

Химическая физика, 2020, T. 39, № 5, стр. 33-42
Физико-химические изменения антиоксиданта фенольного типа при многократных фазовых переходах стабилизированного полиэтилена
Е. В. Воробьёва *
Гомельский государственный университет им. Франциска Скорины
Гомель, Белоруссия
* E-mail: evorobyova@gsu.by
Поступила в редакцию 03.12.2018
После доработки 01.03.2019
Принята к публикации 20.03.2019
Аннотация
В работе представлены результаты исследований методом фурье-ИК-спектропии физико-химических и структурных превращений антиоксиданта фенольного типа ирганокс 1010 в составе полиэтиленовых пленок при фазовых переходах полимера твердое состояние ↔ расплав. Режим прерывистого окисления состоял из циклов, каждый из которых включал в себя три стадии испытаний: нагрев образца до расплавленного состояния, его изотермическую обработку при 150 °С и последующий обратный перевод расплава полимера в твердое состояние. Фурье-ИК-спектры получали в режиме in situ, т.е. без извлечения образца из термоприставки. Установлено, что при каждом цикле термообработки расплава происходит дополнительное сорбционное насыщение полимера антиоксидантом, что сопровождается одновременным расходом части его гидроксильных групп. Кристаллизация полимерного композита сопровождается десорбцией антиоксиданта – из полимера вытесняется частично поврежденный антиоксидант, и поэтому качество антиоксиданта, остающегося сорбированным, несколько “улучшается”. Зафиксированные изменения в структуре и физико-химическом поведении антиоксиданта обусловлены особенностями превращений надмолекулярной структуры полимера.
ВВЕДЕНИЕ
Стабилизация полиолефинов обычно проводится путем введения в их состав ингибиторов или антиоксидантов (АО). Одним из типов промышленных АО являются соединения типа InH, включающие в себя подвижный атом водорода: вторичные амины ряда бензола (PhNH) или замещенные одно- и многоядерные фенолы (PhОH). Считается, что АО, имеющий подвижный атом водорода в основном реагирует с радикалами, ведущими цепи окислительных превращений (РO• и ${\text{PO}}_{2}^{\centerdot }$). В результате происходит обрыв реакционных цепей и образуется неактивный радикал ингибитора [1]. Например, в основной реакции ингибирования окисления АО фенольного типа гидроксил разрушается и образуется неактивный феноксильный радикал:
Эффективность АО, используемых для придания полимерам окислительной стойкости, чаще всего оценивают по величине индукционного периода окисления (периода индукции) полимерного материала [1, 2]. Для ингибированных полимеров такого рода испытания, как правило, продолжаются многие десятки и сотни часов [3]. С целью сокращения времени испытаний исследователи идут на различного рода методические ухищрения или “приемы”, например: уменьшают концентрацию АО, увеличивают давление кислорода или температуру при испытаниях [4]. Обычно период индукции определяют не при рабочей температуре использования полимерных изделий, а в более жестких условиях, например при температуре, превышающей температуру плавления полимера [4, 5]. Однако даже при таком подходе не всегда удается снизить трудоемкость и закончить испытания за один цикл. Эксперимент приходится неоднократно прерывать: образец охлаждают до комнатной температуры, а затем снова возвращаются к испытаниям в расплаве, чтобы в конечном счете путем суммирования продолжительности нахождения образца в расплавленном состоянии определить искомый период индукции.
Режим прерывистого окисления расплава полимера имеет свои особенности, связанные, в первую очередь, с распределением антиокислительной добавки в объеме образца и ее поведением на различных этапах испытаний. Известно, что в случае кристаллизующихся полимеров, например полиэтилена, АО только частично сорбируется или поглощается в аморфной зоне вещества [6, 7]. При этом остальная часть ингибирующей добавки находится в дисперсном виде в межфибрилярных участках внутри надмолекулярных образований – сферолитов и в межсферолитных промежутках. Как показывают исследования ученых [8], АО, как и кислород, практически отсутствует внутри полимерных кристаллитов. Предельное количество АО, которое может быть сорбировано в аморфной зоне полимера, т.е. концентрация насыщения, составляет примерно 10–2 мас.%. В то же время в реальных рецептурах изделий для антиокислительной защиты используют более высокие концентрации добавки: от 0.1 до 0.5 мас.%. [9]. Понятно, что при таких повышенных дозировках некоторое количество АО выпотевает на поверхности образца и далее либо испаряется в атмосферу, либо вымывается технологическим жидкостями. Применение повышенных температур при ускоренных испытаниях интенсифицирует процессы, связанные с поведением АО, – увеличиваются его физические потери вследствие испарения и вымывания, несколько возрастают сорбционное поглощение добавки в аморфной зоне полимера и поглощение АО наполнителями и другими ингредиентами композиций. Увеличения сорбционной емкости для АО можно ожидать при полном плавлении полимера, т.е. когда ранее закристаллизованная часть образца переходит в аморфную фазу. При обратном переводе расплава полимера в твердое состояние процесс сорбционного насыщения должен приобрести противоположную направленность. Теперь главными становятся процессы вытеснения части сорбированного АО и его выделения в виде мельчайших дисперсных частиц (в виде отдельной дисперсной фазы, не смешиваемой с полимером). К сожалению, в литературе отсутствуют систематические исследования физико-химических превращений и расходования АО при циклическом воздействии: плавление – кристаллизация ингибированного полиэтилена. В настоящей работе на примере полиэтилена ставится задача хотя бы частично восполнить этот пробел.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Основным объектом исследований был порошкообразный неингибированный полиэтилен низкого давления (ГОСТ 16338-85, марка 20308-05), в который вводили фенольный АО ирганокс 1010 [пентаэритриттетраокси (3-(3,5-ди-трет-бутил-4-гидроксифенил) пропионат)] производства компании Ciba Specialty Chemicals (IUPAC: [3-[3-(3,5-ditert-butyl-4-hydroxyphenyl)propanoyloxy]]-2,2-bis[3-(3,5-ditert-butyl-4-hydroxyphenyl)propanoyloxymethyl]propyl]-3-(3,5-ditert-butyl-4-hydroxyphenyl)propionate), структурная формула которого имеет вид
Как отмечают исследователи, ирганокс 1010 – один из наиболее распространенных и эффективных АО фенольного типа, так как имеет сложную и разветвленную структуру, обладает высокой молекулярной массой и поэтому практически не испаряется на воздухе из расплава полимера [10, 11].
При ингибировании полиэтилена вначале получают сухие механические смеси АО с порошком полимера, из которых затем методом термического прессования (температура – 150 °С, продолжительность – до 1 мин) формируют пленочные образцы для исследований. Смеси готовят путем смачивания полимерного порошка раствором АО в ацетоне. Далее образовавшуюся жидкую суспензию высушивали на воздухе до полного испарения растворителя, затем проводили сухое смешивание компонентов композиции. В результате получали механическую смесь полимера с АО, в которой ингибитор образует тонкий слой на поверхности частиц полимерного связующего.
В проведенных экспериментах использованы пленки ингибированного ПЭ (толщина – 100 мкм), проплавленные на подложках из КВr, прозрачных в ИК-области спектра. Далее образцы помещались внутрь термостатирующей приставки Р/N 21525, входящей в комплект фурье-ИК-спектрометра Vertex-70 (компания Brüker, Германия). ИК-спектры снимали при различных температурах в диапазоне 20–150 °С и в режиме in situ – без извлечения образца из приставки и прерывания эксперимента.
При анализе ИК-спектров ингибированного полиэтилена, подвергнутого окислительной обработке, использовались пять полос поглощения, три из которых относятся непосредственно к АО: 1) пик поглощения с максимумом при 3640 см–1 – поглощение гидроксильных групп, валентные колебания связи O–H; 2) пик поглощения с максимумом при 1745 см–1 – валентные колебания С=О (карбонила) в сложных эфирах; 3) пик поглощения с максимумом при 1500 см–1 – валентные колебания ароматических колец [12, 13]. Две анализируемые полосы связаны со структурой полимерного материала (пик поглощения при 2019 см–1 характеризует толщину пленки, а пик при 1894 см–1 является показателем закристаллизованности полимера) [14].
Интенсивность поглощенного образцом ИК-излучения определяли по измерению площадей соответствующих полос1)1). Расчет площадей полос вели, используя стандартный пакет программ OPUS 6.5, входящих в комплект фурье-спектрометра; значения площадей выражали в условных единицах (у. е.). Частотные границы полос поглощения, выбранных нами, приведены в работе [15]. В дальнейшем площади полос будем обозначать с помощью буквенного символа “S” с прибавлением к нему в индексе частотного положения того или иного пика поглощения: например, S1745. Площади полос поглощения в ИК-спектрах использовали как абсолютные показатели для нахождения используемых ниже относительных коэффициентов.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
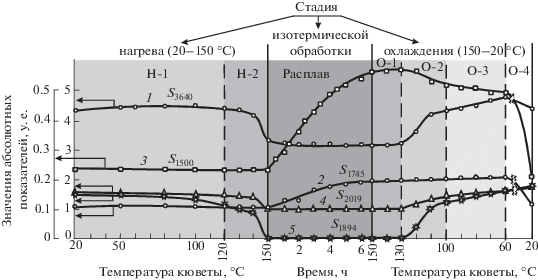
На рис. 1 приведена эволюция изменения величин изменения величин площадей пяти анализируемых полос поглощения на каждой из трех стадий испытаний: 1) нагрев образца от комнатной температуры до температуры окислительного воздействия в расплавленном состоянии (150 °С); 2) изотермическая обработка образца при данной температуре; 3) последующий обратный перевод расплава полимера в твердое состояние при комнатной температуре.
Рис. 1.
Изменение показателей S3640 (1), S1745 (2), S1500 (3), S2019 (4), S1894 (5) в процессе прохождения отдельных стадий испытания образца: нагрева, изотермической обработки (температура кюветы – 150 °С) и охлаждения. Концентрация ирганокса 1010 в пленках – 0.1 мас.%.
Далее трехстадийный цикл испытания образца повторяли до тех пор, пока не заканчивался индукционный период окисления полимерного образца и начиналась автокаталитическая – самоускоряющаяся стадия окисления, легко выявляемая по интенсивному накоплению в полимере карбонильных групп. Период индукции определяли по суммарной продолжительности термообработки образца в расплавленном состоянии до момента дополнительного появления в полимере карбонильных групп – продуктов окисления.
Представленные на рис. 1 данные позволяют в первую очередь отследить, как сказываются процессы плавления и кристаллизации полимера на величинах площадей полос поглощения, относящихся как к АО, так и к полимеру. Наиболее выразительной является динамика изменения величины S1894, характеризующей степень закристаллизованности полимера. Вначале, при нагреве образца в диапазоне от 20 до 100 °С (участок Н-1), происходит относительно медленное снижение величины площади этой полосы поглощения, а затем в диапазоне от 100 до 150 °С (участок нагрева Н-2) она резко уменьшается, и при температуре 150 °С ее значение (S1894) становится равным нулю (рис. 1, кривая 5). Нулевое значение показателя S1894 сохраняется до конца изотермической термообработки образца, что свидетельствует об отсутствии в расплаве полимера кристаллитных образований, фиксируемых методом ИК-спектроскопии. На стадии охлаждения образца, т.е. при переходе полимера из расплавленного в твердое состояние, величина S1894 изменяется в обратном порядке: вначале в интервале температур 150–130 °С (участок О-1) она продолжает оставаться неизменной, затем в диапазоне 130–100 °С скачком увеличивается с 0 до 1.01 у. е. (участок О-2) и далее происходит медленно увеличиваться (при 20 °С до значения S1894, равного 1.52 у. е. – участки охлаждения О-3 и О-4).
Схожая динамика в испытательном цикле “нагрев – изотермическая термообработка – охлаждение образца” характерна и для поведения площади второй полосы поглощения (S2019), относящейся к структурному состоянию полимерного связующего, и, что оказалось самым неожиданным, площади полосы поглощения гидроксильных групп (S3640) АО (рис. 1, кривые 1, 4). При нагреве образца (участок Н-2) величина S3640 резко уменьшается, а в результате кристаллизации при охлаждении полимера (участок О-2) показатель S3640 существенно увеличивается.
Другие же полосы поглощения, относящиеся к АО (полосы с пиками при 1745 и 1500 см–1), наоборот, в такой мере не “ощущают” изменения степени закристаллизованности полиэтилена, и в ходе процессов плавления (участок Н-2) и кристаллизации (участок О-2) полимера их площади остаются постоянными или изменяются незначительно (рис. 1, кривые 2, 3).
В этой связи следует выявить причины и объяснить аномальное поведение показателя S3640 (рис. 1, кривая 1) в температурных диапазонах плавления и кристаллизации полиэтилена. Вначале обратимся к ИК-спектру исходного АО. Такой спектр для тонкого слоя расплава ирганокса 1010 (толщина слоя около – 20 мкм), нанесенного на поверхность пластины из KBr, приведен на рис. 2 (спектр 1). Как видно, спектр имеет одиночную интенсивную полосу поглощения, относящуюся к валентным колебаниям водорода в составе гидроксильных групп АО (пик с максимумом около 3648 см–1). Примерно такое же положение (около 3646 см–1) и форму имеет полоса поглощения гидроксильных групп для АО, растворенного в четыреххлористом углероде (рис. 2, спектр 3). В спектре ингибированного полиэтилена полоса поглощения ИК-излучения, относящаяся к гидроксильным группам ирганокса 1010, принадлежит широкой частотной области: от 3730 до 3580 см–1 (рис. 2, спектр 7). Это полоса с пиком при 3640 см–1, и ее площадь S3640 используется нами для нахождения общего содержания гидроксильных групп в составе АО, введенного в полимер. Форма полосы 3640 см–1 является достаточно сложной – она включает в себя два основных пика: один высокочастотный с максимумом поглощения около 3648 см–1 и второй низкочастотный – около 3606 см–1 (рис. 2, спектр 7). Такая форма полосы поглощения, относящейся к гидроксильным группам, и положение образующих ее пиков позволяют говорить об одновременном присутствии свободных и связанных колебаний водорода в составе гидроксильных групп АО, введенного в полимер. Связанные колебания водорода, по-видимому, возникают из-за образования водородных связей (ассоциатов) между карбонильными и гидроксильными группами, входящими в молекулярную структуру АО. Последнее подтверждает появление смещения низкочастотной полосы поглощения, относящейся к карбонильным группам (см. данные, приведенные в работе [15]).
Рис. 2.
ИК-спектры ирганокса 1010 на KBr (1); четыреххлористого углерода (2); раствора ирганокса 1010 в четыреххлористом углероде (3, 3 '); ацетона (4); раствора ирганокса 1010 в ацетоне (5); разностный спектр 5 – 4 (6); полиэтилена, содержащего ирганокс 1010, при температурах 20 (7) и 150 °С (8); разностный спектр 7 – 8 (9).
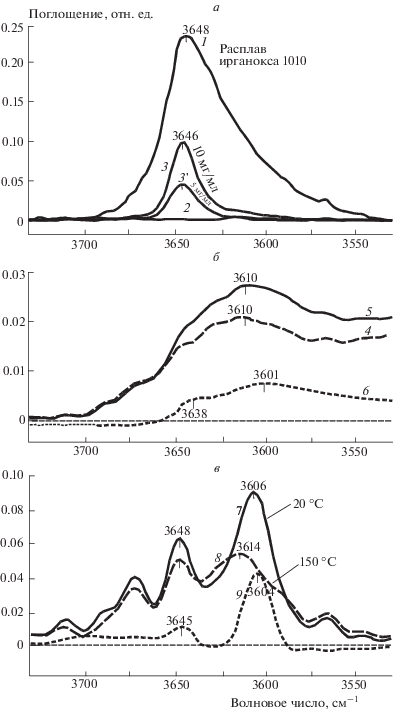
Следует также отметить, что сосуществование свободных и связанных колебаний водорода в гидроксильных группах характерно для раствора ирганокса 1010 в ацетоне – растворителе, имеющем в своем составе карбонильную группу. На рис. 2 представлена разность интенсивностей двух спектров: раствора, содержащего АО, и чистого растворителя – ацетона (спектр 6). Видно, что в растворе имеются как свободные (пик поглощения при 3638 см–1), так и связанные (при 3601 см–1) гидроксильные группы АО. Можно считать, что при сорбции АО полиэтиленом гидроксильные группы ингибитора вступают в образование как внутримолекулярных, так и межмолекулярных водородных связей.
Как было отмечено выше, на стадии нагрева образца величина S3640 уменьшается, при этом основной спад этого показателя осуществляется в интервале температур плавления полимера – участок нагрева Н-2 (при охлаждении образца имеет место обратное увеличение S3640 со скачком в температурном диапазоне кристаллизации полимера – участок охлаждения О-2). На рис. 2в на фоне ИК-спектров в области поглощения гидроксильных групп АО в полиэтилене приведена частотная зависимость разности интенсивностей двух спектров (дифференциальный спектр), снятых при граничных температурах стадии нагрева 20 и 150 °С (рис. 2, спектр 9). Частотная зависимость полосы дифференциального спектра поглощения имеет максимум при 3604 см–1. С переводом полимера в расплав это поглощение исчезает (при обратном переходе в закристаллизованное состояние оно появляется снова), и отсюда можно сделать вывод о том, что спектр 9, представленный на рис. 2, фактически представляет собой частотное распределение интенсивности ИК-излучения, рассеваемого на кристаллитных образованиях полимера. Естественно, что при плавлении кристаллитов в полимере поглощение света на этих образованиях не происходит.
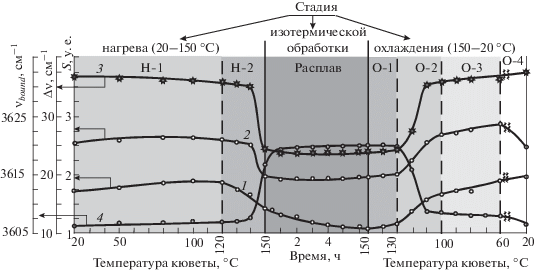
Полученный результат позволяет уточнить интерпретацию структуры полосы поглощения, отнесенной нами к гидроксильным группам (полоса поглощения в частотном диапазоне 3730–3530 см–1). Эта полоса образована наложением двух видов поглощений: собственно поглощений свободных и связанных гидроксильных групп и рассеяния света на кристаллитных образованиях, распределенных в объеме полимера. Если из всего поглощения излучения в рассматриваемой частотной области исключить рассеяние света на кристаллитах (дифференциальный спектр 9 на рис. 2), то оставшееся поглощение будет относиться только к поглощению свободными (высокочастотный пик) и связанными (низкочастотный пик) гидроксильными группами (рис. 2, спектр 8). Так как максимум в частотном распределении рассеянного света лежит в низкочастотной области (около 3604 см–1), то после исключения спектра рассеянного света происходит сдвиг положения пика, относящегося к колебаниям гидроксильных групп, входящих в водородные ассоциаты (частота νbound), в сторону меньших частот. Именно поэтому после плавления кристаллитов, т.е. когда в ИК-спектрах пропадает составляющая, связанная с рассеянием света (рис. 2, спектр 8), происходит скачкообразное уменьшение на 11.3 см–1 разности частот Δν = νfree – νbound; здесь νfree – частота пика, относящегося к свободным колебаниям гидроксильной группы (рис. 3, кривая 3, участок Н-2). Этот скачок определяется, по существу, изменением частоты νbound (рис. 3, кривая 4, участок Н-2). Соответственно, при кристаллизации полимерного образца на стадии охлаждения (в интервале от 130 до 100 °С) также наблюдается скачок величины Δν и опять таки за счет частоты νbound. А частота νbound уменьшается за счет появления рассеяния света из-за образования в полимере кристаллитов (рис. 3, кривые 3, 4, участок О-2). Становится понятным, что возникающие скачки Δν при плавлении или кристаллизации полимера обусловлены изменением формы низкочастотного пика поглощения. При плавлении кристаллитов вследствие снижения уровня рассеяния света частично “обрезается” низкочастотная ветвь ИК-спектра. В результате значение νbound увеличивается, т.е. эта частота приближается к νfree и, как следствие, наблюдается скачок величины Δν. При кристаллизации полимера, т.е. при появлении рассеяния света на кристаллитах, происходят уширение спектра и сдвиг пика поглощения в низкочастотную область. В итоге пиковые значения νbound, как и величина Δν, увеличиваются.
Рис. 3.
Зависимости показателей Sfree (1), Sbound (2) для связанных и свободных гидроксильных групп, а также разности частот ∆ν = νfree – νbound (3) и частоты νbound (4) в процессе прохождения отдельных стадий испытания образца: нагрева, изотермической обработки (температура кюветы – 1590 °С) и охлаждения. Концентрация АО в пленках – 0.1 мас.%.
Стадия изотермической обработки образца в расплавленном состоянии является основной в изучении поведения АО в индукционном периоде окисления. Заметим, что так как на этой стадии процесса рассеяние света на кристаллитах не происходит, то по величине S3640 можно достаточно точно определять содержание гидроксильных групп в составе АО, сорбированного полимером.
В первом цикле испытаний изотермическая обработка образца продолжалась 6.5 ч, что значительно меньше индукционного периода, который при концентрации АО, равной 0.1 мас.%, составлял 31 ч [16, 17]. В ходе экспериментов было установлено, что при термообработке расплава полимера происходит дополнительная сорбция АО. Главная причина этого – увеличение сорбционной емкости полимера в результате плавления кристаллитов. Уже отмечалось [8], что внутри кристаллитных образований кристаллизующихся ингибированных полимеров АО практически отсутствует. Однако после плавления кристаллитов и перевода этой части полимера в аморфное состояние становится возможным дальнейший диффузионный перенос АО в локальные зоны образца, которые раньше входили в состав кристаллитов и не содержали ингибирующей добавки. Подтверждением дополнительной сорбции АО полимером является рост площади полосы поглощения, относящейся к карбонильным группам АО. Так, за 6.5 ч термообработки образца в расплавленном состоянии величина S1745 увеличилась с 1.09 до 1.93 у. е. – на 77% по отношению к начальному моменту, когда образец был нагрет до температуры 150 °С (рис. 1, кривая 2). Подтверждением дополнительного сорбционного поглощения АО расплавом полимера является также рост S1500 – площади полосы поглощения, относящейся к ароматическим структурам антиокислительной добавки (рис. 1, кривая 3). Таким образом, результаты эксперимента однозначно подтвердили факт дополнительной сорбции – поглощения АО, который поступал в аморфную фазу полимера из диспергированного состояния, не наблюдаемого в ИК-спектрах образца. Поступающий в полимер АО главным образом дислоцировался в локальных зонах образца – зонах прежнего содержания кристаллитных образований.
На фоне дополнительного сорбирования АО полимером, связанного с изменением показателей S1745 и S1500, аналогичного прироста площади полосы, относящейся к гидроксильным группам, S3640, не произошло (рис. 1, кривая 1). Более того, за 6.5 ч термообработки в расплаве показатель S3640 даже снизился с 3.29 до 3.11 у. е. Очевидно, что наряду с диффузионным поступлением АО в полимер происходит частичное разрушение гидроксильных групп, входящих в состав антиокислительной добавки. Полученные данные позволяют считать, что наряду с дополнительной сорбцией АО по местам прежнего содержания в полимере кристаллитов развивается второй процесс – разрушение гидроксильных групп ингибитора. Этот процесс является достаточно интенсивным, так как, несмотря на приток АО, общее количество принадлежащих ему гидроксильных групп со временем снижается. Можно попытаться оценить степень разрушения (расходования) гидроксильных групп АО при его дополнительном сорбировании расплавом полимера.
До изотермической обработки образца в расплавленном состоянии относительный коэффициент для гидроксильных групп ${{K}_{{{\text{OH}}}}} = {{{{S}_{{{\text{3640}}}}}} \mathord{\left/ {\vphantom {{{{S}_{{{\text{3640}}}}}} {{{S}_{{1745}}}}}} \right. \kern-0em} {{{S}_{{1745}}}}} = 3.02.$ Как уже отмечалось, к моменту окончания термической обработки в расплаве значения показателей площадей полос поглощения изменились следующим образом: S3640 снизилась до 3.11 у. е., а S1745 увеличилась до 1.93 у. е. Полагая, что во время дополнительной сорбции АО полимером гидроксильные группы не разрушаются, т.е. KOH при термообработке расплава остается постоянным и равным 3.02, можно легко рассчитать ожидаемые текущие значения показателя $S_{{{\text{364}}0}}^{'}$ = KOHS1745, где S1745 – значение показателя площади полосы поглощения карбонильных групп к моменту окончания термообработки. В результате расчетов получаем, что $S_{{{\text{364}}0}}^{'}$ = 3.02 ⋅ 1.92 = 5.83 у. е. На самом же деле произошло не увеличение показателя S3640, а наоборот, его снижение. Отсюда понятно, что к моменту окончания термообработки потеря гидроксильных групп, вызываемая их разрушением, составила ΔS = $S_{{{\text{364}}0}}^{'}$ – S3640 = 5.83 – 3.11 = 2.72 у. е. (что в процентах к ожидаемому – гипотетическому количеству равно $\Delta S = {{(\Delta S} \mathord{\left/ {\vphantom {{(\Delta S} {S_{{3640}}^{{\text{'}}})}}} \right. \kern-0em} {S_{{3640}}^{{\text{'}}})}} \cdot 100 = 46.7\% $). Видно, что практически половина гидроксильных групп АО, наблюдаемого в образце методом ИК-спектроскопии, была разрушена. В условном пересчете получается, что из четырех гидроксильных групп, приходящихся на одну молекулу АО, после окончания стадии термообработки в первом цикле испытаний в среднем остались только две.
Отрыв водорода от гидроксильных групп АО, т.е. их разрушение, в неравной степени затрагивает группы, находящиеся в свободном и связанном состояниях. Частотный диапазон полосы поглощения, относящейся к гидроксильным группам (3730–3530 см–1), был разделен на два равных участка. Первый, высокочастотный участок поглощения (3730–3630 см–1) в основном относился к ИК-излучению, поглощаемому свободными гидроксильными группами; второй, низкочастотный участок (3630–3530 см–1) – к поглощению ИК-излучения водородом в составе связанного гидроксила. В соответствии с принятым делением на участки основной полосы при 3640 см–1 были отдельно рассчитаны площади полос, относящихся к свободным (Sfree) и связанным (Sbound) гидроксильным группам. На рис. 3 (кривые 1, 2) представлено изменение показателей Sfree и Sbound на всех трех стадиях обработки образца в ходе первого цикла испытаний.
При нагреве образца величины Sfree и Sbound изменяются аналогично изменению суммарного показателя S3640, относящегося ко всем гидроксильным группам АО, сорбированного полимером (рис. 1, кривая 1). На участке плавления полимера (температурный интервал Н-2) по причине уменьшения рассеяния света происходит интенсивный спад величин Sfree и Sbound. Однако, при обратном процессе – кристаллизации полимера (температурный участок охлаждения О-2) из-за появляющегося светорассеяния на кристаллитных образованиях величины Sfree и Sbound достаточно быстро возрастают. Заслуживает внимания ход этих зависимостей в процессе изотермической термообработки полимера (рис. 3, срединный участок кривых 1 и 2). Как видно, при термообработке расплава полимера фиксируемое в ИК-спектрах содержание свободных гидроксильных групп в АО постепенно уменьшается, в то время как аналогичный показатель для связанных групп даже несколько возрастает. Следует также иметь в виду, что во время изотермической термообработки происходит дополнительное сорбирование АО расплавом полимера. Соответственно, имеет место приток гидроксильных групп, которые при этом разрушаются (расходуются), дезактивируя возникающие в полимере радикалы – инициаторы дальнейших окислительных превращений. К концу изотермической обработки доля свободных гидроксильных групп в образце становится меньше, чем в начале этой стадии обработки. В отношении связанных гидроксильных групп можно заметить, что их количество к моменту завершения термообработки образца в расплаве увеличивается. В целом такая тенденция – уменьшение количества свободных гидроксильных групп и увеличение групп связанного гидроксила характерна и для стадии изотермического воздействия на образец в других циклах его испытаний. Естественно, что при этом суммарная продолжительность термообработки в расплаве не может превышать времени индукционного периода окисления.
По завершению изотермической обработки к ее повторению мы возвращаемся только после двух следующих стадий обработки образца. Вначале образец охлаждают до комнатной температуры, а затем после некоторой паузы его вновь нагревают до температуры 150 °С, и далее формируется очередная стадия термообработки образца в расплавленном состоянии.
Стадия охлаждения образца была разбита на четыре температурных участка: участок О-1 (температура 150–130 °С) – образец еще продолжает оставаться в расплавленном состоянии, и по данным ИК-спектроскопии нет никаких признаков кристаллизации; участок О-2 (130–100 °С) – в полимере идет интенсивное образование кристаллитной структуры; участок О-3 (100–60 °С) – в полимере в основном формируется надкристаллитная структура; последний участок О-4 (60–20 °С), на котором продолжается формирование надмолекулярной структуры и выдерживается технологическая пауза перед проведением очередного цикла испытаний. В силу специфики программы эксперимента температурный участок О-4 оказался самым продолжительным – до 17 ч, так как он включал в себя нерабочее, ночное время, на котором эксперимент прерывался.
Оказалось, что при проведении промежуточных стадий испытательного цикла – охлаждение и нагрев – в образце продолжают протекать процессы, связанные с перераспределением АО и продуктов его превращений в объеме полимера. Так, если к окончанию изотермической обработки в первом цикле испытаний образца значения показателей S3640 и S1745 составляли 3.11 и 1.93 у. е., то к началу очередной стадии термообработки во втором цикле испытаний значения указанных величин уже составили 3.03 и 1.33 соответственно. Здесь снова обратимся к относительному коэффициенту ${{K}_{{{\text{OH}}}}} = {{{{S}_{{{\text{3640}}}}}} \mathord{\left/ {\vphantom {{{{S}_{{{\text{3640}}}}}} {{{S}_{{1745}}}}}} \right. \kern-0em} {{{S}_{{1745}}}}}.$ Чем выше KOH, тем более высока доля гидроксильных групп в общей массе сорбированного в полимере АО. Так как именно гидроксильные группы обуславливают антиокислительный эффект, то можно говорить, что KOH характеризует состояние АО, т.е. его “качество”. Результаты эксперимента показывают, что после охлаждения и повторного нагрева образца до 150 °С качество сорбированного АО заметно улучшилось – относительный коэффициент KOH вырос с 1.61 до 2.28 отн. ед. Такие изменения в составе АО стали возможными из-за вытеснения при кристаллизации части разрушенной антиокислительной добавки с более низким содержанием гидроксильных групп. В результате проведения двух видов воздействия на образец: охлаждения после первого цикла изотермической обработки в расплаве и нагрева до 150 °С для осуществления новой стадии термообработки, показатели S1745 и S3640 снизились соответственно на 0.60 и 0.08 у. е. При этом отношение KOH для состава вытесненного АО сильно уменьшилось от исходного значения, равного 3.02, до очень низкого – 0.13. Наиболее интенсивное вытеснение отработанного АО происходило на участке охлаждения О-4 (величины S3640 и S1745 снизились соответственно на 0.82 и 0.31 у. е.).
Вытеснение продуктов превращений АО на стадиях охлаждения и нагрева образца убедительно продемонстрировано рис. 4. Здесь в схематичной форме представлено изменение коэффициента KOH в моменты завершения очередной стадии термообработки образца (четные точки на графике) и начала следующего (очередного) цикла термообработки (нечетные точки). Время на рисунке соответствует суммарной продолжительности изотермической обработки образца в расплавленном состоянии. Дадим необходимые пояснения по представленной зависимости.
Рис. 4.
Динамика коэффициента KOH от продолжительности изотермической обработки образца при температуре 150 °С (нечетные точки – значения коэффициента перед началом очередного этапа термообработки, четные – в момент ее завершения на данном этапе; другие пояснения – в тексте).
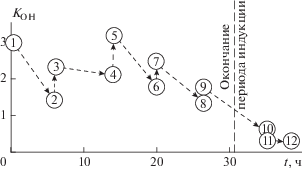
В начальном положении (точка 1 на рис. 4) образец был впервые нагрет до температуры 150 °С, и далее в течение 6.5 ч продолжалась его обработка при этой температуре. В это время происходило дополнительное сорбирование АО и одновременное его расходование. По завершению термообработки значение KOH снизилось (точка 2). Далее, следуя по стрелке, мы переходим в точку 3 – за это время образец прошел стадии охлаждения и повторного нагрева до 150 °С. Значение KOH несколько выросло, так как часть разрушенного АО из сорбированного состояния была вытеснена в дисперсную фазу. В точке 3 начинается второй цикл изотермической обработки образца, в котором, как и при переходе 1 → 2, происходит уменьшение KOH – точка 4. При переходе из 4 → 5, как и при переходе 2 → 3, происходит улучшение качества антиокислительной добавки из-за вытеснения части отработанного АО. Далее, вплоть до окончания индукционного периода (указан на графике вертикальной штриховой линией), цикличность в изменении KOH повторяется. К окончанию периода индукции значение коэффициента KOH значительно снижается, и далее он становится крайне малым из-за накопления в образце продуктов окисления самого полиэтилена в виде карбонильных групп.
Из данных рис. 4 следует, что при 6-7-часовых циклах термообработки суммарный индукционный период окисления образца составил 31 ч. При проведении эксперимента, в котором продолжительность изотермической обработки образца составляла 10 ч за один цикл испытаний, продолжительность индукционного периода окисления образца снизилась и составила всего 28.5 ч, несмотря на одинаковую концентрацию антиоксиданта в составе полимера (0.1 мас.% ирганокса 1010). Обсудим полученный результат более подробно. С увеличением времени термообработки уменьшается число фазовых переходов полимерного образца в кристаллическое состояние. Как было показано выше, переход в кристаллическое состояние ингибированного полимера сопровождается некоторым улучшением “качества” сорбированного антиоксиданта (показатель KOH возрастает). Сокращение числа фазовых переходов закономерно привело к уменьшению времени протекания процесса, приводящего к улучшению качества антиоксиданта, что отразилось на продолжительности суммарного индукционного периода окисления.
ЗАКЛЮЧЕНИЕ
С использованием метода фурье-ИК-спектроскопии изучены физико-химические изменения состояния фенольного антиоксиданта ирганокс 1010 в образцах полиэтилена, подвергнутых окислению в расплавленном состоянии с циклически повторяемыми фазовыми переходами расплав ↔ твердый полимер. Установлено, что при переходе полиэтилена в расплав происходит дополнительное сорбирование антиоксиданта аморфной фазой полимера. При обратном переходе, наоборот, идет десорбция антиокислительной добавки и продуктов ее превращений. Выявлено, что одновременно с дополнительной сорбцией антиоксиданта достаточно интенсивно происходит разрушение части его гидроксильных групп, т.е. имеет место расходование антиоксиданта. При десорбции из аморфной фазы полимерного образца в основном вытесняется более поврежденный продукт (с разрушенными гидроксильными группами), и поэтому “качество” антиоксиданта, остающегося в виде сорбированного полимера, несколько улучшается.
В итоге, при циклическом окислении расплава полимера на стадии периода индукции циклически изменяется физико-химическое состояние антиоксиданта – в ходе термообработки расплава образца уменьшается относительное количество гидроксильных групп антиоксиданта, а для твердой фазы образца это количество увеличивается. Увеличение количества фазовых переходов приводит к увеличению продолжительности суммарного индукционного периода окисления полимерного образца, ингибированного антиоксидантом фенольного типа.
Автор выражает благодарность проф. Лину Дмитрию Григорьевичу (1945–2017) за помощь и поддержку при выполнении и написании работы.
Работа выполнена в рамках государственной программы научных исследований “Физическое материаловедение, новые материалы и технологии” (подпрограмма “Полимерные материалы и технологии”, задание 6.35, № 20160352), а также при финансовой поддержке Белорусского республиканского фонда фундаментальных исследований (договор № Т17-033).
Список литературы
Фойгт И. Стабилизация синтетических полимеров против действия света и тепла. Л.: Химия, 1972.
Richaud E. // Eur. Polym. J. 2013. V. 49. № 8. P. 2223.
Jong-Il Weon // Polym. Degrad. and Stab. 2010. V. 95. № 1. P. 14.
Pimentel Real L.E., Miguel B. Correia // Polym. Test. 2012. V. 31. № 7. P. 963.
Liu D., Pourrahimi A.M., Pallon L.K.H. et al. // Polym. Degrad. and Stab. 2016. V. 125. P. 21.
Lundbäck M., Hedenqvist M.S., Mattozzi A., Gedde U.W. // Ibid. 2006. V. 91. № 7. P.1571.
Richaud E., Monchy-Leroy C., Colin X., Audouin L., Verdu J. // Ibid. 2009. V. 94. № 11. P. 2004–2014.
Шляпников Ю.А. Антиокислительная стабилизация полимеров. М.: Химия, 1986.
Цвайфель Х., Маер Р.Д., Шиллер М. Добавки к полимерам. Справочник. / Пер. с англ. под. ред. Узденского В.Б., Григорова А.О. М.: Профи-Информ, 2010.
Лин Д.Г., Воробьева Е.В., Шаповалов В.М. // Материалы, технологии, инструменты. 2015. № 1. Т. 20. С. 70.
Ghaffari M., Ahmadian V. // Radiat. Phys. and Chem. 2007. V. 76. № 11. P. 1666.
Lambert J.B., Shurvell H.F., Cooks R.G. Organic Structural Spectroscopy Prentice Hall: Upper Saddle River, 1998.
Дехант И. Инфракрасная спектроскопия полимеров. М.: Химия, 1976.
Lobo H., Bonilla J.V. Handbook of plastics analysis. New York: Marcel Dekker, Inc., 2003.
Vorobyova E.V., Lin D.G. // ISJ Theoretical & Appl. Sci. 2016. V. 39. № 7. P. 17.
Lin D.G., Vorob’eva E.V. // Rus. J. Appl. Chem. 2013. V. 86. № 1. P. 82.
Лин Д.Г., Воробьева Е.В. // Полимерные материалы и технологии. 2017. № 2. Т. 3. С. 41.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика