

Химическая физика, 2021, T. 40, № 8, стр. 34-42
Влияние насыпного слоя катализатора на прохождение фронта водородно-воздушного пламени в ограниченном пространстве
С. Н. Козлов 1, *, А. М. Тереза 2, С. П. Медведев 2
1 Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
Москва, Россия
2 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова
Российской академии наук
Москва, Россия
* E-mail: kozlovse@yandex.ru
Поступила в редакцию 12.02.2021
После доработки 18.02.2021
Принята к публикации 22.02.2021
Аннотация
В связи с тяжелыми последствиями, возникающими после аварий на АЭС, вызванных или сопровождающихся взрывами и воспламенениями водородно-воздушных смесей, появилась необходимость более тщательного подхода к изучению сопутствующих им физико-химических процессов. В работе представлен краткий анализ данных ситуаций в свете водородной взрывобезопасности на АЭС и применения катализаторов на основе платины и палладия с целью утилизации водорода. Экспериментально исследовано взаимодействие водородно-воздушного пламени с поверхностью насыпного катализатора и проведено сравнение эффективностей выбранных катализаторов по отношению к гетерогенной реакции окисления водорода. Сделан анализ зависимости экспериментальных результатов от конструкции и места расположения функциональных зон реактора.
ВВЕДЕНИЕ
В связи с присутствием водорода на АЭС исследования по обеспечению безопасности охватывают вопросы генерации водорода, его распространения по помещениям АЭС, воспламенения и горения, включая вопросы устранения последствий сгорания водорода и контроля, т.е. обнаружения и управления процессами его распределения и накопления. Проблема возникла в связи с известными крупными авариями на АЭС “Три-Майл-Айленд” (США), Чернобыльской АЭС (СССР), Фукусимской АЭС (Япония). Когда в процессе аварии на АЭС образуется значительное количество водорода в корпусе водоохлаждаемого реактора, возникает опасность его истечения и последующего воспламенения. Взрыв такой массы водорода непосредственно угрожает целостности элементов и конструкций герметичного ограждения АЭС. В настоящее время уже сделан значительный шаг в изучении источников образования водорода [1, 2] и созданы программы расчета распространения его по помещениям и накопления под защитной оболочкой [3]. В трехкомпонентной смеси водород – воздух – водяной пар под защитной оболочкой более легкий водород может концентрироваться под потолком помещений, что увеличивает вероятность воспламенения. Взрыв смеси водород–воздух может привести к разгерметизации защитной оболочки, выбросу больших количеств радиоактивных материалов и распространению их на значительные расстояния.
Удаление поверхностных защитных оксидных пленок с металлических материалов реактора в результате аварии при больших температурах способствует генерации водорода в процессе его восстановления из молекул воды. Водород при таких авариях накапливается в основном в результате пароциркониевой реакции Zr + 2H2O = = ZrO2 + 2H2, а также при взаимодействии расплава активной зоны с бетоном, что может приводить к выбросу большого количества водорода в защитную оболочку. И это стало важной проблемой обеспечения водородной безопасности при авариях на АЭС.
Главной причиной выделения водорода в первом контуре охлаждения является радиолиз теплоносителя, происходящий в меньшей мере при штатной и в большей мере при нештатной работе АЭС. При наличии в теплоносителе первого контура аммиака идет его разложение с образованием водорода и азота. При отсутствии в теплоносителе аммиака идет разложение воды с образованием водорода, кислорода и перекиси водорода. Образующиеся при радиолизе теплоносителя газы в неповрежденных конструкциях, как правило, находятся в растворенном состоянии.
С учетом высокой текучести водорода появляется опасность его накопления в отдельных технологических и строительных конструкциях, что приводит к превышению его допустимых концентрационных пределов в воздухе. Вследствие этого одной их основных задач в обеспечении пожаровзрывобезопасности на АЭС является проведение мероприятий по удалению и утилизации водорода. Один из способов утилизации – каталитическое превращение водорода и кислорода воздуха в воду. Для этого для АЭС разработаны и применяются каталитические устройства, называемые рекомбинаторами водорода (точнее, пассивными каталитическими рекомбинаторами водорода (ПКРВ)) [4]. Термин “пассивный” в данном случае означает энергонезависимый, не потребляющий энергии. Конструкции рекомбинаторов заполняются катализаторами, позволяющими утилизировать водород, т.е. превращать его в воду в гетерогенной реакции окисления даже при низких температурах (273–293 К) и концентрациях водорода в воздухе (0.5–1.0%). Для этой цели чаще всего применяются катализаторы гетерогенного гидрирования, представляющие собой многокомпонентные каталитические системы на основе платины, палладия и других переходных металлов. В большинстве коммерчески доступных конструкций ПКРВ каталитические активные материалы (обычно это платина или палладий) нанесены на несущие элементы – металлические пластины или пористые керамические элементы с развитой поверхностью контакта (шарики, цилиндры и т.д.). Конструкции ПКРВ размещаются в вертикальных металлических шкафах с перфорированными низом и верхом для конвективной смены воздуха помещений АЭС. В связи с вышеизложенным очень большой интерес представляют исследования влияния катализаторов на прохождение фронта водородно-воздушного пламени над их поверхностью в ограниченном пространстве.
Цели данной работы – изучение влияния насыпного слоя катализаторов на распространение водородно-воздушного пламени и сравнение эффективности катализаторов на основе платины и палладия по скорости гетерогенной каталитической реакции окисления водорода в смеси с воздухом при комнатной температуре и начальном атмосферном давлении.
АНАЛИЗ СОВРЕМЕННОГО СОСТОЯНИЯ ПРОБЛЕМЫ
Несмотря на обширную научную литературу, освещающую каталитическую реакцию водорода с кислородом, в ее детальном механизме еще остается много неясного. Перечислим некоторые, уже принятые положения, относящиеся к данной реакции: преобладание механизма попеременного восстановления–окисления поверхности, имеющего место при обычных температурах катализа; на фоне диссоциативной хемосорбции кислорода и водорода при избытке кислорода происходит взаимодействие молекулярного водорода из газовой фазы с сорбированным атомом кислорода с образованием воды; при избытке водорода взаимодействие молекулярного кислорода с диссоциированным на поверхности водородом приводит к образованию адсорбированных радикалов HO2 и OH-групп. Схема взаимодействия может быть представлена в следующем виде:
В схему включены только начальные стадии и реакции, непосредственно участвующие в получении конечного продукта – воды. Процессы диссоциативной адсорбции исходных веществ и десорбции продуктов реакции подразумеваются. При достаточно медленной реакции и невысокой температуре в механизм также можно включить равновесную адсорбцию реагирующих веществ в молекулярной форме. Реакция идет до тех пор, пока не закончатся реагенты.
Наибольшей активностью обладают платина, палладий, никель. Чаще всего в научной литературе и учебных пособиях рассматривается окисление водорода в присутствии платины [5–7]. На металлической платине, обладающей развитой поверхностью, реакция катализа протекает очень быстро. Платина в ходе катализа испытывает значительное влияние реакционной смеси, которое заключается в растворении кислорода в поверхностном слое металла. Кинетика окисления водорода платиной исследовалась во многих работах, где применялись статический или проточный методы [8, 9].
При избытке кислорода в реакционной смеси предполагается существование двух стационарных режимов, для каждого из которых характерны свои скорость и кинетика окисления водорода [7]. В области высоких скоростей возможен цепной механизм реакции; активными центрами являются лабильные, богатые энергией поверхностные соединения. При снижении температуры и откачке эти соединения разрушаются, и в реакции участвует адсорбированный кислород, реагирующий по обычному (нецепному) механизму с меньшей скоростью. Уменьшение скорости при повышении давления кислорода можно объяснить усилением блокировки поверхности прочносвязанным кислородом.
В работе Г. Эртля [10] с помощью метода сканирующей туннельной микроскопии удалось проследить стадии окисления Н2 на платине. Водород и кислород адсорбируются в виде атомов и образуют далее OH-группы. Также они могут образоваться в ходе реакции [O]s + H2O → 2[OH]s. Малые островки OH-групп концентрируются с образованием кольца, которое расширяется внутрь поверхности, покрытой кислородом. Продукт реакции аккумулируется позади фронта реакции. Общая картина оказалась более сложной, чем предполагали.
Следует отметить, что существует проблема совместимости данных, полученных в области глубокого вакуума и в области реального катализа [11]. Было показано, что окисление водорода является структурно-нечувствительной реакцией. Удельная каталитическая активность и кинетика реакции на изученных гранях платины практически одинаковы и совпадают с данными для поликристаллических образцов. Скорость стационарной реакции определяется стадиями диссоциативной адсорбции водорода и кислорода и не зависит от их дальнейшего превращения вследствие больших констант взаимодействия в адсорбированном слое.
Существует целый ряд веществ, вызывающих потерю активности катализатора – обратимое или необратимое отравление катализатора [12]. При обратимом отравлении катализатора молекулы “яда” адсорбируются на поверхности катализатора, блокируют ее, прекращая доступ молекулам реагирующего вещества к активным центрам. Примером обратимого отравления катализатора в реакции окисления водорода может быть дезактивация платины окисью углерода, этиленом или самим молекулярным кислородом. В работе [13] исследовали скорость гетерогенного окисления водорода в области между вторым и третьим пределом воспламенения на поверхностях реактора из SiO2, MgO и нержавеющей стали. При смене очередности раздельного напуска кислорода и водорода в реактор скорости реакции различались на порядок величины. Когда напускали сначала O2, скорость была намного ниже.
При взаимодействии водорода с палладием, а также с некоторыми другими металлами образуются объемные гидриды. Например, в системе Pd–H2, в которой при температуре 203 К образуется PdH2, а при более высокой температуре (373 К) и при достаточных давлениях водорода над поверхностью – PdH0.6 [5]. Таким образом, часть водорода из водородно-воздушной смеси поглощается палладием и может не участвовать в процессе окисления. При нагревании катализатора водород выделяется и уменьшает степень “гидридности” Pd. То же самое происходит в системе Ti–H2 [14], но только в другом температурном интервале.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исследуемых объектов использовали следующие каталитически активные вещества.
1. Катализатор селективного гидрирования марки РК-415 (КПШ-1.5) состава 1.5% Pd/γ-Al2O3 в виде шарообразных гранул диаметром 4 мм и удельной поверхностью 120 м2/г (далее – катализатор 1).
2. Катализатор очистки газовых выбросов АЭС от водорода и дейтерия марки ОПК-2 состава 0.2% Pt/γ-Al2O3 в виде шарообразных гранул диаметром 4 мм (далее – катализатор 2).
3. Катализатор рекомбинации водорода для систем водородной безопасности марки РК-104 состава 1% Pd/γ-Al2O3 в виде экструдатов (цилиндров) диаметром 4 мм и длиной 2–8 мм (далее – катализатор 3).
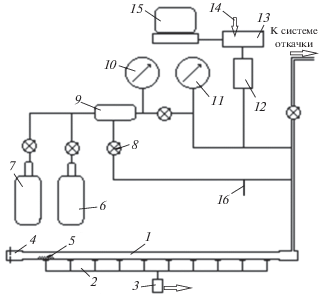
Функциональная схема установки представлена на рис. 1. Откачка установки осуществлялась с помощью форвакуумного насоса 2НВР-5ДМ через ресивер и ловушки с жидким азотом. Исследовано влияние насыпного слоя катализаторов на распространение водородно-воздушного пламени и проведено сравнение трех видов катализаторов по их эффективности для гетерогенной каталитической реакции водородно-воздушной смеси.
Рис. 1.
Схема установки для проверки эффективности действия катализаторов на распространение пламени в реакции водорода с воздухом: 1 – трубчатый кварцевый реактор с внутренним диаметром 12 мм, длиной 130 см и объемом ≈150 см3; 2 – световоды для 10 точек регистрации свечения фронта пламени на длине реактора 90 см; 3 – фотодиод марки C30839 (RCA Solid State Division, USA) со спектральной чувствительностью на длинах волн в диапазоне 200–1000 нм; 4 – искровая камера для поджигания смеси; 5 – расположение катализатора в реакторе; 6 – баллон с водородом; 7 – баллон с воздухом; 8 – газовые вентили; 9 – смесительная камера для газов из нержавеющей стали емкостью 3 л; 10 – манометр образцовый с диапазоном измерения давления до 0.4 МПа; 11 – вакуумметр образцовый с диапазоном измерения давления 0–100 кПа; 12 – датчик давления–разрежения “Сапфир-22 ДВ-2240” с диапазоном измерения давления 100–0 кПа; 13 – электронный блок обработки сигналов с АЦП E14-140-M фирмы “L-CARD”; 14 – вход для сигнала с фотодиода; 15 – компьютер с программным обеспечением PowerGraph Professional; 16 – трубчатый отросток для вымораживания газообразных продуктов реакции.
Проведение экспериментов
В смесительной камере 9 (рис. 1) составляли нужную смесь водорода с воздухом общим давлением в 0.2 МПа. После откачки реактора до давления в 1 Па, в него вводили смесь до атмосферного давления. Зажигание смеси производили с помощью электрической искры в разрядной камере 4 (см. рис. 1). Факт прохождения пламени устанавливали по измерению манометрами начального и конечного давления в реакторе. Визуально и фотоприемником через световоды пламя не было зарегистрировано ввиду слабости свечения. Базовые эксперименты проводили без катализатора при атмосферном давлении смеси водорода с воздухом и начальной концентрации водорода 6%. Поскольку кварцевая трубка-реактор имеет малый внутренний диаметр (12 мм), следовало ожидать, что искровое поджигание смеси при горизонтальном распространении пламени будет эффективным при концентрациях водорода, превышающих 6%. Неустойчивое горение с выгоранием не более 1.5% и погасанием пламени вблизи точки поджигания наблюдали при концентрации водорода в воздухе 8%. Выгоранием будем называть разницу между начальным и конечным значениями давления при проведении эксперимента, выраженную в процентах. При концентрации водорода 10% наблюдали нестабильность в воспроизведении опытов, и выгорание смеси от опыта к опыту изменялось от 2 до 6.5%. Наиболее устойчивое горение смеси наблюдали при концентрации водорода в смеси 12% при стабильных конечных выгораниях от 9.1 до 9.5% от общего давления смеси. Такие, кажущиеся “аномально” большими, выгорания появляются из-за процесса конденсации образующихся в реакции паров воды на стенках реактора при комнатной температуре. Смесь 12% водорода с воздухом была выбрана в качестве “рабочей” для испытания катализаторов.
Влияние катализаторов на распространение пламени
Навеску гранулированного материала катализатора 5 размещали на расстоянии в 20 см от искровой камеры равномерно на длине около 7 см, как это показано на рис. 1. Каждый катализатор в гранулах общим весом 2.4 г вводили в реактор путем насыпки продолговатой формы в один-два слоя, причем высота середины насыпки не перекрывала половины сечения трубки реактора. Влияние катализатора на способность смеси к воспламенению будет зависеть от времени ее выдержки до искрового поджига. При зажигании смеси, сразу после ее напуска в реактор в присутствии катализатора, выгорание по сравнению с реактором без катализатора резко сокращается и составляет не более 2.5% от общего давления смеси.
Через 2–3 мин выдержки смеси в присутствии катализатора прекращается ее искровое воспламенение, и падение давления в реакторе происходит только за счет каталитической реакции окисления водорода. Эти явления вполне понятны. При напуске в реактор смесь на пути к искровой камере 4 (см. рис. 1) проходит зону реактора с катализатором и обедняется водородом. Условия для поджигания смеси в искровой камере ухудшаются, и с течением времени воспламенение смеси становится невозможным.
На рис. 2 представлены экспериментальные данные по регистрации пламени в присутствии катализатора 1. Датчик давления разрежения дает положительный возрастающий сигнал при уменьшении давления от атмосферного до нуля, поэтому для удобства на рис. 2 и 3 представлен инвертированный сигнал датчика.
Рис. 2.
Влияние времени задержки зажигания смеси на ее способность к воспламенению в присутствии катализатора: кривая 1 – опыт 4, кривая 2 – опыт 8, кривая 3 – опыт 7. В соответствии с результатами и параметрами опытов, представленными в табл. 1.
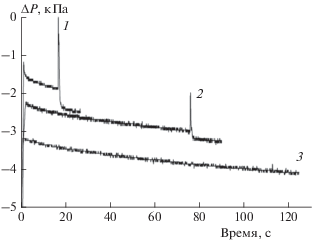
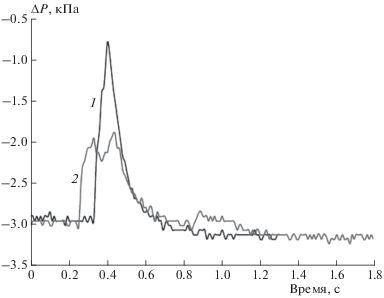
Рис. 3.
Формы импульсов давления при прохождении фронта пламени по реактору, когда реакционный фронт движется только по горизонтальной трубке реактора (1) и когда пламя переходит в конце реактора в перпендикулярную ему подводящую трубку меньшего диаметра с небольшой задержкой по времени (2).
При проведении экспериментов варьируемым параметром является время tign от окончания напуска смеси до инициирования электрического разряда. Как упоминалось выше, при выдержке более 2 мин смесь от искры не воспламеняется и “выгорание” происходит только за счет гетерогенной каталитической реакции. На кривой 3 рис. 2 поджигание смеси произвели на 113-й секунде, и произошла едва заметная вспышка. В табл. 1 представлены результаты опытов по определению критических условий поджигания смеси в реакторе в присутствии катализатора 1.
Таблица 1.
Результаты экспериментов с катализатором 1
| Номер опыта | Время напуска, с | Время до вспышки tign, с | Степень выгорания смеси за счет распространения фронта пламени, кПа | Средняя скорость распространения фронта пламени, м/с |
|---|---|---|---|---|
| 1 | 5 | 7 | 0.67 | 16.25 |
| 2 | 14 | 8 | 0.17 | 18.5 |
| 3 | 5 | 10 | 0.17 | 17.5 |
| 4 | 4 | 16.5 | 0.50 | 16.25 |
| 5 | 7 | 5 | 0.22 | 16.25 |
| 6 | 5 | 42 | 0.12 | 16.25 |
| 7 | 14 | 113 | 0 | 0 |
| 8 | 8 | 73.5 | 0.22 | 16.7 |
Исходя из формы и продолжительности по времени импульса давления в реакторе в зависимости от вспышки и прохождения фронта пламени, можно оценить среднюю скорость распространения фронта пламени по трубчатому реактору. Поскольку фронт пламени движется по реактору до тех пор, пока идет реакция в газовой фазе, которая сопровождается выделением тепла и повышением давления в реакторе, то время тепловыделения и есть время, за которое фронт пламени проходит весь реактор.
На рис. 3 представлены два вида формы импульса давления (данные опыта № 3 и № 8 из табл. 1) для следующих случаев: 1 – когда пламя распространяется только от искровой камеры до конца трубки реактора, 2 – когда пламя переходит в конце реактора в перпендикулярную ему подводящую трубку меньшего диаметра с небольшой задержкой по времени. Когда фронт пламени достигает конца реактора, пламя гаснет. Подводящая трубка (меньшего диаметра) заполнена такой же смесью, но расположена она перпендикулярно направлению трубки реактора, и поэтому не всегда создаются условия для продолжения распространения пламени в ней. Когда эти условия созданы, импульс давления имеет “двугорбый” характер, связанный с задержкой воспламенения в подводящей трубке. Средняя скорость распространения пламени определяется по длительности переднего фронта импульса давления при известной длине реактора. Обратный склон импульса давления соответствует времени тепловой релаксации смеси. Время переднего фронта импульса давления для кривой 1 рис. 3 составило 0.074 с, а для кривой 2 – 0.078 с, и, соответственно, средние скорости распространения пламени будут равны 17.5 и 16.7 м/с при длине реактора 1.3 м. Данные по скорости распространения фронта пламени для других опытов также представлены в табл. 1.
Из проведенных экспериментов следует важный вывод: катализаторы, применяемые в рекомбинаторах, понижают концентрацию водорода в смеси и затрудняют при искровом поджоге воспламенение и распространение пламени водородно-воздушных смесей. Это хорошо видно в реакторах малого объема в статических условиях опыта. Данное свойство катализатора можно использовать для повышения водородной взрывобезопасности. Вблизи катализатора вероятность поджигания смеси будет ниже. Все три катализатора проявляют подобные свойства, но в разной степени. Следует заметить, что в условиях, более благоприятных для прохождения гетерогенной реакции окисления (стехиометрическая смесь водорода с кислородом, отсутствие отвода тепла), катализатор может нагреваться до высоких температур и способствовать самовоспламенению реакционной смеси при концентрации водорода более 5%. Аналогичные явления наблюдались в работах [15, 16], в которых установлено, что воспламенение может произойти от активированной кварцевой поверхности [15] или воздействия светового облучения [16], возможного при авариях на АЭС.
Результаты экспериментов показывают, что, несмотря на разную интенсивность горения во фронте пламени (по степени выгорания), скорости его распространения имеют достаточно близкие значения, и в качестве первого приближения в данных условиях экспериментов скорость можно считать постоянной и не зависящей от степени выгорания смеси и амплитуды импульса давления. Это означает, что из-за разных масштабов времен процессов, происходящих во фронте пламени и при гетерогенной каталитической реакции окисления водорода, их взаимное влияние в одном опыте минимизировано. Время прохождения фронта пламени над насыпкой катализатора составляет ≈4 ⋅ 10–3 с.
Сравнение эффективности катализаторов по скорости гетерогенной реакции
Эффективности катализаторов по отношению к реакции окисления водорода лучше всего сравнивать по ее скорости. Все три катализатора испытывали в одинаковых условиях: температура комнатная, 293 К; смесь 12% водорода с воздухом; начальное давление – 100 кПа. Для непрерывной регистрации кинетики изменения давления в реакторе при гетерогенной каталитической реакции применяли датчик-преобразователь давления разрежения “Сапфир-22ДВ-2240” с диапазоном измерения 100–0 кПа.
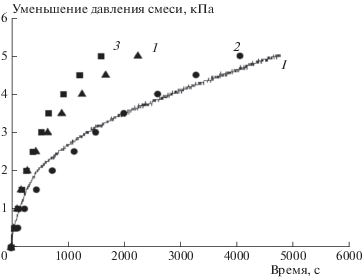
На рис. 4 представлена зависимость изменения давления водородно-воздушной смеси от времени (скорость уменьшения давления смеси можно считать аналогом эффективности катализатора). Непрерывная регистрация кривой I относится к катализатору 1, порция которого подвергалась предварительному воздействию водородно-воздушного пламени и гетерогенной реакции. Точки ▲, ⚫ и ◼ отображают регистрацию давления, а нумерация “кривых” соответствует номеру катализатора. Понижение давления фиксировали во времени через каждые 0.5 кПа на образцовом вакуумметре 11 (см. рис. 1). Каждый образец перед опытом выдерживали 15 мин в реакторе при давлении 1 Па. Катализаторы 1–3, которые использовали при точечной регистрации, не подвергались предварительным испытаниям. Из рис. 4 видно, что активность катализатора 1 (1.5% Pd) изменяется в сторону уменьшения с увеличением времени его обработки реакционной смесью и практически сравнивается с активностью свежего катализатора 2 (0.2% Pt). Практика применения водородных рекомбинаторов на АЭС показывает, что при долговременной эксплуатации катализаторов их свойства стабилизируются, а реанимационные мероприятия для них потребуются реально через 1–2 года (заявленный некоторыми производителями срок службы каталитических блоков ПКРВ – до 5 лет). В наших экспериментальных условиях среди свежих катализаторов лучший результат по активности показал образец 3 (1.0% Pd).
Анализ прохождения гетерогенной каталитической реакции окисления водорода
На рис. 5 показаны непрерывная кинетическая кривая падения давления в результате гетерогенного окисления водорода на катализаторе 1 и момент определения количества паров воды методом вымораживания. В районе 500-й секунды реакции хорошо заметен излом на кинетической кривой, что означает смену механизма (характера) реакции.
Рис. 5.
Определение количества паров воды в газовой фазе на момент времени 6000 с в гетерогенной реакции окисления водорода на образце катализатора 1.
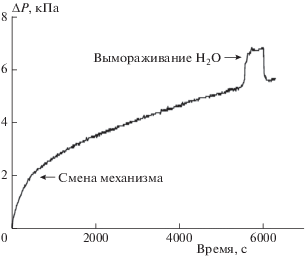
Проанализируем промежуточное состояние реагирующей смеси на 6000-й секунде проведения реакции каталитического окисления водорода на катализаторе 1. Для определения парциального давления паров воды в газовой фазе вымораживаем их в отводе 16 (см. рис. 1) при температуре жидкого азота. Изменение давления в реакторе показано на рис. 5. После этого воду вернули в газовую среду реактора. Изменение давления в реакторе при этом составило 1.3 кПа. Такое количество паров воды присутствовало в реакторе на данный момент времени. Давление насыщенного пара при температуре реактора 293 К составляет 2.34 кПа, т.е. процесс конденсации не происходит (возможные процессы капиллярной конденсации в порах носителя катализатора не учитываем). Изменение давления в реакторе с учетом вымораживания паров воды на 6000-й секунде реакции составило 6.83 кПа. В этом случае при соблюдении стехиометрии реакции на данный момент времени в реакцию должно вступить 4.56 кПа водорода, 2.28 кПа кислорода и образоваться 4.56 кПа воды. Поскольку в газовой фазе реактора присутствует только 1.3 кПа воды, то в сорбированном виде на катализаторе и, возможно, в виде капиллярного конденсата в порах носителя катализатора и на стенках реактора (в малой доле) должен рассредоточиться объем паров воды, эквивалентный 3.25 кПа. При общей поверхности 2.4 г катализатора ≈300 м2 это вполне вероятно. Если принять во внимание, что на 1 нм2 поверхности катализатора приходится четыре посадочных места для адсорбции среднего размера молекул [5], то на 1 м2 можно будет адсорбировать 4 ⋅ 1018 молекул воды, а на 300 м2 – 1.2 ⋅ 1021. Количество молекул, эквивалентное числу молекул в 3.25 кПа паров воды в объеме 200 мл (объем реактора + соединительные трубки), составляет ≈2 ⋅ 1020 молекул. При адсорбции они будут занимать на поверхности катализатора 16% его площади (при условии, что поверхность до этого была свободна от адсорбированных частиц). Конечно, идеальных условий не бывает, но расчет показывает возможность размещения такого количества воды на поверхности катализатора в адсорбированном виде. Если оценка состояния реакционной смеси справедлива, то в данный момент времени реакция прошла на 38%.
Рассмотрим характерные времена диффузии реагентов к месту расположения катализатора из различных участков реакционной трубки. Применим для их оценки формулу Энштейна–Смолуховского для одномерной диффузии: x2 = 2Dτ, где x – средний размер диффузионного пути, D – коэффициент диффузии молекулярного водорода в воздухе при комнатной температуре и атмосферном давлении, τ – характерное время диффузии. Для времени реакции t = 100 с, τ = 100 с, D = 0.64 см2/с [17] получаем, что x = 11.3 см. Это означает, что в реакции в первые 100 с в основном участвуют молекулы со средним расстоянием от центра катализатора, равным 11.3 см. Для t = 500 с это расстояние равно 25.3 см. Для t = 6000 с расстояние составляет 87.6 см. При длине реактора 130 см наши оценки состояния реакционной среды и степени “выгорания” выглядят вполне адекватными. Наши экспериментальные данные уже позволяют с уверенностью сказать, что в закрытых протяженных реакторах скорость каталитического процесса окисления водорода (кроме начальной стадии) лимитируется процессом диффузии компонентов реакционной смеси из дальних зон реактора к месту расположения катализатора. Исходя из вышесказанного, можно сделать вывод о том, что экспериментальные результаты по величине выгорания, способности пламени распространяться по реактору, способности реакционной смеси поджигаться от искры будут зависеть от конфигурации реактора, места расположения насыпного катализатора и искровой камеры зажигания, направления впуска в реактор реакционной смеси. И, кроме этого, общим фоном при проведении экспериментов будет постоянное уменьшение концентрации реагентов по всему реактору из-за взаимодействия с каталитическим материалом.
Начальные стадии гетерогенной реакции
Рассмотрим начальные стадии каталитической реакции окисления водорода применительно к нашим условиям. На рис. 6 представлены экспериментальные данные по кинетике падения давления и скорости рассматриваемой реакции на начальной стадии во временном пределе до 500 с.
Рис. 6.
Гетерогенная реакция окисления водорода в начальный период времени ее развития на катализаторе 1. Кривая 1 – изменение давления в реакторе, 2 – изменение скорости реакции.
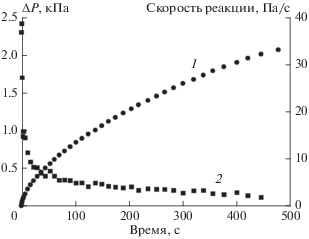
Согласно полученным нами экспериментальным результатам, отображенным на рис. 5 и 6, процессы в реакторе можно условно разделить на три временны́е функциональные зоны: 1 – от 0 до 20 с, 2 – от 20 до 500 с и 3 – от 500 с до окончания реакции.
Первая зона характеризуется быстрым падением давления и скорости реакции в реакторе. Причиной этого может быть повышенная скорость сорбционных процессов в приповерхностных слоях катализатора после длительной выдержки реактора при давлении ≈1 Па.
Вторая зона может характеризоваться скоростью самой каталитической реакции и диффузией реагентов в глубь пористого материала носителя катализатора с последующей адсорбцией и реакцией.
Третья зона характеризуется процессами диффузии реагентов из более дальних районов объема реактора к месту расположения катализатора, диффузией реагентов в глубь пористого катализатора, десорбцией и встречной диффузией продуктов реакции из пор носителя катализатора, самой каталитической реакцией. Скорость указанных процессов лимитируется самым медленным процессом – диффузией реагентов из более дальних частей реактора по отношению к расположению катализатора. Смена определяющего скорость реакции диффузионного механизма транспортировки реагентов к месту расположения катализатора происходит предположительно на 500-й секунде реакции в месте излома кинетической кривой.
ВЫВОДЫ
1. Определена степень взаимодействия фронта водородно-воздушного пламени с поверхностью катализатора. Из-за разных масштабов времен процессов, происходящих во фронте пламени и при гетерогенной каталитической реакции окисления водорода, их взаимное влияние в одном опыте минимизировано.
2. Оценены средние скорости распространения фронта пламени в трубчатом кварцевом реакторе в присутствии насыпного катализатора. Их можно считать, в первом приближении, одинаковыми для разных опытов и не зависящими от степени выгорания смеси и амплитуды импульса давления.
3. Главной причиной прекращения воспламенения смеси является понижение концентрации водорода в районе искровой камеры реактора из-за каталитической реакции окисления водорода.
4. Проведено сравнение активности трех видов катализаторов платиновой группы по отношению к гетерогенной реакции окисления водорода. Наиболее активным при наших испытаниях оказался катализатор 3 (см. раздел “Экспериментальная часть”).
Работа выполнена при частичной финансовой поддержке госкорпорации Росатом (ГК № Н.4ч.241.09.20.1075).
Список литературы
Кириллов И.А., Харитонова Н.Л., Шарафутдинов Р.Б., Хренников Н.Н. // Ядерная и радиационная безопасность. 2017. № 2(84). С. 26; https://nrs-journal.ru/upload/iblock/b84/hydrogen_safety.pdf
Mitigation of hydrogen hazards in water cooled power reactors. IAEA-TECDOC-1196. Vienna: IAEA, 2001.
Григорьев С.Ю. Дис. … канд. физ.-мат. наук. М.: ИБРАЭ РАН, 2016.
Келлер В.Д. // Теплоэнергетика. 2007. № 3. С. 65.
Крылов О.В. // Гетерогенный катализ. М.: Академ-книга, 2004. С. 679.
Моррисон С. Химическая физика поверхности твердого тела. М.: Мир, 1980. С. 488.
Матрос Ю.Ш. Каталитические процессы в нестационарных условиях. Новосибирск: Наука, 1987. С. 228.
Netzer F.R., Kneringer G. // Surface Sci. 1975. V. 51. P. 526.
Acres G.J.K. // Platinum Met. Rev. 1966. V. 10. P. 60.
Ertl G. // Adv. Catal. 1990. V. 37. P. 213.
Катализ и катализаторы. Фундаментальные исследования Института катализа им. Г.К. Борескова / Под ред. чл.-корр. РАН Буянова Р.А. Новосибирск.: ИК СО РАН, 1998.
Захарьевский М.С. Кинетика и катализ. Л.: Изд-во ЛГУ, 1963.
Козлов С.Н., Александров Е.Н., Кузнецов Н.М., Маркевич Е.А. // Хим. физика. 2013. Т. 32. № 11. С. 75.
Козлов С.Н., Сажина Н.Н., Кузнецов Н.М., Александров Е.Н. // Хим. физика. 2005. Т. 24. № 12. С. 3.
Козлов С.Н. // Хим. физика. 2020. Т. 39. № 8. С. 40; https://doi.org/10.31857/S0207401X20080075
Кузнецов Н.М., Козлов С.Н. // Хим. физика. 2019. Т. 38. № 5. С. 29; https://doi.org/10.1134/S0207401X19050078
Физические величины. Справочник / Под ред. Григорьева И.С., Мейлихова Е.З. М.: Энергоатомиздат, 1991.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика