevirostris | Река Черный Иртыш, Иртыш, Обь | 2015 | Kaz | 47.93067, 84.949812 | 4 | 1 | 2 | 2 |
| T. brevirostris | Река Кобдо, Центрально-Азиатский | 2006 | Mon | 49.12000, 90.994450 | 31 | 6 | 12 | 16 |
Выделение ДНК из плавников, фиксированных 96%-ным этанолом, проводили с использованием набора QIAGEN DNeasyTM (“QIAGEN”, Германия) по протоколу производителя. Для амплификации Д-петли использовали последовательности праймеров LRBT-25, LRBT-1195 (Uiblein et al., 2001), для амплификации участка гена CO1 использовали последовательности универсальных праймеров CO1 для рыб (Ivanova et al., 2007). Амплификацию проводили в 20 мкл смеси, содержащей: 2 мкл 10× Taq-буфера, 2.5 мМ Mg2+, 0.8 мM каждого дезоксирибонуклеозидтрифосфата, 1 ед BioHYTaq ДНК полимеразы (“ДИАЛАТ”, Россия), 5 пкM каждого праймера (“Синтол”, Россия), 1.0 мкл выделенной ДНК, до необходимого объема смесь доводили деионизованной водой. Амплификацию фрагментов проводили в следующем режиме: первичная денатурация – 95°С 5 мин, затем 35 циклов по: 20 с – 95°С, 30 с – 56°С (CR) или 52°С (СО1), 1 мин – 72°С; заключительная элонгация 72°С – 10 мин. Для контроля ПЦР-продукта проводили горизонтальный гель-электрофорез, амплификат разделяли в 1.5%-ном агарозном геле. Очистку продуктов ПЦР проводили методом осаждения смесью ацетата аммония с 96%-ным этанолом с последующей промывкой 70%-ным этанолом. Для сиквенс-реакции использовали смесь терминаторов BigDye Terminator v.3.1 Cycle Sequencing Kit (“Applied Biosystems”, США), анализ нуклеотидной последовательности проводили в генетическом анализаторе ABI Prism 3500 (“Applied Biosystems”). Для секвенирования контрольного региона использовали внутренние праймеры (5'–3'): In(429)F – TCAACTAACACGAGCTCTGTCTCT; In(928)R – GGGGGTTTGTCGAGTAGAAGCC; In(737)F – TCAGCTTGCATCTCACAGTG.
Первичную обработку и множественное выравнивание нуклеотидных последовательностей проводили в программе Geneious® 6.0.5 (“Biomatters Ltd”), определение количества гаплотипов (H), разнообразия гаплотипов (Hd), разнообразия нуклеотидов (Pi) проводили в программах FaBox (Villesen, 2007), DnaSP v.5 (Librado, Rozas, 2009). Двухпараметрическая модель Кимуры K2P (Kimura, 1980) была применена для расчета матрицы попарных дистанций гаплотипов CO1. Для построения дистанционных матриц использовали программу Mega X (Kumar et al., 2018). Информационный критерий Акаике (AIC) был применен для определения наилучшей модели замен (Lefort et al., 2017). Модель GTR + G + I (I = 0.665, Υ = 0.712) была применена для построения деревьев: методом присоединения соседей (NJ) с бутстреп поддержкой (10 000 итераций) в программе Mega X (Kumar et al., 2018), BioNJ дерево методом максимального правдоподобия в программе PhyML 3.0. (Guindon, Gascuel, 2003; Guindon et al., 2010), методом байесовской статистики (цепочки длиной 400 000 генераций с отбрасыванием первых 10%) в программе MrBayes (Huelsenbeck, Ronquist, 2001) в плагине, встроенном в Geneious® 6.0.5. MSN алгоритм был использован для построения сети гаплотипов в программе PopArt (Bandelt et al., 1999). График соответствия распределения частот попарных нуклеотидных различий модели экспансии вида построен в программе DnaSp v.5 (Librado, Rozas, 2009). Для оценки соответствия моделям численного и пространственного расширения вида (Rogers, Harpending, 1992) рассчитывали сумму квадратичных отклонений (SSD) и индекс Харпендинга r (raggedness index), а также индексы, отражающие демографические события, Таджимы (D) и Фу (FS) в программе Arlequin v. 3.5.2.2 (Excoffier, Lischer, 2010). Для построения дерева гаплотипов и матрицы дистанций были использованы последовательности, депонированные в базе нуклеотидных последовательностей GenBank® (NCBI), содержащие исследованные участки мтДНК: NC012928 (Thymallus thymallus), NC012929 (T. arcticus), NC027409 (Thymallus tugarinae), NC027412 (T. brevirostris), KJ866482 (Thymallus baicalolenensis) KT867089 (T. arcticus mertesii), KU674353 (Thymallus grubii flavomaculatus). Полученные последовательности гаплотипов были депонированы в GenBank под номерами: MW036546 – MW036553; MW048650 – MW048665.
Для уточнения вопросов видовой принадлежности мы ориентировались на критерии, принятые в проекте паспортизации видов BOLD (Ratnasingham, Hebert, 2007).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
После выравнивания был исследован участок гена CO1 (длиной 699 п. н.), всего было выявлено 17 вариабельных сайтов, из них 16 информативных, 15 транзиций и 2 трансверсии, все замены – синонимичные. Среди 62 исследованных последовательностей было выделено 8 гаплотипов, из них 6 гаплотипов у монгольского хариуса из реки Кобдо, 1 гаплотип у хариусов из Черного Иртыша, отличающийся на 3 замены от массового гаплотипа Mon_1, и 1 гаплотип у хариусов из верховьев Енисея, отличающийся на 11 замен от массового гаплотипа Mon_1 (табл. 1, рис. 2б, прил. 1). После выравнивания 60 последовательностей, в проанализированном участке длиной 1066 п. н., содержащем полную последовательность контрольного региона (CR) мтДНК, было выявлено 22 вариабельных информативных сайта, из них 18 транзиций и 4 трансверсии. Всего было выделено 16 гаплотипов с инделами и 13 без инделов, из них 12 гаплотипов с инделами принадлежали T. brevirostris, 5 из них были выявлены впервые. Два новых гаплотипа (Kaz_1 и Kaz_2), отличающихся на 3 замены от гаплотипов из реки Кобдо, выделили у рыб из реки Черный Иртыш (табл. 1, рис. 2а, прил. 1). Один из гаплотипов хариусов из Верхнего Енисея совпал с последовательностями CR из Генбанка (EU676291, -2, -4), принадлежавшими T. svetovidovi из Шишкид Гол, второй гаплотип – новый, все гаплотипы отличались на одну замену – транзицию. После объединения двух фрагментов (CO1 + CR) и выравнивания участка длиной 1765 п. н. было выявлено 20 гаплотипов с инделами (табл. 1). Параметры генетического разнообразия были значительно выше для монгольского хариуса, чем для верхнеенисейского (табл. 2).
Рис. 2.
MST сеть гаплотипов T. brevirostris (серый – р. Кобдо, черный – р. Черный Иртыш), T. svetovidovi (белый). Насечки – количество замен между гаплотипами, диаметры пропорциональны количеству гаплотипов. (а) – CR мтДНК, (б) – СО1 мтДНК, (в) – объединенные фрагменты мтДНК CR + CO1.
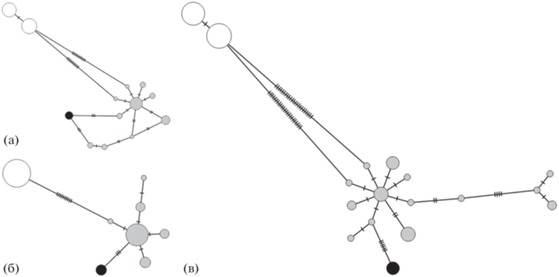
Таблица 2.
Характеристики генетического разнообразия T. brevirostris и T. svetovidovi на основе фрагментов мтДНК, n – количество исследованных образцов, h – количество гаплотипов, s – число сегрегирующих сайтов, k – среднее число нуклеотидных различий, Pi – нуклеотидное разнообразие, Hd ± SD – разнообразие гаплотипов и стандартное отклонение
| Участки мтДК | Вид | n | h | s | k | Pi | Hd ± SD |
|---|---|---|---|---|---|---|---|
| CO1 | T. brevirostris | 34 | 7 | 8 | 1.429 | 0.0024 | 0.722 ± 0.073 |
| T. svetovidovi | 28 | 1 | 0 | – | – | – | |
| Всего | 62 | 8 | 17 | 6.206 | 0.00888 | 0.708 ± 0.045 | |
| CR | T. brevirostris | 30 | 14 | 11 | 2.645 | 0.00249 | 0.867 ± 0.041 |
| T. svetovidovi | 28 | 2 | 1 | 0.516 | 0.00048 | 0.516 ± 0.029 | |
| Всего | 60 | 16 | 22 | 8.322 | 0.00786 | 0.859 ± 0.024 | |
| CO1 + CR | T. brevirostris | 31 | 18 | 19 | 4.06 | 0.00231 | 0.927 ± 0.024 |
| T. svetovidovi | 28 | 2 | 1 | 0.516 | 0.00029 | 0.516 ± 0.029 | |
| Всего | 59 | 20 | 39 | 14.528 | 0.00826 | 0.873 ± 0.026 |
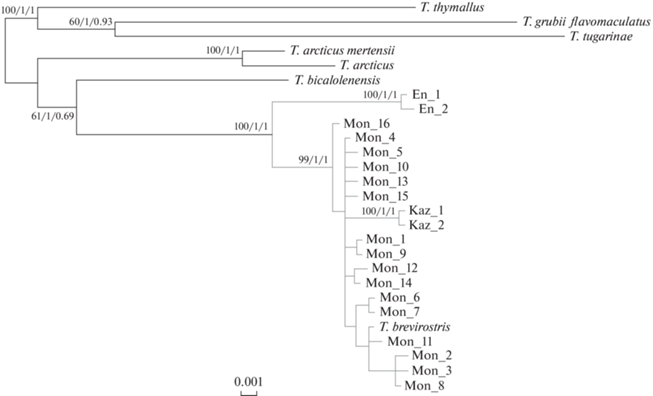
В структуре сетей гаплотипов монгольского хариуса прослеживается звездообразная топология (рис. 2), наиболее четко такая структура сети выявляется по гену СО1 с центральным наиболее часто встречающимся гаплотипом Mon_1 (рис. 2б). При построении деревьев разными методами топология основных узлов сохранялась (рис. 3). Гаплотипы, полученные из выборки Черного Иртыша, образовали внутреннюю ветвь в кластере монгольского хариуса (рис. 3). Гаплотипы верхнеенисейского хариуса образуют сестринскую ветвь по отношению к монгольскому с максимальной поддержкой (100/1/1) в узле ветвления, ближайшую ветвь к кластеру монгольско-верхнеенисейского хариусов образует байкалоленский хариус (T. baicalolenensis) (рис. 3).
Рис. 3.
Дерево гаплотипов, построенное на основе объединенных последовательностей CR и CO1 мтДНК. Значения бутстреп поддержки и вероятности указаны в соответствующих узлах. Mon – гаплотипы T. brevirostris из р. Кобдо, Kaz – T. brevirostris из р. Черный Иртыш, En – T. svetovidovi из р. Большой Енисей.
В матрице дистанций К2Р по баркодинговому участку гена CO1 наибольшая дистанция (0.064) была между T. tugarinae и T. thymallus, наименьшие дистанции (0.006) – между T. arcticus и T. arcticus mertensii и между T. brevirostris и T. brevirostris (Kaz), дистанция между T. brevirostris и T. svetovidovi – 0.017 (табл. 3).
Таблица 3.
Матрица средних попарных дистанций Кимуры (K2P) между гаплотипами CO1. T. brevirostris (Kaz) – хариус из реки Черный Иртыш (Казахстан)
| 1 | T. svetovidovi | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| 2 | T. brevirostris | 0.017 | |||||||
| 3 | T. brevirostris (Kaz) | 0.017 | 0.006 | ||||||
| 4 | T. baicalolenensis | 0.034 | 0.027 | 0.028 | |||||
| 5 | T. arcticus_mertesii | 0.042 | 0.035 | 0.032 | 0.028 | ||||
| 6 | T. grubii_flavomaculatus | 0.051 | 0.051 | 0.049 | 0.043 | 0.048 | |||
| 7 | T. thymallus | 0.043 | 0.046 | 0.046 | 0.042 | 0.043 | 0.051 | ||
| 8 | T. arcticus | 0.041 | 0.038 | 0.035 | 0.028 | 0.006 | 0.048 | 0.043 | |
| 9 | T. tugarinae | 0.063 | 0.057 | 0.057 | 0.048 | 0.054 | 0.052 | 0.064 | 0.057 |
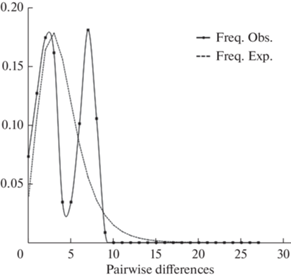
На диаграмме распределения частот попарных различий гаплотипов монгольского хариуса наблюдается бимодальное распределение (рис. 4). Расчет индексов популяционной экспансии для монгольского хариуса – SSD = 0.0132 (P > 0.05), r = 0.0185 (P > 0.05), D = −0.504 (P > 0.05), FS = = −7.3998 (P < 0.01), суммарно эти значения указывают на численную экспансию вида.
Рис. 4.
График соответствия распределения частот попарных нуклеотидных различий T. brevirostris модели экспансии вида.
В данном исследовании для большинства попарных сравнений дистанции между видами хариусов выше, чем 0.027 (табл. 3). Рассчитанные попарные дистанции были ниже формальных для вида (1%) для двух сравнений: пары T. brevirostris из реки Кобдо и T. brevirostris (Kaz) из Черного Иртыша; пары T. arcticus и T. arcticus mertensii – по 0.006 (табл. 3). При этом гаплотипы хариусов из реки Черный Иртыш находятся внутри кластера гаплотипов монгольского хариуса и не формируют внешнюю самостоятельную ветвь на дендрограмме (рис. 3). Таким образом, на основе анализа мтДНК выборки из Черного Иртыша относятся к монгольскому хариусу T. brevirostris.
Анализ мтДНК выборок из притоков Верхнего Енисея показал, что выявленные гаплотипы были идентичны или отличались на одну замену от уже известных для этого вида. При этом, для верхнеенисейского хариуса известно экстремально низкое число гаплотипов: только три гаплотипа, вместе с полученными в нашей работе. Все это свидетельствует об “эффекте основателя” и “бутылочном горлышке”, т.е. значительном сокращении вида и происхождении от небольшого количества предков после разделения ареалов с монгольским хариусом и относительно недавнем формировании вида.
В выборке монгольского хариуса выявлен высокий уровень генетического разнообразия (табл. 1, 2), и при сопоставимом с верхнеенисейским объеме выборки у исследованных рыб было выявлено значительно большее количество гаплотипов. При этом звездообразная топология сети гаплотипов, особенно по СО1 (рис. 3), и соответствие рассчитанных параметров модели численной экспансии вида (рис. 4) свидетельствуют о произошедшем в прошлом сокращении, а затем увеличении численности монгольского хариуса. В исследованных нами выборках хариусов показатели генетического разнообразия были почти в полтора-два раза выше для контрольного региона, чем для фрагмента CO1 (табл. 2). При этом количество замен между гаплотипами монгольского и верхнеенисейского хариусов по обоим фрагментам были сопоставимы (рис. 2). Рассчитанная в нашем исследовании дистанция между верхнеенисейским и монгольским хариусами составила 0.017 и совпала с дистанциями, полученными на основе CR в работе Вайса с коллегами Weiss et al., 2020, что выше, чем формальные внутривидовые границы. Кроме того, высокий уровень поддержки в узле ветвления монгольского и верхненисейского хариусов подтверждает хорошее разделение этих видов.
Таким образом, анализ мтДНК выборок хариуса из притоков Большого Енисея показал обитание верхнеенисейского хариуса в российской части ареала. В верховьях Оби (река Черный Иртыш, Казахстан) подтверждено обитание монгольского хариуса.
Благодарности. Авторы искренне признательны О.О. Колачевской, Н.В. Гордеевой и Г. Шалгимбаевой за помощь в сборе материала.
Список литературы
Берг Л.С. Рыбы пресных вод СССР и сопредельных стран. 4-е изд. Ч. 1. М.; Л.: Изд-во Акад. наук СССР, 1948. 465 с.
Гундризер А.Н. О новом подвиде сибирского хариуса из бассейна Большого Енисея // Учен. зап. /Том. ун-т/. 1967. Вып. 53: Биология и почвоведение. С. 79–94.
Гундризер А.Н. Зубастый сибирский хариус – Thymallus arcticus Dentatus Subsp. Nova // Новые данные о фауне и флоре Сибири. Томск: Изд-во Том. ун-та, 1979. С. 15–22.
Книжин И.Б., Вайс С.Дж. Новый вид хариуса Thymallus svetovidovi sp. nova (Thymallidae) из бассейна Енисея и его положение в роде Thymallus // Вопросы ихтиологии. 2009. Т. 49. № 1. С. 5–14.
Световидов А.Н. Европейско-азиатские хариусы (genus Thymallus Cuvier) // Тр. Зоол. ин-та АН СССР. 1936. Т. 3. С. 183–301.
Bandelt H.J., Forster P., Röhl A. Median-joining networks for inferring intraspecific phylogenies // Mol. Biol. Evol. 1999. V. 16. P. 37–48.
Dyldin Yu.V., Hanel L., Romanov V.I., Plesnik J. A review of the genus Thymallus (Pisces: Salmoniformes, Salmonidae, Thymallinae) with taxonomic notes // Bulletin Lampetra. 2017. V. 8. P. 103–126.
Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. // System. Biology. 2010. V. 59. Iss. 3. P. 307–321.
Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood // System. Biology. 2003. V. 52. P. 696–704.
Excoffier L., Lischer H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows // Mol. Ecol. Res. 2010. V. 10. P. 564–567.
Froufe E., Knizhin I., Weiss S. Phylogenetic analysis of the genus Thymallus (grayling) based on mtDNA control region and ATPase 6 genes, with inferences on control region constraints and broad-scale Eurasian phylogeography // Mol. Phylogen. Evol. 2005. V. 34. P. 106–117.
Ivanova N., Zemlyak T., Hanner R., Hebert P. Universal primer cocktails for fish DNA barcoding // Mol. Ecol. Notes. 2007. V. 7. P. 544–548.
Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences // J. Mol. Evol. 1980. V. 16. P. 111–120.
Kottelat M. Fishes of Mongolia: a check-list of the fishes known to occur in Mongolia with comments on systematics and nomenclature. World Bank Report (NEMO). Washington, D.C.: World Bank Group, 2006. 103 p.
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms // Mol. Biol. Evol. 2018. V. 35. P. 1547–1549.
Lefort V., Longueville J.-E., Gascuel O. SMS: Smart Model Selection in PhyML // Mol. Biol. Evol. 2017. V. 34. Iss. 9. P. 2422–2424.
Librado P., Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data // Bioinformatics. 2009. V. 25. P. 1451–1452.
Ratnasingham S., Hebert P.D.N. BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). // Mol. Ecol. Notes. 2007. V. 7. P. 355–364.
Rogers A.R., Harpending H. Population growth makes waves in the distribution of pairwise genetic differences // Mol. Biol. Evol. 1992. V. 9. P. 552–569.
Romanov V.I. On the taxonomic composition of the graylings (Thymallidae) from the Ob and Yenisei River Basins // Intern. J. Environ. Studies. 2017. V. 74. P. 845–853.
Uiblein F., Jagsch A., Honsig-Erlenburg W., Weiss S. Status, habitat use, and vulnerability of the European grayling in Austrian waters // J. Fish Biology. 2001. V. 59. P. 223–247.
Villesen P. FaBox: an online toolbox for fasta sequences // Mol. Ecol. Notes. 2007. V. 7. Iss. 6. P. 965–968.
Weiss S., Grimm J., Gonçalves D.V., Secci-Petretto G., Englmaier G.K., Baimukanov M., Froufe E. Comparative genetic analysis of grayling (Thymallus spp. Salmonidae) across the paleohydrologically dynamic river drainages of the Altai-Sayan mountain region // Hydrobiologia. 2020. V. 847. P. 2823–2844.
Дополнительные материалы
- скачать ESM.docx
- Приложение 1. Распределение гаплотипов с номерами Генбанка по выборкам
Инструменты
Известия РАН. Серия биологическая