

Известия РАН. Серия физическая, 2020, T. 84, № 5, стр. 651-653
Молекулярно-динамическое моделирование возбуждения валентного колебания OH-группы фенола
Г. П. Михайлов *
Федеральное государственное бюджетное образовательное учреждение высшего образования
“Уфимский государственный авиационный технический университет”
Уфа, Россия
* E-mail: gpmihailov@mail.ru
Поступила в редакцию 28.11.2019
После доработки 19.12.2019
Принята к публикации 27.01.2020
Аннотация
Методом молекулярной динамики c использованием квантово-классического подхода проведены расчеты времени перераспределения кинетической энергии атомов по внутримолекулярным колебательным модам (нижней границы V-V-обмена энергией) молекулы фенола в зависимости от энергии возбуждения валентного колебания гидроксильной группы.
ВВЕДЕНИЕ
Эффект внутримолекулярного колебательного перераспределения (intramolecular vibrational redistribution, IVR) – фундаментальное явление, присущее любой многоатомной молекуле, если она достаточно сильно возбуждена [1]. Несмотря на определяющую роль экспериментальных методов исследования IVR [2–4], важны методы молекулярной динамики (МД) позволяющие получать уникальную информацию о процессах IVR [5–8]. Результаты расчетов методом МД зависят от выбора внутримолекулярных силовых полей, и поэтому важно использование квантово-классических подходов. Цель работы – исследование временной эволюции флуктуаций кинетической составляющей колебательной энергии молекулы фенола от степени колебательного возбуждения валентного колебания гидроксильной группы и оценка нижней границы времени внутримолекулярного обмена энергией между модами (τVV).
МЕТОДИКА РАСЧЕТОВ
C использованием программного пакета Hyper Chem [9] был реализован квантово-классический подход, состоящий в решении уравнения Шредингера методом Хартри–Фока в полуэмпирической параметризации PM3 на каждом временном шаге метода МД. Предварительно проводилось моделирование при T = const (термостат Берендсена, значение постоянной времени релаксации 0.1 пс и временной шаг 0.5 фс) оптимизированной структуры молекулы фенола для достижения температуры 300 K. Далее задавали начальное возмущение длины связи O–H и в режиме сохранения полной энергии рассчитывали суммарную кинетическую энергию колебаний Ek всех атомов (энергии поступательного движения молекулы и вращения вокруг центра масс устанавливались равными нулю) в течение нескольких десятков пикосекунд. Флуктуации полной энергии не превышали 0.01% от равновесного значения. Cреднеквадратичная относительная флуктуация энергии Ek определяется выражением [10]
(1)
${{{{{\left( {D\left[ {{{E}_{k}}} \right]} \right)}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}} \mathord{\left/ {\vphantom {{{{{\left( {D\left[ {{{E}_{k}}} \right]} \right)}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}} {\left\langle {{{E}_{k}}} \right\rangle }}} \right. \kern-0em} {\left\langle {{{E}_{k}}} \right\rangle }} = {{{{{\left( {{{C}_{V}}k{{T}^{2}}} \right)}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}} \mathord{\left/ {\vphantom {{{{{\left( {{{C}_{V}}k{{T}^{2}}} \right)}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}} {\left\langle {{{E}_{k}}} \right\rangle }}} \right. \kern-0em} {\left\langle {{{E}_{k}}} \right\rangle }},$(2)
${{N}_{{vib}}}(t) = {{\left( {2{{{\left\langle {{{E}_{k}}} \right\rangle }}^{2}}} \right)} \mathord{\left/ {\vphantom {{\left( {2{{{\left\langle {{{E}_{k}}} \right\rangle }}^{2}}} \right)} {D\left[ {{{E}_{k}}} \right]}}} \right. \kern-0em} {D\left[ {{{E}_{k}}} \right]}}.$Разбивая временной интервал на подынтервалы и рассчитывая D[Ek], на каждом из них можно получить зависимость числа возбужденных колебательных мод от времени [11] и провести оценку момента времени, при котором возбуждено полное число колебательных мод 3N – 6, т.е. получить минимальное значение времени τVV.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В процессе возбуждения выделяемого колебания после превышения некоторой граничной величины Est (Est – граница стохастизации, см., например, [3]) запасенная в нем энергия равновесным образом перераспределяется по остальным колебаниям молекулы. Анализ матрицы нормальных координат молекулы фенола показал, что вклад растяжения связи O–H в ν(OH) (3880 см–1) составляет 92.8%. Изменение длины связи O–H позволяет задавать значение энергии возбуждения, используя зависимость энергии связи O–H от межатомного расстояния (рис. 1). Полученная зависимость хорошо аппроксимируется ангармоническим потенциалом Морзе:
где De = 4.1 эВ – энергия диссоциации, α = = 2.17 Å–1 – параметр, определяющий кривизну потенциала в минимуме. С течением времени длина связи O–H убывает и стремится к равновесному значению. С другой стороны, наблюдается возбуждение связей C–H с заметной задержкой по времени. С течением времени дисперсия суммарной кинетической энергии колебаний уменьшается (рис. 2а) и увеличивается число возбужденных колебаний (рис. 2б), рассчитанное по формуле (2). Анализ зависимости числа возбужденных колебаний от времени позволяет выделить две характерные области, а именно, область возбуждения 3N – 6 колебаний и переход в область колебательного квазиконтинуума, в которой происходит стохастизация колебательного движения. Оценена энергия стохастизации Est для молекулы фенола, примерно 1.0 эВ, т.е. низкоэнергетическая граница перехода в область колебательного квазиконтинуума. Для частично замещенной дейтерием молекулы фенола энергия стохастизации равна 1.2 эВ. Найденные значения энергии стохастизации молекулы фенола находятся в соответствии с величинами Est для многоатомных молекул (4–15 атомов), которые составляют (3–8) ∙ 103 см–1 (0.4–1 эВ) [3]. В табл. 1 представлены рассчитанные значения времени перераспределения кинетической энергии по внутримолекулярным колебательным модам молекул фенола C6H5OH и частично замещенного дейтерием фенола C6D5OH в зависимости от энергии колебательного возбуждения связи O–H, которая определялась как разница полных энергий 〈Etot〉 возбужденной молекулы и молекулы при T = 300 K. С увеличением энергии ∆U время τVV для молекул C6H5OH, C6D5OH уменьшается. Времена IVR из высокочастотных валентных колебаний связи O–H молекул C6H5OH и C6D5OH, полученные методом двойного ИК-УФ-резонанса с временным разрешением, равны, соответственно, 14 и 80 пс [12]. Различие между рассчитанными значениями τVV и экспериментальными временами IVR обусловлено, вероятнее всего, регистрацией в эксперименте дополнительных каналов перераспределения колебательной энергии, а именно, колебательно-поступательного (VT-) и колебательно-вращательного (VR-) обменов.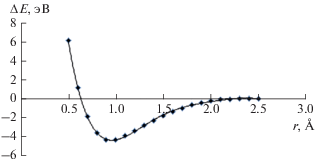
Рис. 2.
Зависимость дисперсии кинетической энергии атомов (а) и числа возбужденных колебательных мод в молекуле C6H5OH (б) от времени (энергия возбуждения ∆U = 1 эВ).

Таблица 1.
Рассчитанные энергии возбуждения ∆U, равновесные значения кинетической 〈Ek〉, потенциальной 〈Epot〉, полной 〈Etot〉 энергий (энергии приводятся в эВ), колебательной температуры 〈Tvib〉 и времени перераспределения кинетической энергии по колебательным модам τVV
| Молекула | ∆U | 〈Tvib〉, K | 〈Ekin〉 | 〈Epot〉 | 〈Etot〉 | τVV, пс |
|---|---|---|---|---|---|---|
| C6H5OH | – | 300 | 0.5 | –61.2 | –60.7 | – |
| 1.0 | 580 | 1.0 | –60.7 | –59.7 | 12 ± 1 | |
| 1.4 | 730 | 1.2 | –60.5 | –59.3 | 11 ± 1 | |
| 2.1 | 910 | 1.5 | –60.2 | –58.6 | 9.6 ± 1 | |
| 2.5 | 1030 | 1.7 | –60.0 | –58.2 | 5.0 ± 1 | |
| С6D5OH | – | 300 | 0.5 | –61.5 | –61.0 | – |
| 1.3 | 600 | 1.0 | –60.7 | –59.7 | 27 ± 2 | |
| 1.8 | 745 | 1.3 | –60.4 | –59.2 | 14 ± 2 | |
| 2.4 | 900 | 1.5 | –60.2 | –58.6 | 11.5 ± 2 |
ЗАКЛЮЧЕНИЕ
Проведено исследование временной эволюции флуктуаций кинетической составляющей колебательной энергии на примере молекулы фенола. Установлено, что время τVV фенола уменьшается при росте энергии колебательного возбуждения OH-группы в результате ангармоничности и интерференции колебаний и значительно увеличивается для частично замещенной дейтерием молекулы фенола.
Работа выполнена при финансовой поддержке Минобрнауки России в рамках реализации проекта № 16.1969.2017/4.6.
Список литературы
Макаров А.А., Малиновский А.Л., Рябов Е.А. // Изв. РАН. Сер. физ. 2008. Т. 75. № 5. С. 734; Makarov A.A., Malinovsky A.L., Ryabov E.A. // Bull. Russ. Acad. Sci. Phys. 2008. V. 72. № 5. P. 698.
Yoo H.S., McWhorter D.A., Pate B.H. // J. Phys. Chem. A. 2004. V. 108. P. 1380.
Макаров А.А., Малиновский А.Л., Рябов Е.А. // УФН. 2012. Т. 182. С. 1047; Makarov A.A., Malinovsky A.L., Ryabov E.A. // Phys. Usp. 2012. V. 55. P. 977.
Компанец В.О., Лохман В.Н., Пойдашев Д.Г. и др. // ЖЭТФ. 2016. Т. 149. С. 723; Kompanets V.O., Lokh-man V.N., Poydashev D.G.// JETP. 2016. V. 122. P. 621.
Käb G., Schröder C., Schwarzer D. // Phys. Chem. Chem. Phys. 2002. V. 4. P. 271.
Bastida A., Soler M.A., Zunig J. et al. // J. Phys. Chem. A. 2010. V. 114. P. 11450.
Nguyen P.H., Park S., Stock G. // J. Chem. Phys. 2010. V. 132. Art. № 025102.
Farag M.H., Bastida A., Ruiz-Lopez M.F. et al. // J. Phys. Chem. B. 2014. V. 118. P. 6186.
http://www.hyper.com/.
Ландау Л.Д., Лифшиц Е.М. Статистическая физика. Ч. 1. М.: Физматлит, 2001. 616 с.
Селезнев А.А., Алейников А.Ю., Бригинас И.В. // Хим. физика. 2008. Т. 27. С. 5; Selezenev A.A., Aleinikov A.Yu., Briginas I.V. // Russ. J. Phys. Chem. В. 2008. V. 2. P. 147.
Yamada Y., Ebata T., Kayano M., Mikami N. // J. Chem. Phys. 2004. V. 120. P. 7400.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Серия физическая