

Известия РАН. Серия физическая, 2020, T. 84, № 9, стр. 1246-1250
Релаксационные процессы при тепловом воспламенении в реакторе проточного типа
А. Т. Пономаренко 1, *, В. Г. Шевченко 1
1 Федеральное государственное бюджетное учреждение науки Институт синтетических полимерных материалов имени Н.С. Ениколопова Российской академии наук
Москва, Россия
* E-mail: anapon@ispm.ru
Поступила в редакцию 10.04.2020
После доработки 29.04.2020
Принята к публикации 27.05.2020
Аннотация
Продемонстрирована возможность определения кинетических параметров реакции, протекающей в реакторе с неподвижным слоем катализатора, по критическим условиям воспламенения. Детально описано устройство измерительного реактора и следствия явления воспламенения. Предложенный подход применим также к соответствующим вариантам синтезов Фишера–Тропша с целью синтеза углеводородов, а также других синтезов с неподвижным слоем катализатора и высокими энергиями активации, с его помощью легко определять активность катализаторов.
ВВЕДЕНИЕ
Каталитические синтезы из СО и водорода в настоящее время рассматриваются как альтернативные варианты получения топлив, смазок, присадок из попутных при добыче нефти газов, выделяемых до 100 м3/т добываемой нефти и, по мнению Д.И. Менделеева “сжигание продуктов нефтедобычи то же самое, что топить ассигнациями”. Не удивительно поэтому, что открытый в 1926 г. Ф. Фишером и Г. Тропшем синтез углеводородов из СО и водорода в настоящее время находится на четвертом поколении развития и интенсивно продолжаются работы по его совершенствованию [1]. Отличительной особенностью этих процессов является их высокая энергонапряженность и по этой причине весьма важны исследования макрокинетики таких синтезов. Уместно привести суждение Д.А. Франк-Каменецкого о том, что “ряд важных каталитических процессов наивыгоднейшим образом протекают в неустойчивых режимах, поскольку скорость реакции в нижнем устойчивом режиме очень мала, а в верхнем устойчивом режиме велик выход нежелательных продуктов реакции. Поэтому целесообразно проводить синтез в среднем неустойчивом температурном режиме, применяя принудительное регулирование технологического процесса [2]. Для реализации такого подхода необходимо проведение предварительных исследований с тем, чтобы на основании полученных результатов осуществить аппаратурное оформление процесса в большем масштабе.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Особенностью рассматриваемого в работе подхода является использование в качестве лидирующего параметра температуры газовой смеси в ядре потока, выходящего после слоя катализатора, а также ее нарастание после достижения критической температуры воспламенения. При этом проводился непрерывный хроматографический анализ газа на содержание СО и метана, поскольку содержание последнего в выходящем из реактора газе является индикатором селективности процесса. Ранее этот подход предложен для гипотетических реакций нулевого порядка [3], а также для реакций, с порядком отличным от нулевого [4]. Отметим также, что представленные аппаратурные особенности методики исследования и некоторые наблюдения ранее в литературе не освещались.
На рис. 1 изображен узел реактора, с помощью которого проводились указанные измерения. Узел состоит из термопары, введенной в реактор высокого давления с помощью специального устройства из органического стекла, в которое впрессовывались выводы. На керамическую трубку термопары прикреплена полочка (8) с фиксатором для размещения восстановленного катализатора, который загружался в трубчатый реактор в токе углекислого газа. В ряде случаев осуществлялось довосстановление катализатора в токе водорода при 50 атм и температуре 450°С. Объем катализатора выбирался таким, чтобы расстояния между верхним и нижним спаями термопары и катализатором находилось на уровне 0.5–0.7 см. Использовалось 3 комплекта термопар с разными расстояниями между верхним и нижним спаями, что позволяло менять объем загружаемого катализатора от 1.3 до 5.1 см3.
Рис. 1.
Фрагмент реактора для измерения температуры воспламенения в реакторе высокого давления 200 атм. 1 – Узел ввода термопар; 2 – кожух реактора, 3 – теплоизоляция, 4 – реакторная трубка, 5 – спираль для элекрообогрева, 6 – термопара в блоке, 8 – полочка с отверстиями, 9 – алюминиевый блок, 10 – нижний спай термопары.
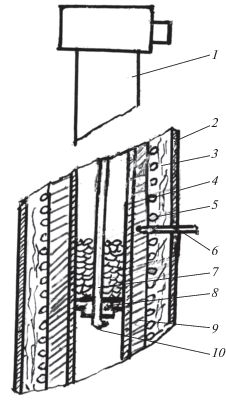
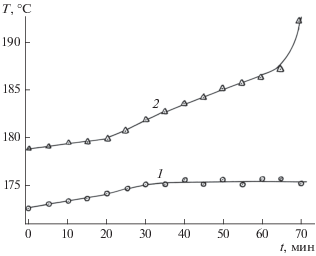
График изменения температуры в блоке реактора и в ядре потока газовой смеси после прохождения слоя катализатора в окрестности температуры воспламенения представлен на рис. 2. Температуру измеряли и регистрировали с частотой 5–10 с. После каждого ее повышения на 1°С делали выдержку ≈5 мин, после которой вновь увеличивали температуру. Видно, что при приближении к температуре воспламенения температура газа после слоя катализатора стремительно возрастает (рис. 2). С помощью хроматографического анализа установлено, что при этом резко возрастает скорость реакции и меняется селективность синтеза в сторону образования воды и метана, что можно рассматривать как характерный для подобных гетерогенных экзотермических реакций релаксационный процесс.
МОДЕЛЬНЫЕ ПРЕДСТАВЛЕНИЯ О ВОЗМОЖНОСТИ ОПИСАНИЯ ВОСПЛАМЕНЕНИЯ ПО ТЕМПЕРАТУРЕ ГАЗА В ЯДРЕ ПОТОКА ПОСЛЕ СЛОЯ КАТАЛИЗАТОРА
Полное обоснование применимости подхода, согласно которому минимальная температура газа в ядре потока при которой происходит воспламенение, является параметром, определяющим кинетические характеристики гетерогенных каталитических реакций в проточном реакторе, приведено в [3]. Количество тепла, выделяемого в единицу времени на единице поверхности катализатора составляет
где Q – тепловой эффект реакции, ${{k}_{0}}{{e}^{{ - \frac{E}{{TT}}}}}$ – зависящая от температуры константа скорости реакции, E – энергия активации реакции, k – постоянная Больцмана. Количество тепла, отводимого в единицу времени с этой поверхности без учета лучеиспускания и термодиффузииВ формулах (1) и (2) T – температура поверхности катализатора, T0 – температура газа в ядре потока, α коэффициент теплопередачи.
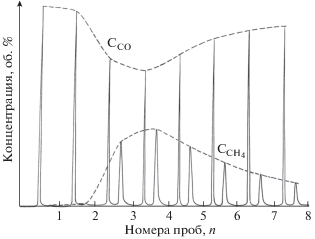
Рис. 4.
Хроматограмма газовой смеси до воспламенения, в начале воспламенения (резкое повышение концентрации метана) и после дезактивации катализатора (повышение концентрации оксида углерода и уменьшение концентрации метана).
Условие стационарности можно записать в виде:
Приняв в качестве температурного масштаба ${{RT_{0}^{{\text{2}}}} \mathord{\left/ {\vphantom {{RT_{0}^{{\text{2}}}} E}} \right. \kern-0em} E},$ можно записать разогрев θ поверхности катализатора в безразмерной форме в виде
где R – газовая постоянная. ТогдаУчитывая зависимости (4) и (5), получим
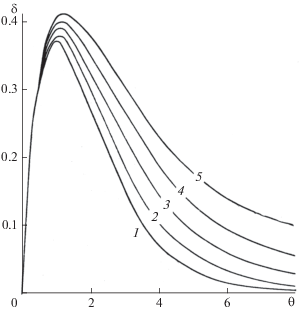
где безразмерный параметр δ включает все величины, характеризующие свойства системыУравнение (6) характеризует зависимость стационарного разогрева поверхности катализатора в безразмерной форме от безразмерных параметров δ и b. График функции (6) представлен на рис. 3, из которого следует, что имеется максимум в точке М. Обозначив ее координаты через θ и δкр, можно убедиться, что решения имеют физический смысл для части кривой, ограниченной значениями 0 ≤ θ ≤ θ*, где θ* – максимально возможный в условиях эксперимента разогрев поверхности, при котором сохраняется нулевой порядок. Нижний предел соответствует отсутствию реакции, а верхний – границе нулевого порядка – переходу в диффузионную область или полному превращению реагирующего вещества. Так, при δ < δкруравнение (6) имеет одно решение. В области δ(θ*) ≤ δ < δкр существуют два решения, соответствующим двум стационарным разогревам поверхности. Анализ графика показывает, что для реакции нулевого порядка устойчивый стационарный режим реализуется при δ < δкр, т.е. левая часть кривых до максимума соответствует устойчивым стационарным режимам, а правая ветвь – неустойчивым стационарным режимам. Следует подчеркнуть, что при протекании реакции нулевого порядка в устойчивом стационарном режиме состояние системы выражается точкой на левой ветви кривой, а в точке максимума реализуется максимально возможные для стационарных состояний значения параметров δ > δкр. При δ > δкр стационарные режимы исключаются. При этом q+ > q– и dq+/dT > dq–/dT и имеет место прогрессирующий разогрев поверхности (тепловое воспламенение).
ОПРЕДЕЛЕНИЕ КИНЕТИЧЕСКИХ ПАРАМЕТРОВ СИНТЕЗА ИЗ ОКСИДА УГЛЕРОДА И ВОДОРОДА
Как отмечалось ранее [5], разработанная методика определения кинетических параметров гетерогенных каталитических реакций по критическим условиям воспламенения применена для изучения синтеза высших спиртов из СО и Н2. В этом случае используется зависимость температуры воспламенения $Т_{0}^{'}$ от α при изменении массовой скорости υ.
Анализ показал, что при развитой турбулентности и высокой теплопроводности слоя катализатора его внешняя поверхность может рассматриваться как равнодоступная. Это позволяет считать идентичность критических условий воспламенения на одиночном зерне и в слое катализатора для изотермического и адиабатического режимов работы реактора. Примем во внимание, что степень превращения оксида углерода до воспламенения в ядре газового потока мала при изменении температуры на 20–25°С, а концентрация реагентов у поверхности в критических условиях постоянна, т.к. разность концентраций в ядре газового потока и у внешней поверхности катализатора практически не зависит от массовой скорости, размера зерен и других условий эксперимента.
Кажущуюся энергию активации находили из критического условия воспламенения, которое может быть записано в виде:
(8)
$\delta = {{\delta }_{{{\text{кр}}}}} = \frac{{Q{{k}_{0}}{{e}^{{ - \frac{E}{{RT_{0}^{'}}}}}}E}}{{\alpha 'R{{{(T_{0}^{'})}}^{2}}}},$(9)
$E = \frac{{RT_{{0(1)}}^{'}T_{{0(2)}}^{'}\left( {\ln \frac{{\alpha _{{(1)}}^{'}}}{{\alpha _{{(2)}}^{'}}} + 2\ln \frac{{T_{{0(1)}}^{'}}}{{T_{{0(2)}}^{'}}}} \right)}}{{T_{{0(1)}}^{'} - T_{{0(2)}}^{'}}}.$На практике кажущуюся энергию активации определяют из зависимости температуры воспламенения от массовой скорости газового потока при постоянных значениях давления и концентрации реагентов.
Массовую скорость меняли в интервале 5–20 моль/ч, что соответствовало изменению скорости газа на выходе из реактора в интервале 115–450 л/ч. Построенные в координатах 0.65lg υвх + + 2lg$Т_{0}^{'}$ от 1/T графики позволили определить из 3 серий опытов величину Е = 20.9 ккал/моль.
Порядок реакции по оксиду углерода находили из зависимости температуры воспламенения от концентрации СО при постоянном давлении и 200 атм и постоянной массовой скорости 10 моль/ч. Концентрацию оксида углерода варьировали в интервале 5–14.3 об. %. Оказалось, что средняя температура воспламенения находится в интервале $Т_{0}^{'}$ от 452.8 до 453.4–453.9 К, что соответствует нулевому порядку при проведении этой серии экспериментов.
Для определения порядка реакции по водороду варьировали парциальное давление H2 от 50 до 100 атм, а постоянство коэффициента теплопередачи α обеспечивали изменением значений массовой скорости в соответствии с изменением состава газовой смеси. Парциальное давление водорода составляло от 50 до 100 атм при давлении в реакторе от 71.8 до 117.7 атм. Из полученных результатов следует, что порядок реакции по водороду равен 0.65. Отметим один важный результат, полученный при давлении в реакторе 70.3 атм, концентрации СО 28.5 об. % и парциальном давлении водорода равном 50.3 атм. После достижения температуры воспламенения, как и в других опытах, селективность синтеза менялась в сторону образования метана (см. рис. 4). Затем содержание метана начинало убывать (сравните пробы 1–2 и 3‒4, которые отбирались с интервалом 3–5 мин), что может свидетельствовать о выходе из режима воспламенения за счет дезактивации катализатора. После прекращения опыта и разгрузки реактора, на термопаре были обнаружены гирлянды волокон. По сегодняшним представлениям это могли быть углеродные нанотрубки, поскольку во время проведения рассматриваемых экспериментов такого понятия не существовало.
Определение порядка реакции по общему давлению осуществляли варьированием давления в реакторе в интервале от 100 до 200 атм при постоянном составе газовой смеси и постоянной массовой скорости исходного газа. Величина температуры воспламенения менялась соответственно от 465.2 до 454.5 К и оказалось, что порядок равен 0.64, что практически совпадает с найденным порядком реакции по водороду. В [6] приведено сопоставление результатов, полученных разными методами.
Приведенные результаты свидетельствуют о целесообразности применения рассмотренного метода определения кинетических параметров по температурам воспламенения, которые важны для выбора соответствующих методов автоматического регулирования таких процессов [1, 7].
В заключение авторы искренне благодарят чл.-корр., проф. А.Н. Башкирова, д. х. н. Ю.Б. Кагана, д. х. н., проф. А.Я. Розовского, чл.-корр., д. х. н. М.Г. Слинько и д. х. н., проф. С.М. Локтева за сотрудничество при изучении кинетики синтеза высших спиртов из СО и водорода в 60-е гг. прошлого века и последующие годы и за совместную разработку приведенного в докладе метода, а также коллективу механиков, активно содействовавших проведению экспериментов под руководством И.А. Антощука.
Список литературы
Мордкович В.З., Синева Л.В., Кульчанская Е.В., Асалиева Е.Ю. // Катализ. в пром. 2015. Т. 15. № 5. С. 23.
Сливинский Е.В., Клигер Г.А., Кузьмин А.Е. и др. // ЖРХО им. Д.И. Менделеева. 2003. Т. 47. № 6. С.12.
Франк-Каменецкий Д.А. Диффузия и теплопередача в химической кинетике. М.: Наука, 1967. 491 с.
Каган Ю.Б., Розовский А.Я., Слинько М.З., Пономаренко А.Т. // Кинет. и катализ. 1964. Т. 5. № 5. С. 920.
Каган Ю.Б., Розовский А.Я., Слинько М.З., Пономаренко А.Т. // Кинет. и катализ. 1964. Т. 5. № 6. С. 1111.
Каган Ю.Б., Пономаренко А.Т., Розовский А.Я. // Кинет. и катализ. 1966. Т. 7. № 4. С. 679.
Каган Ю.Б., Пономаренко А.Т., Розовский А.Я. и др. // Нефтехим. Т. 5. № 1. С. 82.
Кипнис M.A. // Кинет. и катализ. 2017. Т. 17. № 4. С. 266.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Серия физическая