

Известия РАН. Серия физическая, 2020, T. 84, № 7, стр. 1049-1056
Влияние высокоэнергетического помола на фазово-структурное состояние и магнитные свойства смеси порошков железа и нитрида бора
В. П. Менушенков 1, *, И. О. Минкова 1, И. В. Дорофиевич 1, И. В. Щетинин 1, Д. Г. Жуков 1, Ю. Н. Пархоменко 1, Е. А. Скрылева 1, А. Г. Савченко 1
1 Федеральное государственное автономное образовательное учреждение высшего образования
“Национальный исследовательский технологический университет МИСиС”
Москва, Россия
* E-mail: menushenkov@gmail.com
Поступила в редакцию 18.02.2020
После доработки 16.03.2020
Принята к публикации 27.03.2020
Аннотация
Исследовано влияние высокоэнергетического помола (механического сплавления, МС) смеси порошков железа и гексагонального нитрида бора (h-BN) при весовом отношении компонентов Fe : BN = 1 на фазовый состав и магнитные свойства синтезированного материала. Показано, что высокоэнергетический помол и последующий отжиг МС-порошка при 600°С приводят к существенному изменению его фазового состава, структуры и магнитных характеристик.
ВВЕДЕНИЕ
Среди известных соединений железа с азотом: Fe8N, Fe4N, Fe3N, Fe2N, FeN первые три нитрида являются ферромагнетиками и обладают высокими значениями намагниченности. Более того, магнитные моменты железа в упорядоченной фазе α''-Fe16N2 и гипотетическом соединении Fe3N4 (3.2µВ [1] и 3.26µВ [2] соответственно, µВ – магнетон Бора), заметно превышающие магнитный момент чистого железа (2.22µВ), обусловливают повышенный интерес исследователей к этим нитридам как к перспективным материалам для изготовления постоянных магнитов.
Широко применяемая схема азотирования, основанная на взаимодействии тонкой пленки или дисперсных частиц железа с газовой средой NH3 или Н2/NH3, обладает серьезным недостатком, связанным с локализацией зоны с повышенным содержанием азота в приповерхностном слое образцов. Для получения объемных материалов на основе соединений железа с азотом альтернативным методом азотирования может служить спекание брикетов из смесей порошков Fe и нитридов BN, AlN или Si3N4 при температурах до 1550°С. В работах [3–5] показано, что совместное влияние азота и бора на формирование нитридов и боридов железа в процессе спекания позволяет получать магнитные материалы, обладающие высокими значениями магнитных характеристик, в том числе коэрцитивной силой.
Еще одним способом введения азота в сплавы на основе железа является высокоэнергетический помол (механическое сплавление, МС) смесей порошков железа или исходного сплава с порошком нитрида BN [6–8]. При измельчении таких смесей в высокоэнергетической мельнице происходит не только уменьшение размера частиц, но и развиваются механохимические процессы, приводящие к формированию нитридов и боридов железа. Например, в работе [6] при измельчении смеси Fe и нитрида бора в атмосфере аргона был получен нанокомпозит ε-FexN/BN, состоящий из нитрида железа, внедренного в немагнитную аморфную матрицу из нитрида бора. При этом, по мнению авторов, критический размер частиц α-Fe, вступающих в реакцию с азотом, составляет ≈8 нм. Аналогичный метод азотирования был использован в работах [7, 8], результаты которых отличаются тем, что в [7] был получен борид железа, а не нитрид [8]. Следует отметить, что результаты, приведенные в статьях [6–8], были получены при различных комбинациях состава смесей, длительности МС и режимов последующих отжигов: в частности, весовое соотношение порошков Fe : BN изменялось в диапазоне от 1 : 1 [7] до 1 : 12.5 [8], отношение веса шаров к весу порошка – от 15 : 1 [6, 8] до 6 : 1 [7], а энергетические условия помола приведены лишь в работе [7]. Использование различных режимов МС затрудняет проведение анализа и сопоставление полученных результатов и, как следствие, мешает выявлению закономерностей формирования фазового состава и свойств синтезированных материалов. Более того, в работах [6–8] не приводятся магнитные характеристики полученных образцов, хотя одной из целей этих исследований было получение и исследование магнитного материала на основе нитридов железа с особыми магнитными свойствами.
В свете вышесказанного, целью настоящей работы является исследование фазового состава, структуры и магнитных свойств материала, полученного путем механического сплавления в высокоэнергетической планетарной шаровой мельнице смеси порошков Fe и h-BN с весовым отношением Fe : BN = 1 после извлечения его из кармана мельницы на воздух и после отжига этого порошка при температуре 600°С в атмосфере азота.
МАТЕРИАЛ И МЕТОДИКА ИСЛЕДОВАНИЯ
В качестве исходных материалов были использованы: железный порошок ПЖР 3.200.28 (ГОСТ 9849-86), нитрид бора гексагональный марки Т (состав: h-BN – 97%; B2O3 – 0.2%). Измельчение смеси порошков и последующий отжиг проводили в азоте особой чистоты (99.996%), аргоне высшего сорта или в вакууме.
Высокоэнергетический помол чистых порошков Fe и h-BN, а также их смесь с весовым отношением Fe : BN = 1 (в табл. 1 приведены состав смеси в массовых и атомных долях), проводили в высокоэнергетической планетарной шаровой мельнице “Активатор 2S”. Отношение веса шаров к весу порошка – 15 : 1, длительность помола составляла 1–60 ч. Прессование МС-порошков проводили в металлической прессформе при давлении 40 МПа. Пресс-заготовки из МС-порошков отжигали в вакуумной печи сопротивления в атмосфере азота при температуре 600°C в течение 2 ч.
Таблица 1.
Химический состав исходной смеси порошков Fe и BN
| Вес. отношение Fe/BN |
Fe, масс. доля | B, масс. доля | N, масс. доля | Fe, ат. доля | B, ат. доля | N, ат. доля |
|---|---|---|---|---|---|---|
| 1 | 50.0 | 21.8 | 28.2 | 18.2 | 40.9 | 40.9 |
Химический состав МС-порошков определяли на рентгенофлуоресцентном спектрометре PRIMUSII. Рентгеноструктурный фазовый анализ (РФА) проводили на дифрактометре ДРОН-4 в CoKα-излучении. Для проведения качественного и количественного фазового анализа использовали пакет программных продуктов [9 ] .
Микроструктуру и элементный состав МС-порошков изучали на прессованных образцах с помощью растрового электронного микроскопа (РЭМ) JEOL JSM-6610LV и энергодисперсионного спектрометра (ЭДС). Исследование химического состава поверхности порошковых образцов проводили методом рентгеновской фотоэлектронной эмиссии (РФЭС) на приборе PHI 5000 Versa ProbeII с монохроматическим источником AlKα-излучения. Для очистки поверхности и смещения области анализа в объем использовали травление ионами аргона с энергией 2 кэВ в течение 8 мин (скорость травления, определенная на слое SiO2, составляла 10 нм/мин). Атомные концентрации элементов рассчитывали по обзорным спектрам методом факторов относительной элементной чувствительности, химическое состояние элементов определяли по спектрам высокого разрешения, снятым при энергии пропускания анализатора 11.75–29.35 эВ с шагом 0.25 эВ.
Измерения магнитных характеристик проводили при комнатной температуре на вибрационном магнетометре VSM 250 (Dexing Magnet Tech. Co, Ltd) в полях напряженностью до 2.5 Тл с точностью ±2%.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Структура МС-порошка Fe–BN после механосплавления и отжига
На рис. 1 показаны дифрактограммы чистых порошков h-BN и Fe, а также смеси Fe:BN = 1 после МС длительностью 60 ч и после 2 ч отжига МС-порошка Fe:BN = 1 при 600°С. МС порошка чистого Fe приводит к уширению линий и небольшому изменению периода а решетки α-Fe. Так, после MС 60 ч период а увеличивается с 0.2866 до 0.2869 нм (рис. 1а).
Рис. 1.
Дифракционные спектры порошков после МС 60 ч: (а) чистого Fe, (б) чистого BN, (в) порошка Fe:BN = 1 после МС 60 ч и (г) после МС 60 ч и отжига при 600°С. Вертикальные штриховые линии – положение линий α-Fe.
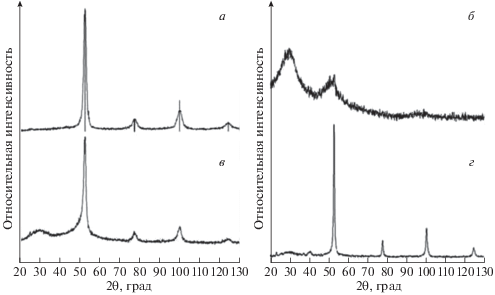
В спектре измельченного порошка BN видны два гало в окрестности углов θ = 30° и 50° (рис. 1б), свидетельствующие о формировании в процессе 60 ч МС рентгено-аморфной фазы (РАФ). В структуре этого образца присутствует также небольшое количество Fe, образовавшегося в результате намола от шаров и стенок стакана мельницы.
В спектре МС-порошка Fe:BN = 1 (рис. 1в) присутствуют только линии α-Fe и гало в области θ ≈ 30°. Период решетки α-Fe в порошке Fe:BN = 1 после 60 ч МС увеличился до 0.2872 нм, что может быть связано с присутствием в решетке Fe не только межузельных атомов (N), но также и атомов B. Ассиметричное уширение линии (110) Fe со стороны малых углов свидетельствует о начальной стадии формирования нитридов Fe.
Отжиг МС-порошка при 600°С в течение 2 ч, как это видно на рис. 1г, приводит к уменьшению количества РАФ (высота и площадь соответствующего гало уменьшаются), сужению линий α-Fe и к появлению в интервале малых углов слабых линий, возможно, от нитридов железа.
На рис. 2 приведены фотографии микроструктуры порошков чистого Fe и чистого h-BN после МС 60 ч, а также смеси порошков Fe:BN = 1 после 60 ч МС, и после 60 ч МС и 2 ч отжига при 600°С, полученные с помощью СЭМ на прессованных образцах. Образец чистого Fe после МС 60 ч (рис. 2а) состоит из частиц со средним размером 1–3 мкм, часть из которых имеет анизотропную форму. В структуре порошка h-BN после МС 60 ч (рис. 2б) видны равноосные частицы, размер которых составляет всего 200–300 нм. Порошок Fe:BN = 1 после 60 ч МС (рис. 2в) характеризуется присутствием слабо различимых частиц Fe (средний размер ~100 нм) на фоне аморфно-подобной матричной фазы. После отжига при 600°С (рис. 2г) порошок характеризуется однородной по размеру частиц структурой, как на рис. 2б.

Элементный состав и химическое состояние элементов в порошках Fe:BN = 1 после механосплавления и отжига
Изучение элементного состава исследуемых порошков после МС с помощью энергодисперсионного спектрометра показало присутствие в них большого количества кислорода: в чистых Fe и BN – около 20 ат. % О, в МС-порошке Fe:BN = 1 (60 ч МС) – около 30 ат. % О. Хотя эти результаты имеют скорее качественный характер, обнаружение присутствия значительного количества кислорода во всех порошках после МС свидетельствует о том, что после их извлечения из стакана шаровой мельницы на активированной поверхности частиц происходит интенсивная адсорбция кислорода и паров воды из атмосферы, сопровождающаяся экзотермическим эффектом.
Подтверждением этому служат результаты рентгеновской фотоэлектронной спектроскопии, полученные на порошке Fe:BN = 1 в исходном, после 60 ч МС состоянии, а также после отжига МС-порошка при 600°С в течение 2 ч. В табл. 2 представлены атомные концентрации элементов, полученные в результате обработки обзорных спектров с использованием значений факторов элементной чувствительности из базы данных программного обеспечения PHI. Концентрация кислорода на поверхности образцов до и после отжига превышает 35 ат. % как в исходном состоянии, так и после ионного травления, которое использовали для очистки поверхности от адсорбированных примесей. Для сравнения в табл. 2 приведены данные для исходного порошка h-BN, в котором содержание адсорбированного кислорода даже без очистки составляет всего 2 ат. %.
Таблица 2.
Концентрации элементов (ат. %) на поверхности МС-порошка Fe:BN = 1 (МС 60 ч) и исходного порошка h-BN
| Образец | Состояние поверхности | Концентрации, ат. % | ||||
|---|---|---|---|---|---|---|
| С | B | N | O | Fe | ||
| Fe/BN = 1 (МС 60 ч) | Исходная | 11.4 | 26.2 | 18.0 | 38.4 | 6.0 |
| После ионного травления | 2.3 | 32.7 | 16.1 | 35.3 | 13.6 | |
| Fe/BN = 1 (МС 60 ч) после отжига | Исходная | 28.1 | 20.2 | 10.8 | 35.2 | 5.7 |
| После ионного травления | 22.8 | 22.5 | 9.6 | 36.6 | 8.4 | |
| Порошок h-BN | Исходная | 6.0 | 45.5 | 46.5 | 2.0 | – |
Анализ спектров высокого разрешения азота (N1s), бора (B1s) и железа (Fe2p), рис. 3 и 4, позволил определить химическое состояние элементов и объяснить высокое содержание кислорода в МС-порошке Fe:BN = 1.
Рис. 3.
Спектры высокого разрешения на исходной поверхности: (a) B1s и (б) N1s порошка h-BN и порошков Fe:BN = 1 до и после отжига при 600°С; (в) спектры Fe2p3 порошков Fe:BN = 1 до и после отжига при 600°С. Сплошные линии – экспериментальные кривые, прерывистые линии – кривые синтетических пиков (красные – Fe3+, зеленые – Fe2+, синие – Fe0).
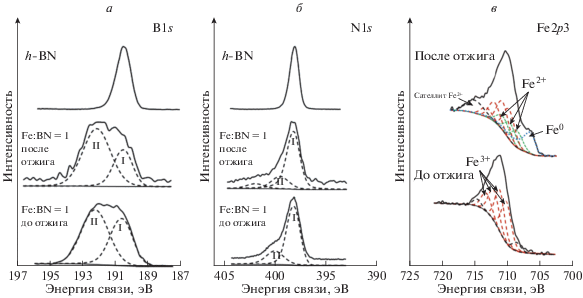
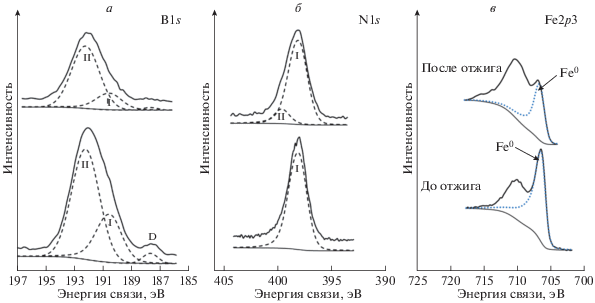
Рис. 4.
Спектры высокого разрешения порошков Fe:BN = 1 после ионного травления: (a) B1s до отжига (верхний) и после отжига при 600°С (нижний), (б) N1s до отжига (верхний) и после отжига при 600°С (нижний); (в) спектры Fe2p3 до и после отжига при 600°С. Сплошные линии – экспериментальные кривые, прерывистые линии – кривые синтетических пиков (синие – Fe0).
Анализ спектров бора B1s
Сравнение концентраций бора и азота в порошке h-BN и в порошках Fe:BN = 1, табл. 2, показывает, что в МС-порошках Fe:BN = 1 до и после отжига отношение концентраций N/B намного меньше, чем в исходном порошке h-BN. При этом заметно изменился и спектр бора B1s (рис. 3а): в обоих МС-порошках Fe:BN = 1 помимо пика I (190.5 эВ) от BN, присутствует пик II в области 192.2 эВ, который, согласно справочным данным [10], можно отнести к оксиду бора B2O3. Причем, интенсивность пика II от оксида бора превышает интенсивность пика I от BN в МС-порошках Fe:BN = 1 до и после отжига (рис. 3а, табл. 3). Ионное травление (рис. 4а), наоборот, приводит к его увеличению, что, свидетельствует об окислении бора, находящегося не только на поверхности, но и в объеме частиц Fe.
Таблица 3.
Энергии связи Есв(эВ) и относительные интенсивности пиков (%) МС-порошка Fe:BN = 1 и исходного порошка h-BN
| Образец | Состояние поверхности | Есв/интен-сивность | N1s | B1s | ||||
|---|---|---|---|---|---|---|---|---|
| I | II | III | D | I | II | |||
| Fe/BN = 1 (МС 60 ч) | Исходная | Есв, эВ | 398.1 | 399.9 | – | – | 190.5 | 192.2 |
| % | 73 | 27 | – | – | 40 | 60 | ||
| После ионного травления | Есв, эВ | 398.1 | – | – | 187.8 | 190.5 | 192.2 | |
| % | 100 | – | – | 3 | 30 | 67 | ||
| Fe/BN = 1 (МС 60 ч) + + отжиг 600оC, 2 ч в азоте | Исходная | Есв, эВ | 398.1 | 399.5 | 401.9 | – | 190.5 | 192.1 |
| % | 68 | 24 | 8 | – | 30 | 70 | ||
| После ионного травления | Есв, эВ | 398.1 | 399.7 | – | 187.8 | 190.5 | 192.2 | |
| % | 88 | 12 | – | 2 | 21 | 77 | ||
| Порошок h-BN | Исходная | Есв, эВ | 398.1 | – | – | – | 190.5 | – |
Анализ спектров азота N1s
На спектрах N1s от поверхности исходного МС-порошка Fe:BN = 1, рис. 3б, кроме пика I (398.1 эВ) от BN присутствует относительно слабый пик II (399.9 эВ), связанный, предположительно, с поверхностными примесями (например, группами –NH, –NH2 и/или CN), так как пик II исчезает после ионного травления МС-порошка или заметно уменьшается после отжига при 600°С.
Анализ спектров железа Fe2p
Высокая концентрация кислорода в исходном МС-порошке Fe:BN = 1 может быть связана не только с окислением бора, но также и с окислением железа. Спектр Fe2p3 от поверхности МС-порошка (рис. 3в) хорошо описывается GS-мультиплетом, известным для метагидроксида FeO(OH) [11, 12], состоящим из 4 основных пиков (710.2; 711.2; 712.1, 713.2 эВ), пика Surf. Peak (714.6 эВ) и пика Pre-Peak (708.7 эВ).
Спектр Fe2p3 от поверхности МС-порошка Fe:BN = 1 после отжига при 600°С расширяется в область более низких энергий и на нем появляется интенсивный пик-сателлит в окрестности Есв = 715 эВ, характерный для оксида Fe3O4 (рис. 3г). Применение GS-мультиплета для Fe2+ (3 пика), GS-мультиплета для Fe3+ (4 пика), пика-сателлита Fe2+, а также одиночного асимметричного пика от Fe0, позволяет достаточно точно описать спектр Fe2p3 (рис. 3в, 3г) и дает количественное распределение долей Fe0, Fe2+ и Fe3+ в соотношении 18, 35 и 47%, соответственно.
После ионного травления (рис. 4в, 4г) часть спектра Fe2p3, соответствующая окисленному состоянию железа, заметно снижается: в МС-порошке Fe:BN = 1 до отжига – до 30% (рис. 4в) и в меньшей степени в отожженном МС-порошке – до 60% (рис. 4г), т.е. сохраняется та же тенденция, что и наблюдавшаяся ранее при окислении бора.
Пик Fe0 (706.8 эВ) соответствует металлическому Fe и после ионного травления МС-порошка Fe:BN = 1 его доля до и после отжига составляет 70 и 40%, соответственно. Однако сопоставление этого пика с чистым Fe не совсем корректно, во-первых, потому, что появление в спектрах B1s после ионного травления слабого пика с Есв = = 187.8 эВ (рис. 4a) может быть связано с образованием борида FeB [10], и во-вторых, возможно образование химических связей Fe–N. Согласно данным [13] Есв(Fe2p3) в Fe8N равна 706.8 эВ, что в точности совпадает с положением пика Fe0.
Дополнительным подтверждением наличия связей Fe–N в МС-порошке Fe:BN = 1 служит оценка отношений атомных концентраций азота и бора (NI/BI), вычисленных из общих концентраций с учетом относительных интенсивностей пиков I в спектрах N1s и B1s. Полученные значения отношения NI/BI превышают номинальную величину, соответствующую порошку h-BN, в 1.3–1.5 раза как на исходных поверхностях МС-порошков Fe:BN = 1 до и после отжига, так и после ионного травления. Это означает, что концентрация азота, рассчитанная по пику I B1s, относится не только к нитриду бора. Можно предположить, что избыточный по отношению к BN азот образует связи Fe–N, указывающие на присутствие в образце Fe8N.
Из данных, приведенных в табл. 3, видно также явное превышение концентраций бора и азота над концентрацией Fe. Причиной этого может быть неоднородность состава по объему частиц, ядро которых состоит из Fe, а оболочку образуют оксид бора B2O3 (или оксинитрид BOxNy [14]) и BN.
Таким образом, анализ вышеприведенных результатов РФС указывает на то, что в процессе МС и последующего взаимодействия МС-порошка с атмосферным кислородом и парами воды, содержащимися в воздухе, в структуре его частиц, помимо α-Fe, присутствуют BN, B2O3 (или BOxNy), FeO(OH), FeB, Fe3O4, а также сохраняются химические связи Fe–N (Fe8N). После отжига при 600°С при распаде метагидроксида FeO(OH) в приповерхностном слое частиц α-Fe формируется оксид Fe3O4.
Обобщение полученных результатов, позволило нам предложить гипотетическую схему структурообразования в процессе МС и последующего отжига МС-порошков Fe:BN = 1 (рис. 5). Во-первых, происходит островковое (в окрестности точек соударения частиц Fe и BN) внедрение в поверхностный слой частиц Fe нитрида бора. Во-вторых, ионы азота из частично разложившегося BN (в процессе диссипации энергии соударения) диффундируют к центру частиц Fe с образованием пересыщенного твердого раствора внедрения в α-Fe, а высвободившийся при разложении BN бор вступает в химическое взаимодействие с поверхностными атомами Fe. Наконец, в-третьих, происходит не только измельчение, но и деформация и накопление дефектов в кристаллических частицах BN, что трансформирует их в рентгено-аморфную фазу.
Рис. 5.
Схемы микроструктуры порошков Fe:BN = 1: (а) после МС 60 ч, (б) после извлечения порошка на воздух, (в) после ионного травления МС-порошков, (г) после отжига МС-порошков при 600°С в азоте.
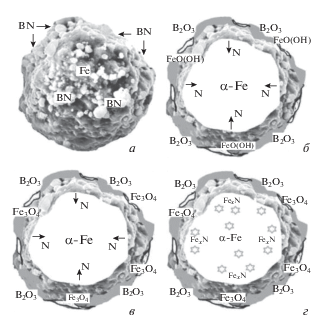
После завершения МС и извлечения МС-порошка Fe:BN = 1 из стакана шаровой мельницы он активно взаимодействует с атмосферным кислородом и парами воды (идет экзотермическая реакция), образуя на поверхности частиц железа оксид (Fe3O4), “шубу” из метагидроксида железа FeO(OH) и островки (возможно, аморфного) оксида бора (B2O3). В процессе ионного травления и отжига при 600°С МС-порошка слабо связанные с поверхностью частиц Fe примесные группы (–NH, –NH2 и/или –CN) удаляются, метагидроксид железа FeO(OH) разлагается с образованием оксида Fe3O4 и воды, и на ней остаются только оксид железа, островки оксида бора, которые на дифракционных спектрах проявляются в виде аморфного гало, а также нитриды Fe, сформировавшиеся в приповерхностном слое частиц железа.
Гистерезисные свойства порошков Fe:BN = 1 после МС и отжига
Значения коэрцитивной силы Hc порошка Fe:BN = 1 после высокоэнергетического помола длительностью 14, 30 и 60 ч, а также размер областей когерентного рассеяния 〈D〉 и величина микродеформации решетки 〈ε〉 железа приведены в табл. 4. Как видно, с увеличением длительности измельчения от 14 до 60 ч коэрцитивная сила монотонно растет и достигает максимума Hc = = 32.5 кА ∙ м–1 (410 Э), при этом величина 〈D〉 монотонно уменьшается от 12 до 8 нм, а 〈ε〉 меняется в пределах 0.4–1.1.
Таблица 4.
Коэрцитивная сила Hc, размер областей когерентного рассеяния 〈D〉 и величина микродеформации решетки железа 〈ε〉 в зависимости от длительности МС порошков Fe:BN = 1
| Hc, кА/м (Э) | 〈D〉, нм | 〈ε〉, % | Hc, кА/м (Э) | 〈D〉, нм | 〈ε〉, % | Hc, кА/м (Э) | 〈D〉, нм | 〈ε〉, % | |
|---|---|---|---|---|---|---|---|---|---|
| МС 14 ч | МС 30 ч | МС 60 ч | |||||||
| Fe:BN = 1 | 24.7 (310) | 12 | 0.4 | 29.5 (370) | 11 | 0.6 | 32.5 (408) | 8 | 1.1 |
На рис. 6 показаны петли магнитного гистерезиса МС-порошков Fe:BN = 1 в исходном состоянии и после 2 ч отжига в азоте при 600°С. Как видно, после отжига наблюдается возрастание всех гистерезисных характеристик: коэрцитивной силы Hc – с 32.5 (408 Э) до 42 кА ∙ м–1 (525 Э), удельной намагниченности насыщения σs – с 40.6 до 65.7 А · м2 ∙ кг–1, удельной остаточной намагниченности σr – с 9 до 15 А · м2 ∙ кг–1. Судя по среднему размеру частиц Fe (≈100 нм) в МС-порошке Fe:BN = 1 (рис. 2а), на порядок превышающему критический размер однодоменности для чистого железа (dкр = 15–25 нм [15]), механизм их магнитного твердения может быть связан с закреплением доменных границ (ДГ), о чем свидетельствует вид кривых намагничивания и форма петель гистерезиса (рис. 5).
Рис. 6.
Петли гистерезиса порошков Fe:BN = 1: (а) после МС 60 ч, (б) после отжига МС-порошка при 600°C в азоте.
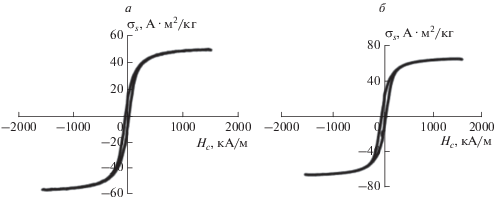
Основываясь на результатах проведенного выше фазово-структурного анализа МС-порошков Fe:BN = 1, можно заключить, что основными причинами магнитного твердения порошка магнитомягкого железа (Hc = 3.3 кА ∙ м–1 (40 Э) до МС) в результате его МС совместно с порошком BN, включая последующее взаимодействие МС-порошка с атмосферным кислородом и парами воды при его извлечении из стакана шаровой мельницы, являются преимущественно неравновесные дефекты структуры (дислокации, вакансии, внутренние напряжения, поверхностные примеси), появляющиеся в процессе МС, а также изменение фазово-структурного состояния, приводящее к формированию на поверхности и приповерхностном слое частиц железа центров закрепления доменных границ.
В МС-порошках после отжига при 600°C, согласно приведенной схеме (рис. 5), доминирующими центрами закрепления доменных стенок служат наночастицы оксидов Fe, появляющиеся в результате разложения метагидроксида FeO(OH), и, что более вероятно, наночастицы нитридов Fe, образующиеся в результате распада пересыщенного твердого раствора азота в Fe и располагающиеся преимущественно в приповерхностном слое частиц Fe.
ЗАКЛЮЧЕНИЕ
Выполненные в работе исследования показывают, что высокоэнергетический помол длительностью до 60 ч в планетарной шаровой мельнице смесей порошков Fe и h-BN приводит к развитию двух процессов: 1) физическому измельчению частиц Fe и BN (средний размер частиц Fe в МС-порошке Fe:BN = 1 составляет ≈100 нм); 2) взаимодействию механоактивированных частиц Fe и BN друг с другом, а также с кислородом и парами воды, содержащимися в воздухе, в процессе извлечения порошка из стакана мельницы. Это взаимодействие приводит, во-первых, к формированию на поверхности частиц Fe химических связей типа Fe–N, Fe–B и Fe–O, которые можно ассоциировать с формированием нитридов и боридов Fe, а также метагидроксида FeО(OH). Кроме того, вследствие взаимодействия бора с кислородом происходит образование аморфного оксида B2O3, а в результате распада гидроксида FeО(OH) в процессе отжига МС-порошка могут образовываться оксиды железа FeOх (в том числе Fe3O4).
На порошке Fe:BN = 1 после МА 60 ч получена Hc = 32.5 кА ∙ м–1 (410 Э), которая после отжига при 600°С возрастает до 42 кА ∙ м–1 (525 Э). Анализ кривых намагничивания и формы петель гистерезиса показывает, что механизм перемагничивания МС-порошков связан с закреплением ДГ на дефектах структуры (неравновесные дефекты структуры после МС), или на наночастицах оксидов и нитридов, формирующихся в приповерхностном слое частиц железа в процессе отжига МС-порошков.
Работа выполнена при финансовой поддержке Российского научного фонда в рамках мероприятия “Проведение исследований научными группами под руководством молодых ученых” Президентской программы исследовательских проектов, реализуемых ведущими молодыми учеными (проект № 18-72-10161).
Список литературы
Chen S.K., Jin S., Kammlott G.W. // J. Magn. Magn. Mater. 1992. V. 110. P. 65.
Ching W.Y., Xu P.Y. // J. Appl. Phys. Lett. 2002. V. 80. № 2. P. 2904.
Минкова И.О., Менушенков В.П., Савченко Е.С., Железный М.В. // МиТОМ. 2018. № 8. С. 52.
Minkova I.O., Menushenkov V.P., Savchenko A.G., Minkov O.B. // Mater. Res. Innovat. 2019. V. 23. № 7. P. 422.
Комков Н.Т., Менушенков В.П., Минкова И.О. и др. Способ легирования железа азотом. Пат. РФ № 2665658. 2018.
Liu L., Bin Y., Hongyanet W. et al. // Chin. Sci. Bull. 1998. V. 43. № 6. P. 467.
Bokhonov B., Korchagin M., Borisova Yu. // J. Alloys Compounds. 2004. V. 372. P. 141.
Tao J.G., Yao B., Yang J.H. et al. // J. Alloys Compounds. 2004. V. 384. P. 268.
Шелехов Е.В., Свиридова Т.А. // МиТОМ. 2000. № 8. С. 16.
http://srdata.nist.gov/xps .
Gupta R.P., Sen S.K. // Phys. Rev. B. 1975. V. 12. № 1. P. 15.
Grosvenor A.P., Kobe B.A., Biesinger M.C., McIntyre N.S. // Surf. Interface Anal. 2004. V. 36. P. 1564.
Torres J., Perry C.C., Bransfield St.J., Fairbrother D.H. // J. Phys. Chem. B. 2003. V. 107. P. 5558.
Grafouté M., Petitjean C., Diama A. et al. // Surf. Coat. Techn. 2015. V. 272. P. 158.
Кондорский Е.И. // Изв. АН СССР. Сер. физ. 1978. Т. 42. № 8. С. 1638.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Серия физическая