

Известия РАН. Серия физическая, 2022, T. 86, № 5, стр. 666-672
Электрофоретическое осаждение пленок нанографита и их исследование методами структурного анализа
Д. Н. Савельев 1, Е. А. Грушевский 1, М. А. Смирнова 1, Л. А. Мазалецкий 1, Н. Г. Савинский 2, В. И. Бачурин 2, *, А. Б. Чурилов 2
1 Федеральное государственное бюджетное образовательное учреждение высшего образования
“Ярославский государственный университет имени П.Г. Демидова”
Ярославль, Россия
2 Федеральное государственное бюджетное учреждение науки
“Физико-технологический институт имени К.А. Валиева Российской академии наук” Ярославский филиал
Ярославль, Россия
* E-mail: vibachurin@mail.ru
Поступила в редакцию 13.12.2021
После доработки 24.12.2021
Принята к публикации 21.01.2022
- EDN: GSVJJH
- DOI: 10.31857/S0367676522050210
Аннотация
Методами рентгеновской дифрактометрии, энерго-дисперсионного рентгеновского анализа, спектроскопии комбинационного рассеяния, электронной микроскопии исследованы химические, структурные, морфологические свойства эксфолиированного нанографита и пленок нанографитов, сформированных методом электрофореза. Проведены оценки степени дефектности эксфолиированного нанографита. Полученные данные демонстрируют возрастание расчетных величин плотности числа дефектов эксфолиированного графита по сравнению со значением в исходной фольге.
ВВЕДЕНИЕ
Метод электрофоретического осаждения является привлекательной стратегией для изготовления структурированных тонких пленок нанографитов (НГ) [1] для широкого спектра применений, например, солнечных элементов [2], накопителей энергии [3], полевых эмиссионных приборов [4, 5], защита от электромагнитных помех [6], антибликовых и коррозионно-стойких покрытий [7‒9], доставки лекарств и тканевой инженерии [10], материалов твердых смазок [11]. Он имеет много преимуществ при изготовлении нано графитового покрытия, таких как: высокая скорость осаждения и контролируемая толщина слоя, меньшие ограничения по материалу или геометрии подложки, низкая стоимость и простота масштабирования [12]. Однако, изначально графитовая чешуйка гидрофобна, трудно диспергируется в растворе и, что еще хуже, по своей сути, она не заряжена; поэтому осаждение графитовых покрытий электрофоретическим методом все еще остается сложной задачей [13].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Образцы НГ, в том числе, пленки НГ осажденные электрофорезом и условия их синтеза и обработки представлены в табл. 1. С целью получения заряда на чешуйке графита для осаждения пленок методом катофореза была использована техника электрохимической эксфолиации. В наших более ранних исследованиях [14], было обнаружено, что наряду с расщеплением графита в нанографит, наблюдаются процессы окисления графеновой кристаллической решетки, с формированием кислородсодержащих окисленных форм углерода в виде карбоксилат ионов, оксиирановых функциональных групп, ковалентно связанных с кристаллической решеткой графита. Изменением условий проведения эксфолиации возможно варьировать степень окисления продуктов эксфолиации графита, осуществляя процесс в окислительной или восстановительной среде. Элементный состав образцов (табл. 2, рис. 1), выполненный энерго-дисперсионным рентгеновским методом позволяет определить соотношение содержания кислородных групп в углеродной матрице O/С. В табл. 3 представлены результаты условий эксфолиации графита в водном растворе электролита сульфата аммония.
Таблица 1.
Условия подготовки образцов графито-углеродных материалов (ГУМ)
| Номер образца | Название образца | Условия обработки образца |
|---|---|---|
| 1 | Графитовая фольга | Без обработки |
| 2 | Эксфолиированный нанографит (ЭГ) |
Электрохимическая эксфолиация электрод: катод Pt анод графитовая фольга, электролит 0.05 М (NH4)2 SO4 |
| 3 | Эксфолиированный нанографит (ЭНГ) |
Электрохимическая эксфолиация электрод: катод Pt анод графитовая фольга 3 М (NH4)2SO4; 0.08 мас. % диметилсульфоксида (ДМСО) |
| 4 | Механическия эксфолиация Образца 3 | Механическая эксфолиация образца 3 в воде, 0.01 мас. % ПАВ “синтанол 23Е4P5”, с использованием ротационно-импульсного аппарата (кавитационное воздействие) в течение 5 ч. Далее классификации в центрифуге при 1500 об./мин, в течение 10 мин |
| 5 | Механическия эксфолиация Образца 3 |
Механическая эксфолиация образца 3 в воде, 0.01 мас. % ПАВ “синтанол 23Е4P5”, с использованием ротационно-импульсного аппарата (кавитационное воздействие) в течение 5 ч. Далее классификации в центрифуге при 5000 об./мин, в течение 5 ч |
| 6 | Ультразвуковая эксфолиация Образца 4 | Фильтрат образца 4 подвергался ультразвуковой обработке (20 кГц, 60 Вт) в течение 8 ч. Осадок отделялся центрифугированием 7000 об./мин в течение 25 мин, с последующей сушкой в сушильном шкафу 50°С в течение 24 ч |
| 7 | Пленки ГУМ, осажденные электрофорезом | Электрофоретическое осаждение пленок ГУМ из суспензия углеродного материала: фильтрата образца 6 в концентрации 11 г/л и 5% сульфата аммония в воде проводится в близкой к нейтральной среде (pH 6.7) в интервале напряженности поля от 1.9 до 2.5 В/см в двухэлектродной системе. Осаждение производится на медный катод, анодом является графитовая фольга |
Таблица 2.
Результат энерго-дисперсионного рентгеновского анализа (ЭДРА) образца 6
| Элемент | Условная концентрация | Массовый, % | Массовый, % Сигма |
Атомный, % |
|---|---|---|---|---|
| C (K) | 215.40 | 96.42 | 0.16 | 97.53 |
| O (K) | 2.42 | 2.94 | 0.16 | 2.23 |
| S (K) | 0.67 | 0.64 | 0.05 | 0.24 |
| Суммарно | 100.00 |
Таблица 3.
Соотношение углерода/кислорода С/O в продуктах электрохимической эксфолиации в зависимости от условий процесса
| Образец | Условия процесса эксфолиации | Соотношение C/O, ат. % |
|---|---|---|
| Оригинальная графитовая фольга “Графлекс” | 48.11 | |
| Эксфолиированный графит | Электрод: нержавеющая сталь 0.05 М (NH4)2 SO4 |
12.71 |
| Эксфолиированный графит | Электрод: Pt 0.05 М (NH4)2 SO4 | 16.95 |
| Эксфолиированный графит | Электрод: Pt 3 М (NH4)2SO4; 0.08 мас. % диметилсульфоксида (ДМСО) | 43.70 |
Эксфолиация графита проводилась в двухэлектродной ячейке, в водном растворе сульфата аммония концентрацией 3 моль/л, в качестве анода использовалась графитовая фольга “Графлекс” производства РФ, а в качестве катода платиновая проволока. После эксфолиации полученный образец промывался на нутч-фильтре деионизованной водой от ионов.
Суспензия нанографита для электрофоретического осаждения графитовых пленок имеет состав: фильтрат образца 3 с содержанием углеродного материала 11 г/л и 5% сульфата аммония в воде. Электрофоретическое осаждение проводится в близкой к нейтральной среде (pH 6.7) в интервале напряженности поля от 1.9 до 2.5 В/см в двухэлектродной системе, осаждение производится на медный катод, анодом является графитовая фольга. Исследования структуры пленок проводились с использованием EnSpectr R532 спектрометра комбинационного рассеяния, электронного микроскопа Zeiss SUPRA-40 с рентгеновским энергодисперсионным анализатором INCA Energy Oxford Instruments, порошкового рентгеновского дифрактометра ARL XTRA Termo Techno.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для оценки числа атомарных слоев чешуйки НГ был использован метод, основанный на анализе рентгеновской дифракции [14]. Обработка полученных результатов показала, что поперечный размер кристаллитов LZ исходной фольги составляет 37 нм и для всех образцов после процесса эксфолиации находится в диапазоне 12–15 нм и практически не зависит от условий обработки материала. При величине числа слоев графита более 10, а в нашем случае это 30–40 слоев, согласно номенклатуре [1], материалы отнесены не к классу графенов, а к другой категории графито-углеродных материалов – к графитовым наноплатам или нанографитам.
Спектроскопия комбинационного рассеяния (КР) является мощным инструментом исследования углеродных структур [15]. КР спектроскопия по аналогии с ИК спектроскопии чувствительна к химической природе связи скелета углеродной органической молекулы т.н. “область отпечатков пальцев”. Особенно информативна КР спектроскопия для проведении исследований сложных систем с пониженной (0D, 1D, 2D) размерностью и различных нанообъектов, при проведении анализа химического и фазового состояния различных объектов и их структуры, для неразрушающего контроля разнообразных процессов в режиме on-line; бесконтактного анализа, не требующего подготовки пробы, возможности картографирования образцов с высоким латеральным разрешением до 1 мкм (в рамановских спектрометрах с конфокальным микроскопом); возможности сканирования по глубине образца, прозрачного в выбранном диапазоне, с проникновением вглубь от 0.1 до 100 мкм (в зависимости от частоты источника излучения).
На рис. 2 представлен типичный КР спектр образца 3. Количественная оценка дефектности структуры для широкого семейства систем с sp2 гибридизацией, включающей графеновые структуры, имеет решающее значение, как для понимания фундаментальных свойств, так и для технических приложений, в первую очередь подвижности носителей. Общепринято считать, что пик в области частот 1580 см–1 связан с высокочастотным колебанием оптических фононов E2g решетки для графитов и нанографитов, получивший название G (graphite) пика. Этот пик, названный G (от графита), наблюдается в КР-спектре любого числа слоев графена при 1580 см–1 и связан с плоскостью растяжения связей С–С углеродного скелета молекулы.

Полоса G КР спектров рассеяния для sp2 атомов углерода наблюдается в виде одиночного или мультипликативного пика для всех углеродных структур sp2 гибридизации. Дефектные зоны с sp3 гибридизацией, характеризуются мультипликативным пиком в области 1350 см–1, называемым D (disordered) [16, 17]. Его наличие обуславливает “дыхательная” мода шестиатомного ароматического кольца A1g и требует дефекта для его активации в первой зоне Бриллюэна вблизи точки K или K1. КР-спектр неупорядоченного графита показывает два новых резких пика около 1350 см–1 пик D и 1620 см–1 пик D'. Эти междолинные D и внутри долинные D' дефект-индуцированные резонансные процессы рассеяния, в основном, ответственны за декогеренцию электронa в оптических переходах sp2 атомов углеродов. 2D (или G') полоса также определяется sp2 углеродными атомами в виде одиночного пика или мультипликативной структурой в диапазоне 2500–2800 см–1. Чувствительность 2D пика к деталям sp2 структуры делает эту полосу мощным индикатором для определения числа графеновых слоев в пачке. В то время как полосы G и D' имеют только один пик для любого количества слоев графена, полоса G' состоит, соответственно, из одного, четырех, шести, а полоса D из двух пиков для монослоя, двухслойного, трехслойного и многослойного графена. Так как абсолютная интенсивность редко используется в КР-спектроскопии, нормализованную интенсивность ID/IG широко используют для количественных измерений степени дефектности (беспорядка).
Было замечено [18], что отношение интенсивностей D к G изменяется обратно пропорционально размеру кристаллитов, I(D)/I(G) ∞ 1/LA. В основном, количество беспорядка в нанокристаллите определяется количеством границ (одномерных дефектов) по отношению к общей площади кристаллитов Lsp2. Влияние изменения LD на ширину пика, частоту, интенсивность и интегральную площадь для многих пиков комбинационного рассеяния в однослойном графене было изучено в [19] и расширено до нескольких слоев графена в [20] с использованием одной лазерной линии Ar + EL = 2.41 эВ (λ = 514.5 нм). В графене с нулевыми точечными дефектами расстояние между дефектами, LD, является мерой количества беспорядка.
(1)
${{L}_{{sp2}}} = \left( {2.4 \cdot {{{10}}^{{ - 10}}}} \right) \cdot {{\lambda }}_{{Laser}}^{4}{{\left( {{{{{I}_{D}}} \mathord{\left/ {\vphantom {{{{I}_{D}}} {{{I}_{G}}}}} \right. \kern-0em} {{{I}_{G}}}}} \right)}^{{ - 1}}}\,\,\left( {{\text{нм}}} \right)~,$(2)
$L_{D}^{2} = \left( {2.4 \cdot {{{10}}^{{ - 9}}}} \right) \cdot {{\lambda }}_{{Laser}}^{4}{{\left( {{{{{I}_{D}}} \mathord{\left/ {\vphantom {{{{I}_{D}}} {{{I}_{G}}}}} \right. \kern-0em} {{{I}_{G}}}}} \right)}^{{ - 1}}}\,\,~\left( {{\text{н}}{{{\text{м}}}^{2}}} \right),$(3)
${{n}_{D}} = {{\left( {2.4 \cdot {{{10}}^{{22~}}}} \right)} \mathord{\left/ {\vphantom {{\left( {2.4 \cdot {{{10}}^{{22~}}}} \right)} {{{\lambda }}_{{Laser}}^{4}}}} \right. \kern-0em} {{{\lambda }}_{{Laser}}^{4}}}\left( {{{{{I}_{D}}} \mathord{\left/ {\vphantom {{{{I}_{D}}} {{{I}_{G}}}}} \right. \kern-0em} {{{I}_{G}}}}} \right)~\,\,~\left( {{\text{c}}{{{\text{м}}}^{{ - 2}}}} \right)~.$Эти уравнения – (1) [21], (2) [22] и (3) [23], полученные для определения размера между дефектами кристаллических областей графена LD и плотности дефектов nD, или иными словами, расстояния между дефектами кристаллической решетки графена в зависимости от энергии используемого возбуждающего излучения в видимом диапазоне, где лазерное возбуждение используется в терминах длины волны в нм. Для лазера с длиной волны 532 нм плотность дефектов nD может быть представлена уравнением 3. Для сотовой идеальной решетки число атомов углерода составляет (уравнение 4),
если нормировать число дефектов в уравнении (4) на nc, можно получить концентрацию дефектов на мкм2 в миллионных долях (ppm). Это соотношение обозначается в литературе как ID/IG или I(D)/I(G), в то время как соответствующее соотношение площадей, т.е. интегральное отношение интенсивности по частоте, как AD/AG или A(D)/A(G). Для случая больших нарушений графеновой структуры иными словами “беспорядка” гораздо более информативно разделить данные об интенсивности пика и его полной ширине на половине максимума полосы. Последнее, обозначаемое в литературе как FWHM или Γ, является мерой структурного беспорядка, в то время как интегральная интенсивность отражает фононные моды, молекулярные колебания, участвующие в наиболее резонансных комбинационных процессах.
В табл. 4 представлены результаты обработки КР спектров образцов 1–7. Интенсивность полосы (I) определяется как FWHM или Γ (полная ширина пика на полувысоте максимума полосы).
Таблица 4.
Результат обработки КР спектров образцов 1, 2 и 3 в соответствии с уравнениями (1)–(4)
| № образца | ID/IG | ${{L}_{{sp{\text{2}}}}}$, нм | LD, нм | nD × 1011, см–2 | nD/nC, ppm |
Число графеновых слоев |
|---|---|---|---|---|---|---|
| 1 | 0.24 | 77.0 | 28 | 0.719 | 50 | 105 |
| 2 | 1.16 | 4.2 | 13 | 3.48 | 431 | 30 |
| 3 | 0.33 | 7.6 | 24 | 0.99 | 86 | 30 |
| 4 | 0.160 | 115.0 | 34 | 0.48 | 20 | 30 |
| 5 | 0.26 | 71.0 | 27 | 0.75 | 49 | 30 |
| 6 | 0.25 | 74.0 | 27 | 0.75 | 49 | 30 |
| 7 | 0.25 | 74.0 | 27 | 0.75 | 49 | 30 |
Значительное возрастание расчетных величин плотности числа дефектов nD и nD/nC симбатно поведению экспериментальных отношения интенсивностей пиков ID/IG эксфолиированного графита (образца 2) по сравнению со значением в исходной фольге (образец 1), а расчетные величины Lsp2 демонстрируют снижение размера sp2 доменов в базальной плоскости кристаллической решетки графена более чем на порядок величины и уменьшение размера дефектов LD. Такое поведение легко объяснить процессами расщепления слоев кристаллической решетки графита со 105 до 30 слоев во время процесса электро-эксфолиации. Однако, при изменении условий проведения процедуры эксфолиации (образец 3), характер поведения вышеуказанных величин хотя и не изменился, но темп изменения значительно упал. Это связано, в первую очередь, с введением в реакционную массу веществ антиоксидантов, которые блокируют процессы окисления.
Процессы дальнейшего измельчения нанографита механической эксфолиацией (образцы 4 и 5) и ультразвуком (образец 6) демонстрируют, согласно данным таблицы, практически неизменные экспериментальные и расчетные величины, свидетельствующие об отсутствии эффективных процедур измельчения. Некоторые особенности присутствуют для образца 4. Так, по показателям дефектности, это самый низко дефектный образец с относительно большим размером кристаллического домена Lsp2 более 100 нм. Образец 7 – пленка, выращенная на меди методом катофореза, демонстрирует фактически показатели, аналогичные исходному нанографиту образца 6. Это, косвенно, свидетельствует об отсутствии негативных побочных процессов технологии катофореза.
Тем не менее, окончательное заключение о неэффективности используемых процессов эксфолиации графита на основании представленных методов анализа, делать преждевременно. Все процессы пробоподготовки для анализа осуществлялись осаждением из раствора или прессованием из порошков. Известно, что графеновые чешуйки весьма склонны к агрегации, и при седиментационном осаждении из жидкости, в условиях удаления жидкости между слоями, могут вновь формировать многослойные структуры.
Осаждение нанографитовых пленок методом электрофореза обычно осуществляется под действием постоянного электрического поля в растворе электролита [24, 25]. В данном методе положительно заряженная частица нанографита осаждается на катод (катофорез). Частичный положительный заряд на частице нанографита формируется за счет нейтрализации ионами электролита ${\text{NH}}_{4}^{ + }$ частично окисленного образца, имеющего на поверхности углеродного скелета карбонильный анион.
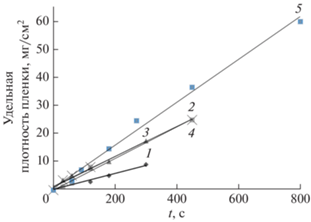
На рис. 3 представлен процесс электрофореза. В целом, процесс осаждения нанографита методом электрофореза протекает по линейному закону во времени. При концентрации графита в суспензии 11 г/л, эта линейная зависимость сохраняется приблизительно 300–500 с проведения процесса, далее график процесса выходит на насыщение и идет с меньшей скоростью. Такое поведение типично для процессов электрофореза и может быть описано в рамках модели Гамакера (5) [25].
где W – осажденная масса, кг; t – время осаждения, с; µ – электрофоретическая подвижность, м2(В ⋅ с)–1; E – приложенный потенциал, В ⋅ м–1; А – площадь поверхности электрода, м2; СS – концентрация частиц в коллоиде, кг ⋅ м–3; f – коэффициент эффективности. Коэффициент эффективности учитывает долю частиц, которые вступают в контакт с поверхностью электрода и впоследствии осаждаются на металлических подложках, что может быть использовано в дальнейшем для количественной оценки влияния подвижности частиц. На начальном этапе роста, до 15 с заметен рост слоя пленки из мелких отдельных частиц нанографита менее 100 нм в диаметре. Затем формируется слоистая структура после 30 с осаждения с толщиной слоя от 300 нм до нескольких микрон. Наконец, при временах роста 800 с и более начинают формироваться столбчатые структуры с развитым рельефом рис. 4.Рис. 3.
Кинетика процесса катофореза при различных значениях напряжения и силы тока: (1) I = 2 А, V = 4 В; (2) I = 1 А, V = 3.8 В; (3) I = 1 А, V = 4 В; (4) I = 1 А, V = 3.8 В; (5) I = 4 А, V = 5 В. Условия проведения катофореза: двухэлектродная ячейка катод–медная фольга, анод графитовая фольга, концентрация суспензии углеродного материала 11г/л, концентрация электролита сульфата аммония в воде 5 мас. %, pH 6.7.

ЗАКЛЮЧЕНИЕ
Разработан процесс электрохимической эксфолиации графитовой фольги в нанографитовую суспензию с регулируемой степенью окисления углеродной решетки, в водном растворе электролита сульфата аммония. Данные анализа КР спектров образцов эксфолиированного нанографита позволили оценить размера бездефектных кристаллических областей графена LD и плотности дефектов nD после процесса эксфолиации. Полученные экспериментальные данные демонстрируют возрастание расчетных величин плотности числа дефектов nD эксфолиированного графита по сравнению со значением в исходной фольге. Осуществлен процесс катофоретического осаждения пленок нанографита из водного раствора электролита сульфата аммония на медную фольгу. Показано, что кинетика электрофоретического осаждение нанографитового слоя описывается в рамках модели Гамакера.
Исследование выполнено при поддержке Министерства науки и высшего образования Российской Федерации в рамках темы государственного задания Ярославского филиала Физико-технического института Российской академии наук имени К.А. Валиева № 0066-2019-0003. Некоторые экспериментальные результаты получены с использованием оборудования Центра коллективного пользования “Диагностика микро и нано структур” при поддержке Министерства науки и высшего образования Российской Федерации.
Список литературы
Bianco A., Cheng H., Enoki T. et al. // Carbon. 2013. V. 65. P. 1.
Chang D.W., Choi H.J., Filer A. et al. // J. Mater. Chem. A. 2014. V. 2. No. 31. Art. No. 12136.
Liu J., Galpaya D., Notarianni M. et al. // Appl. Phys. Lett. 2013. V. 103. No. 6. Art. No. 063108.
Lee K.E., Hwang J.O., Kim S.O. et al. // Proc. IEEE NMDC-2011. (Jeju, 2011). P. 303.
Bose S., Kuila T., Kim N.H. et al. // In: Graphene: properties, preparation, characterization and devices. Woodhead Publishing Limited, 2014. P. 81.
Hong S.K., Kim K.Y., Kim T.Y. et al. // Nanotechnol. 2012. V. 23. No. 45. Art. No. 455704.
Aneja K.S., Bohm S., Khanna A.S. et al. // Nanoscale. 2015. V. 7. No. 42. Art. No. 17879.
Mohsin A.R., AliGhauria F., Aslama A. et al. // Surf. Coat. Technol. 2017. V. 332. P. 112.
Singh B.P., Nayak S., Nanda K. et al. // Carbon. 2013. V. 61. P. 47.
Geetha Bai R., Muthoosamy K., Manickam S. et al. // Progr. Mater. Sci. 2018. V. 91. P. 24.
Shen B., Hong H., Chen S. et al. // Carbon. 2019. V. 145. P. 297.
Mani D., Fam D.W.H., Boccaccini A.R. et al. // Progr. Mater. Sci. 2016. V. 82. P. 83.
Yifei M., Jiemil H., Mei W. et al. // J. Materiomics. 2018. V. 4. P. 108.
Савинский Н.Г., Мелесов Н.С., Паршин Е.О. и др. // Изв. РАН. Сер. физ. 2020. Т. 84. № 6. С. 887; Savinsky N.G., Melesov N.S., Parshin E.O. et al. // Bull. Russ. Acad. Sci. Phys. 2020. V. 84. No. 6. P. 732.
Jorio A., Saito R., Dresselhaus G. Raman spectroscopy in graphene related systems. Weinheim: WILEY-VCH Verlag GmbH & Co. KGaA, 2011. 354 p.
Andrianova N.N., Borisov A.M., Vysotina E.A. // J. Surf. Invest. X-ray, Synchrotron Neutron Techn. 2021. V. 15. No. 2. P. 230.
Andrianova N.N., Borisov A.M., Mashkova E.S. // Vacuum. 2021. V. 188. Art. No. 110177.
Tuinstra F., Koenig J.L. // J. Chem. Phys. 1970. V. 53. No. 3. P. 1126.
Ferreira E.H.M., Moutinho M.V.O., Stavale F. // Phys. Rev. B. 2010. V. 82. Art. No. 125429.
Cançado L.G., Takai K., Enoki T. et al. // Appl. Phys. Lett. 2006. V. 88. Art. No. 163106.
Cancado L.G., Jorio A.M., Ferreira E.H. et al. // Nano Lett. 2011. V. 11. P. 3190.
Sharma R., Chadha N., Saini P. // Indian J. Pure Appl. Phys. 2017. V. 55. P. 625.
Yu P., Lowe S.E., Simon G.P. et al. // COCIS. 2015. V. 20. No. 5–6. P. 329.
Chakrabarti M.H., Hashim M.A., Hussain M.A. et al. // Electrochim. Acta. 2013. V. 107. P. 425.
Hwang M.-J., Kim M.-G., Kim S. et al. // Carbon. 2019. V. 142. P. 68.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Серия физическая