

Химическая физика, 2021, T. 40, № 1, стр. 30-36
Влияние полярности растворителя на скорость термического разложения нитропиразолов
Г. М. Назин 1, В. В. Дубихин 1, И. Л. Далингер 2, А. И. Казаков 1, *, А. В. Набатова 1
1 Институт проблем химической физики Российской академии наук
Черноголовка, Россия
2 Институт органической химии им. Н.Д. Зелинского Российской академии наук
Москва, Россия
* E-mail: akazakov@icp.ac.ru
Поступила в редакцию 27.12.2019
После доработки 15.06.2020
Принята к публикации 22.06.2020
Аннотация
На примере реакций термического разложения нитропиразолов в растворе изучена возможность идентификации согласованного механизма разложения С-нитросоединений по зависимости скорости реакции от полярности растворителя.
ВВЕДЕНИЕ
Предпринятое в данной работе исследование термического разложения нитропроизводных пиразола в растворах связано в основном с необходимостью решения актуальной для С-нитросоединений проблемы – нахождением новых способов идентификации согласованного механизма разложения этих соединений. Результаты квантовохимических расчетов энергии активации [1] и обширный экспериментальный материал (см. обзор [2] и более поздние работы [3, 4]) показывают, что при разложении алифатических и ароматических С-нитросоединений в основном реализуются два механизма: простой, заключающийся в разрыве связи C–N, и более сложный, согласованный распад, при котором одновременно разрываются и образуются две связи. Простой механизм изучен более детально, хотя согласованные реакции распространены не менее широко. Согласно работам [1] и [2] к ним относятся следующие типы реакций.
1. Элиминирование HNO2 из мононитроалканов, имеющее циклическое пятичленное переходное состояние:
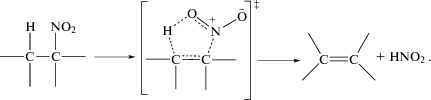
2. Внутримолекулярное окисление нитроалкенов, идущее через переходное состояние типа оксазет:
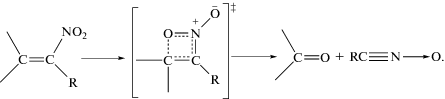
3. Перегруппировка в аци-форму через 1,5-сигматропный сдвиг водорода нитроалкенов с заместителями ОН, NH2, CH3 в цис-положении:
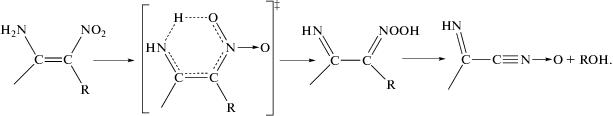
4. Эта же перегруппировка о-нитропроизводных анилина, фенола и толуола.
5. Разложение полинитропроизводных бензола с орто-расположением нитрогрупп по 2-му типу реакций. Фактически реакции 2-го и 3-го типов охватывают все непредельные алифатические и орто-замещенные ароматические нитросоединения.
Экспериментальное доказательство механизма этих реакций основано на значениях кинетических параметров. Согласно теории мономолекулярных реакций [5] разрыв связи C–N, сопровождающийся образованием двух больших групп, должен иметь предэкспоненциальный множитель А > 1014 с–1 и энергию активации близкую к энергии диссоциации связи С–N, Eакт = D(C–N). Такое сочетание кинетических параметров у согласованных реакций не встречается. Все они имеют А < 1013 c–1 и Еакт < D(C–N).
Для нескольких нитроалканов и одного нитроолефина, PhC=C(CH3)NO2, согласованный механизм распада был доказан по составу первичных продуктов разложения [2]. Однако точное определение кинетических параметров и тем более анализ продуктов требуют трудоемких исследований, которые к тому же не всегда выполнимы.
Еще одна возможность идентификации согласованного механизма связана с изучением влияния полярности среды на скорость реакции при проведении реакции в растворах. Влияние полярности на скорость обусловлено неспецифической сольватацией дипольных частиц. При этом адекватной мерой полярности считается относительная диэлектрическая проницаемость ε [6]. В переходном состоянии простых реакций происходит лишь небольшое удлинение связи C–N, дипольный момент и сольватация молекулы при этом изменяются мало, и скорость реакции от ε не зависит. В переходном состоянии согласованных реакций из-за сильного изменения пространственного расположения атомов и разной полярности разрывающихся и образующихся связей дипольный момент может измениться, и тогда скорость будет зависеть от ε.
Систематические исследования указанных выше С-нитросоединений в растворах не проводились, они изучались только в газовой фазе. Перенос этих реакций в раствор с целью отработки новой методики затруднен, в основном из-за высокой температуры разложения. Сравнительно недавно было найдено [7, 8], что распад ди- и тринитропроизводных пиразола тоже протекает по согласованному механизму. Причем эти реакции идут в жидкой фазе при значительно более низких температурах, чем у других нитросоединений. Именно по этой причине в данной работе, которая, по-видимому, является первой попыткой изучения влияния растворителя на скорость согласованных реакций при высоких температурах, в качестве объектов исследований выбраны нитропроизводные пиразола. В этом классе соединений, отличающемся большим разнообразием структур, встречаются реакции 2-го и 3-го типов. В работе планировалось прежде всего экспериментально проверить: сохраняется ли при температурах выше 200 °С ряд полярности растворителей, установленный по величине ε при низкой температуре, какие растворители пригодны для работы при высоких температурах и на какие типы согласованных реакций влияет полярность среды.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве объектов исследования были использованы 3,3',4,4'-тетранитро-2H,2H'-3,3'-бипиразол-5,5' (I), 1-метил-3,4,5-тринитропиразол (II), 3,3',5,5'-тетранитро-1H,1H'-бипиразол-4,4' (III) и 4-амино-3,5-динитро-1H-пиразол (IV).
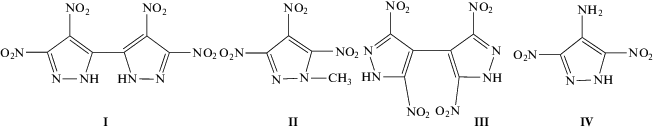
Эти соединения синтезированы по методикам, приведенным в работах [9–11].
Растворители, использованные для создания ряда полярности, приведены в табл. 1. Все эти соединения доступны, стабильны по крайней мере до 350 °С, имеют очень высокие критические температуры и в максимально возможной степени охватывают интервал изменения ε.
Таблица 1.
Характеристики соединений, использованных в качестве растворителей
| Соединение | Ткип, °С | ε (при 25 °С) |
|---|---|---|
| Дифенил | 296 | 2.0 |
| о-Дихлорбензол | 180 | 5.62 |
| Бензонитрил | 191 | 25.2 |
| Нитробензол | 210 | 36.0 |
| Сульфолан | 285 | 41.0 |
| м-Динитробензол | 302 | – |
Кинетику реакций измеряли манометрическим методом. Использовалась описанная ранее [7] установка, позволяющая измерять давление до 40 атм компенсационным методом в стеклянных ампулах. Для удобства сравнения температуру опытов выбирали так, чтобы значения констант скорости лежали в интервале 10–4–10–6 с–1. Скорости газовыделения измерены при концентрации растворов 1 мас.%. Примеры кинетических кривых разложения соединений I–IV в нитробензоле и дифениле приведены на рис. 1. Во всех случаях наблюдается первый порядок реакции. Константы скорости первого порядка k1 приведены в табл. 2. В случае разложения соединений I и III в дифениле и нитробензоле значения k1 определены из двух параллельных опытов. Расхождение не превышает 10%.
Рис. 1.
Зависимость глубины разложения (η) соединений I–IV в нитробензоле (○) и дифениле (∆). Концентрация – 1.0 мас.%. Перечислены: соединение и температура (°С): 1 – II, 220; 2 – I, 230; 3 – III, 260; 4 – IV, 240; 5 – IV, 240; 6 – III, 300; 7 – I, 230; 8 – II, 220.
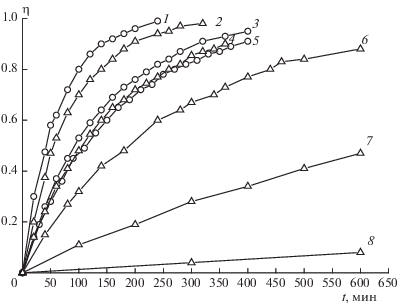
Таблица 2.
Значения констант скорости разложения нитропроизводных пиразола в растворах
| Соединение | Растворитель | Т, °С | k1, с–1 | kотн | E, кДж/моль | lg(A, c–1) |
|---|---|---|---|---|---|---|
| I | Дифенил | 210 | 4.2 ⋅ 10–6 | 1.0 | 147.3 | 10.68 |
| 230 | 1.8 ⋅ 10–5 | 1.0 | ||||
| о-Дихлорбензол | 210 | 1.3 ⋅ 10–5 | 3.2 | 131.4 | 9.34 | |
| 230 | 4.9 ⋅ 10–5 | 2.2 | ||||
| Бензонитрил | 210 | 3.8 ⋅ 10–5 | 9.0 | 96.1 | 9.63 | |
| 230 | 1.5 ⋅ 10–4 | 8.3 | ||||
| Нитробензол | 210 | 5.6 ⋅ 10–5 | 13.5 | 95.2 | 10.2 | |
| 230 | 2.0 ⋅ 10–4 | 11.3 | ||||
| Сульфолан | 210 | 2.6 ⋅ 10–5 | 6.2 | 114.4 | 12.1 | |
| 230 | 1.2 ⋅ 10–4 | 6.7 | ||||
| м-Динитробензол | 230 | 1.2 ⋅ 10–4 | 6.7 | – | – | |
| II | Дифенил | 220 | 2.6 ⋅ 10–6 | 1.0 | – | – |
| о-Дихлорбензол | 220 | 7.2 ⋅ 10–6 | 2.8 | – | – | |
| Бензонитрил | 220 | 5.4 ⋅ 10–4 | 207 | – | – | |
| Нитробензол | 220 | 2.7 ⋅ 10–4 | 102 | – | – | |
| III | Дифенил | 260 | 4.0 ⋅ 10–6 | 1.0 | 128.6 | 11.65 |
| 300 | 6.3 ⋅ 10–5 | – | ||||
| Нитробензол | 230 | 1.7 ⋅ 10–5 | – | 108.5 | 10.52 | |
| 260 | 1.2 ⋅ 10–5 | 31 | ||||
| IV | Дифенил | 240 | 1.1 ⋅ 10–4 | 1.0 | – | – |
| Нитробензол | 240 | 1.0 ⋅ 10–4 | 0.9 | – | – |
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Прежде чем анализировать зависимость константы скорости k1 от полярности растворителя, необходимо показать надежность методики измерения k1 в растворах и обосновать правильность механизма, выбранного для каждого из изученных соединений. В связи с этим полезно также провести сравнение результатов, полученных в растворах, с литературными данными по разложению в расплаве.
Основное исследование выполнено на примере соединения I, имеющего в ряду динитропроизводных пиразола самую высокую скорость разложения. Распад I в расплаве исследован в работе [12]. Реакция протекает по уравнению 1-го порядка и имеет Eакт = 161.4 кДж/моль и lg(A, с–1) = 13.23. Расчет с использованием этих величин дает для k1 значения 6.4 ⋅ 10–5 с–1 при 210 °С и 3.2 ⋅ 10–4 с–1 при 230 °С. По данным табл. 2 в нитробензоле при этих же температурах константы k1 равны соответственно 5.6 ⋅ 10–5 с–1 при 210 °С и 2.0 ⋅ 10–4 с–1 при 230 °C. Наблюдается неплохое соответствие данных для расплава и нитробензола. По-видимому, значения диэлектрической проницаемости расплава I и нитробензола тоже близки.
Главным инструментом при выборе конкретного механизма согласованной реакции служит квантовохимический расчет энергии активации для разных каналов распада и выявление реакции, для которой эта величина является наименьшей. Расчеты Еакт, выполненные для нитропиразолов [13], не охватывают всех возможных путей превращения и не дают следовательно полного представления о конкуренции разных путей разложения. Поэтому при выборе механизма изученных соединений мы ориентировались на более полные данные, полученные для мононитроолефинов и о-динитробензола – непредельных нитросоединений, к типу которых относятся нитропиразолы. В обзорной работе [1] показано, что наиболее выгодным механизмом разложения нитроолефинов и о-динитробензола является внутримолекулярное окисление нитрогруппой соседнего атома углерода (реакция (2)), а для нитроолефинов с водородсодержащими заместителями (NH2, OH, CH3) при двойной связи и аналогичных орто-производных нитробензола таким процессом является перегруппировка в аци-форму через 1,5-сигматропный сдвиг водорода (реакция (3)). Поэтому можно принять, что распад соединений I, II и III идет через окисление, а соединение IV в первом акте изомеризуется в аци-форму.
При переходе от дифенила к нитробензолу наблюдается увеличение константы скорости разложения I примерно в 10 раз как при 210 °С, так и при 230 °С (табл. 2). Это означает, что переходное состояние реакции имеет бóльший дипольный момент и сольватируется сильнее, чем исходная молекула, что приводит к уменьшению Eакт и росту k1 при повышении полярности среды. Действительно, в реакциях типа 2 дипольный момент формирующейся связи С–О должен быть значительно больше, чем дипольный момент разрывающейся связи O–N.
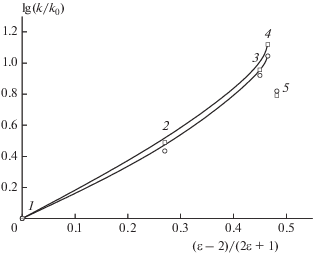
Данные по разложению соединения I в координатах уравнения Кирквуда представлены на рис. 2. Видно, что даже в ряду ароматических растворителей это уравнение описывает реакцию весьма приближенно. Сильный рост констант наблюдается в области самых больших значений ε. Этот результат не является неожиданным. Даже для сольволиза сложных эфиров − классических реакций гетеролитического типа, идущих при низких температурах, это уравнение выполняется редко [6]. Поэтому для учета влияния среды широко используются эмпирические шкалы полярности, построенные по lgk самих реакций. Но и они имеют ограниченное применение.
Рис. 2.
Зависимость lg(k/k0) от параметра уравнения Кирквуда для реакции разложения соединения I при 230 °С (⚪) и 210 °С (◻). Константа скорости k0 относится к дифенилу. Растворители: 1 – дифенил, 2 – о-дихлорбензол, 3 – бензонитрил, 4 – нитробензол, 5 – сульфолан.
Из рис. 1 и табл. 2 видно, что плавный рост константы скорости при увеличении ε нарушается при переходе от ароматических растворителей к сульфолану. В этом растворителе, имеющем при комнатной температуре полярность более высокую, чем нитробензол, константа скорости не растет по сравнению с нитробензолом, а уменьшается примерно в 2 раза. Возможно, что это связано с непропорционально сильным по сравнению с ароматическими соединениями уменьшением диэлектрической проницаемости сульфолана при увеличении температуры. Во избежание возможной путаницы сульфолан в качестве растворителя в дальнейшем не использовался.
Для того чтобы обнаружить согласованный механизм, не обязательно заниматься построением шкалы полярности растворителей. Важно знать, что при высоких температурах расположение соединений в ряду полярности сохраняется. Для выявления полярного эффекта вообще не требуется использовать растворители со средними значениями ε. В практическом плане достаточно применить упрощенный тест, заключающийся в измерении скорости разложения в одном слабо полярном растворителе (например, в дифениле или хлорбензоле) и в одном сильно полярном растворителе, например в нитробензоле или м-динитробензоле. Последний менее летуч, чем нитробензол, а по влиянию на скорость реакций всего в 2 раза уступает нитробензолу (табл. 2).
Термическое разложение соединения II в расплаве ранее исследовали в нескольких работах. В работах [7, 8] было установлено, что в отличие от динитропроизводных пиразола разложение II в жидкой фазе протекает с сильным ускорением. Промежуточным продуктом является диоксид азота, который, по-видимому, и вызывает увеличение скорости распада. Кинетика разложения соединения II в расплаве исследована в работе [14] методом дифференциальной сканирующей калориметрии (ДСК) в потоке инертного газа. Измеренная константа скорости оказалась на два порядка выше, чем в нитробензоле. Следует, однако, подчеркнуть, что метод калориметрии в потоке совершенно не пригоден для изучения кинетики разложения летучих соединений, к числу которых относится соединение II. Этим методом одновременно фиксируются три процесса: испарение вещества и реакции разложения в газовой и конденсированной фазах. При достаточно большой упругости паров испарение сильно искажает пик тепловыделения и делает невозможным правильное определение константы скорости разложения. Таким образом, расхождение с литературными данными в случае соединения II связано с некорректностью проточной методики ДСК.
Эффект полярности среды у соединения II выражен значительно сильнее, чем у I. При переходе от дифенила к нитробензолу скорость меняется в 100 раз. Это является следствием большого положительного заряда на реакционном центре (атом углерода в положении 4) в исходной молекуле. Из-за индукционного влияния соседних нитрогрупп этот центр изначально имеет пониженную электронную плотность, что обеспечивает большой дипольный момент в структуре типа оксазет.
Однако самая большая скорость разложения соединения II наблюдается в бензонитриле, что связано, по-видимому, с побочной реакцией окисления этого растворителя посредством диоксида азота, выделяющегося при разложении II. Очевидно, что бензонитрил нельзя использовать в качестве растворителя при изучении веществ, распад которых сопровождается выделением NO2. Таким образом, это уже второе соединение из табл. 1, которое целесообразно удалить из ряда полярности растворителей.
При разложении соединения III, которое отличается повышенной устойчивостью и изучалось при более высоких температурах (230–260 °С), повторяются закономерности, обнаруженные у соединения I. Скорость разложения III в нитробензоле в 30 раз выше, чем в дифениле и связано это с уменьшением энергии активации.
В случае соединения IV реализуется третий тип механизма распада нитросоединений – изомеризация непредельных нитросоединений в аци-форму (нитроновую кислоту) путем 1,5-сигматропного сдвига водорода. Выбор этого механизма для IV основан, как уже отмечалось, на его подобии с орто-нитроанилином и нитроолефинами, имеющими аминогруппу в качестве заместителя в цис-положении. Квантовохимический расчет [1] дает для изомеризации этих соединений самую низкую энергию активации по сравнению с другими каналами распада. Следует отметить, что для второй стадии распада аминонитроалкенов, т.е. разложения нитроновой кислоты, расчетная энергия активации оказалась значительно выше, чем для первичной изомеризации. Поэтому этот путь распада был признан в работе [1] тупиковым. Однако запрет, справедливый для газовой фазы, легко снимается в случае, если реакция идет в конденсированном состоянии вещества. Известно (см. обзоры в работах [2, 15]), что нитроновые кислоты являются неустойчивыми соединениями. Выделенные в свободном состоянии, они быстро разлагаются уже при 100 °С. Из-за частичной изомеризации в аци-форму скорость разложения жидкого нитрометана на пять порядков превышает скорость гомогенной реакции в газовой фазе [16]. Поэтому представляется наиболее вероятным, что распад соединения IV идет по типу реакции (3).
В экспериментальной работе [17] методом ИК-спектроскопии определили, что при разложении IV (таблетированная смесь с KBr) происходит одновременное уменьшение интенсивностей полос поглощения амино- и нитрогрупп, а первым продуктом, который появляется в газовой фазе, является вода. Эти факты согласуются с распадом IV по механизму, представленному реакцией (3). Согласно этому механизму исчезновение NH2 и одной группы NO2 происходит уже в первом элементарном акте, а H2O образуется в быстрых вторичных реакциях.
Кинетика разложения соединения IV в расплаве была исследована в работе [17] методом ДСК в плоской ячейке с прижатой крышкой (т.е. при очень большом m/V) при атмосферном давлении. Найденные параметры реакции, Eакт = 107 кДж/моль и lg(A, с–1) = 10.7, дают значение константы скорости при 140 °С 1.4 ⋅ 10–3 с–1, что на три порядка превышает k1 в растворе нитробензола (табл. 2). В 7 раз меньше чем представлено в работе [17] оказалась константа скорости, полученная в работе [12] тем же методом в ячейках с проколотой крышкой, но тоже при внешнем давлении в 1 атм. Такие большие скорости распада, измеренные методом ДСК, связаны с необычным характером реакции разложения соединения IV. В работе [12] показано, что распад IV в твердой фазе протекает с ярко выраженным индукционным периодом, после которого наблюдается сильнейший рост скорости разложения. Реакция напоминает автокаталитический процесс с большим отношением констант скоростей каталитической и начальной стадий, k2/k1. При повышении температуры период индукции сокращается и при приближении к точке плавления (~186 °С) становится равным 0. Поэтому возможно, что при исследовании жидкофазного распада методом ДСК регистрируется пик тепловыделения, целиком обусловленный каталитическим процессом, и определяется константа скорости k2, а не k1.
Пример соединений II и IV убедительно показывает преимущество использования методики исследования в разбавленных растворах для получения характеристик реакции разложения, не осложненной испарением, примесными, каталитическими или другими побочными процессами. Судя по фактическому равенству скоростей в дифениле и нитробензоле (рис. 1, табл. 2), полярность среды на скорость разложения IV не влияет. Это означает, что дипольный момент в процессе изомеризации меняется мало. Можно предположить (как это уже было сделано при обсуждении соединения I), что изменение дипольного момента зависит главным образом от разности дипольных моментов рвущейся и образующейся связи. В случае соединения IV разрывающаяся связь N–H имеет дипольный момент μ = 1.66 D, а для связи O–H, которая появляется, μ = 1.58 D. Эти величины близки, и поэтому изменение полярности при образовании переходного состояния невелико. Если такие приближенные оценки полярности справедливы, то можно ожидать, что растворитель не будет влиять на скорость согласованных реакций 3-го и 5-го типов в тех случаях, когда заместителем является не только NH2-группа, но и группа OH. Поэтому вероятно, что о-нитроанилин и о-нитрофенол во всех растворителях будут иметь одинаковую скорость разложения. Однако в случае о-нитротолуола из-за большой разницы дипольных моментов связей С–Н (0.4D) и О–Н (1.58 D) влияние полярности среды должно проявиться.
Таким образом, предлагаемый способ идентификации согласованного механизма может быть полезен, как и предполагалось, только в случае реакций, сопровождающихся изменением дипольного момента при образовании переходного состояния. Однако и при таком ограничении его можно использовать для трактовки механизма многих реакций, идущих при повышенных температурах. В группу этих реакций попадает распад гетероциклических соединений. В частности, до сих пор остается открытым вопрос о детальном механизме распада фуразанов, фуроксанов, изоксазолинов, оксадиазолов и некоторых производных тетразола. Идут ли эти реакции по двухступенчатому бирадикальному или одностадийному согласованному механизмам? Чаще всего ни по кинетическим параметрам, ни по составу продуктов различить механизмы распада этих гетероциклов невозможно.
ЗАКЛЮЧЕНИЕ
Скорости согласованных реакций разложения С-нитросоединений разных типов зависят от полярности растворителя во всех случаях, когда при образовании переходного состояния происходит изменение дипольного момента. Расположение растворителей в ряду полярности, установленное по величине диэлектрической проницаемости при комнатной температуре, не меняется при высоких температурах. Лучшими растворителями для работ при температурах до 350 °С являются ароматические углеводороды, хлор- и нитропроизводные бензола. Разработанный метод полезно использовать для изучения механизма распада гетероциклических соединений, если эти реакции идут при температурах ниже 350 °С.
Работа выполнена на средства ИПХФ РАН по теме 0089-2019-0005 “Фундаментальные и проблемно-ориентированные исследования в области создания энергетических конденсированных систем (ЭКС) различного назначения” (номер госрегистрации АААА-А19-119100800130-0) при финансовой поддержке программой Президиума РАН “Перспективные физико-химические технологии специального назначения”).
Список литературы
Храпковский Г.М., Шамов А.Г., Николаева Е.В. и др. // Успехи химии. 2009. Т. 78. № 10. С. 980; https://doi.org/10.1070/RC2009v078n10ABEH004053
Манелис Г.Б., Назин Г.М., Рубцов Ю.И. и др. Термическое разложение и горение взрывчатых веществ и порохов. М.: Наука, 1996.
Гончаров Т.К., Дубихин В.В., Назин Г.М. и др. // Хим. физика. 2006. Т. 25. № 7. С. 33.
Неделько В.В., Захаров В.В., Корсунский Б.Л. и др. // Хим. физика. 2015. Т. 34. № 12. С. 39.
Бенсон С. Термохимическая кинетика. Пер. с англ. / Под ред. Ениколопяна Н.С. М.: Мир, 1971.
Пальм В.А. Основы количественной теории органических реакций. М.: Химия, 1967.
Дубихин В.В., Назин Г.М., Прокудин В.Г. и др. // Хим. физика. 2015. Т. 34. № 3. С. 28; https://doi.org/10.7868/S0207401X15030036
Дубихин В.В., Назин Г.М., Прокудин В.Г. и др. // Изв. АН. Сер. хим. 2015. Т. 64. № 1. С. 126.
Dalinger I.L., Shkinyova T.K., Shevelev S.A. et al. // Proc. 29th Intern. Annual Conf. ICT. Karlsruhe, Germany, 1998. P. 57.
Dalinger I.L., Vatsadze I.A., Shkineva T.K. et al. // Mendeleev Commun. 2010. V. 20. № 5. P. 253; https://doi.org/10.1016/j.mencom.2010.09.003
Dalinger I.L., Vatsadze I.A., Shkineva T.K. et al. // Synthesis. 2012. № 13. P. 2058; https://doi.org/10.1055/s-0031-1291134
Sinditskii V.P., Smirnov S.P., Egorshev V.Yu. et al. // Thermochim. Acta. 2017. V. 651. P. 83; https://doi.org/10.1016/j.tca.2017.02.019
Dalinger I., Shevelev S., Korolev V. et al. // J. Therm. Anal. Calorim. 2011. V. 105. № 2. P. 509; https://doi.org/10.1007/s10973-010-1213-y
Ravi P., Gore G.M., Sikder A.K. et al. // Thermochim. Acta. 2012. V. 528. P. 53; https://doi.org/10.1016/j.tca.2011.11.001
Химия нитро- и нитрозогрупп / Под ред. Фойера Г. Пер. с англ. М.: Мир, 1972.
Храпковский Г.М., Столяров П.Н., Дорожкин В.П. и др. // Хим. физика. 1990. Т. 9. С. 648.
Wang Y.L., Zhao F.Q., Ji Y.P. et al. // J. Anal. Appl. Pyrolysis. 2012. V. 98. P. 231; https://doi.org/10.1016/j.jaap.2012.08.014
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика