

Химическая физика, 2022, T. 41, № 2, стр. 70-77
Углеродные волокна на основе пека: состояние производства и модификация свойств
И. В. Клименко *
Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
Москва, Россия
* E-mail: inna@deom.chph.ras.ru
Поступила в редакцию 27.07.2021
После доработки 10.08.2021
Принята к публикации 20.08.2021
- EDN: EBGOOS
- DOI: 10.31857/S0207401X22020054
Аннотация
В работе проведен анализ состояния производства углеродных волокнистых материалов, в том числе углеродных волокон (УВ) на основе пека. Показано, что в Российской Федерации имеются большие предпосылки для создания промышленных производств пековых углеродных волокон. Значительное внимание уделяется возможным способам модификации пековых УВ в целях улучшения их электрических свойств. Показана эффективность применения газообразного брома для снижения электрического сопротивления пековых УВ. На основании анализа данных рентгеноструктурного анализа и спектроскопии комбинационного рассеяния света показано, что уменьшение электрического сопротивления в процессе бромирования в 7 раз связано с изменениями, происходящими в структуре волокна.
ВВЕДЕНИЕ
В последние годы в связи с открытием новых форм углерода (фуллерены, нанотрубки, графен) и увеличением объемов потребления промышленностью композиционных материалов с контролируемо изменяемыми свойствами возобновлен интерес к исследованию свойств углеродных волокнистых материалов (УВМ) [1–12]. Последнние являются армирующей основой при производстве современных конструкционных материалов, композитов и углепластиков (композиционных материалов, состоящих из нитей углеродного волокна (УВ) диаметром преимущественно 5–15 мкм, расположенных в матрице из полимерных смол, обычно эпоксидных и полиэфирных). Работы ведутся в основном по трем направлениям: а) исследование физико-химических свойств новых волокнистых материалов и тщательное изучение связи между строением материала и его электронными свойствами с целью расширения областей применения, в том числе создание материалов для применения в медицине; б) поиск путей снижения стоимости волокон; в) поиск способов использования волокон в различных композиционных материалах, в том числе в углепластиках, где в качестве связующих применяются разнообразные термореактивные синтетические смолы и термопласты [13, 14].
Отрасли промышленности, в которых применяются УВМ, различны: электротехническая, химическая, атомная, строительная, аэрокосмическая, а также машиностроение и металлургия. Углеродные волокнистые материалы используются при производстве авиационной и ракетной техники, спортивного инвентаря, товаров медицинского и бытового назначения и т.д. Благодаря отсутствию токсичности и наличию биосовместимости углерода с биологическими тканями эндопротезы из углепластиков начинают широко применяться в хирургии; полые УВ используются как дренажное средство в нейрохирургии, офтальмологии и кардиологии, повязки из графитированной ткани – при лечении глубоких ожоговых и других открытых ран. Такой широчайший спектр применения УВМ объясняется уникальным сочетанием их механических и тепловых свойств, стойкостью при высоких температурах и в агрессивных средах, малым удельным весом и хорошими электро- и теплопроводностью при высокой прочности и жесткости, высокой биосовместимостью, а также возможностью получения различных текстильных форм, например, непрерывных нитей, тканых или нетканых материалов, жгутов, пряжи, ровингов, лент, холстов и т.д.
Основные виды сырья для сложного и высокотехнологичного производства УВМ – это полиакрилонитрил (ПАН), гидратцеллюлоза и пек из продуктов переработки каменного угля и нефти. Углеродные волокна на основе ПАН обладают хорошими прочностными и модульными свойствами, тогда как волокна на основе пека имеют более высокий модуль упругости, но меньшие прочностные характеристики. Основные объемы УВМ производят Япония, США и Германия [15]. В Российской Федерации УВМ производят ОАО “НПК “Химпроминжиниринг” (г. Москва), ООО “Аргон” и ООО “Балаково Карбон Продакшн” (Саратовская область, г. Балаково), ООО “Композит-Волокно” (ХК “Композит”, г. Саратов), “Алабуга-волокно” (ХК “Композит” и Госкорпорация “Росатом”, Татарстан), ООО “Завод углеродных и композиционных материалов” (г. Челябинск), ООО СНВ (г. Саратов), АО “Препрег-СКМ” (в том числе Препрег-Дубна) (г. Москва), ООО “Порше современные материалы” (Калужская обл.) и др. Количество производимого углеродного волокна на этих производствах составляет менее 1% от его мирового производства (~400 т) [13, 16], и в настоящее время наблюдается дефицит УВМ, который в большей степени покрывается импортом.
Стоит заметить, что сегодня в Российской Федерации нет действующих промышленных установок для получения нефтяных пеков, при этом в мировом масштабе доля пекового сырья в производстве УВМ составляет 30% [16]. Вместе с тем нефтяные пеки в отличие от каменноугольных обладают рядом достоинств, например меньшей канцерогенностью и высокой реакционной способностью в термохимических процессах [17, 18]. Использование пеков в качестве сырья при производстве УВ является большим преимуществом благодаря их низкой стоимости, высокому содержанию углерода в них и, как следствие, большому выходу конечного продукта. Это, в свою очередь, приводит к экономии энергоресурсов и ускорению технологического процесса. Но, тем не менее, из-за вредных факторов процесса производства, недостатка оборудования, способного минимизировать выбросы пекококсовых заводов в атмосферу, производство каменноугольного пека также неуклонно сокращается.
В настоящее время УВ из нефтяных пеков получают в Японии, США и некоторых странах Западной Европы. В России пековое волокно производят очень небольшими опытными партиями, однако имеются большие предпосылки для создания промышленных производств пековых углеродных волокон, а именно, огромные сырьевые ресурсы, производственные мощности нефтехимических предприятий, большой научный потенциал [16].
Пек как сырье для производства УВМ представляет собой сложную смесь ароматических углеводородов, которая включает в себя структуры с тремя–восьмичленными кольцами с боковыми алкильными группами со средней молекулярной массой 300–400 [19]. Согласно [20], пек состоит из четырех основных классов химических соединений – низкомолекулярные алифатические соединения, низкомолекулярные нафтеновые ароматические углеводороды, полярные ароматические соединения с более высокой молекулярной массой и гетероциклической природой и асфальтены – фракция пека с наивысшими молекулярной массой и ароматичностью, являющаяся наиболее термически стабильной. Из-за низкого молекулярного веса пеки имеют невысокую температуру плавления и низкую вязкость в расплавленном состоянии, поэтому сформовать из пека можно только хрупкое грубое волокно. Для придания пеку волокнообразующих свойств он подвергается термической и термоокислительной обработке (окислительная стабилизация на воздухе, карбонизация и графитация) с целью повышения молекулярного веса и удаления летучих низкомолекулярных соединений. Содержание углерода в волокне в зависимости от конечной температуры карбонизации составляет 80–99%. Графитация волокна проводится при температурах 2600–3000 °С, и содержание углерода достигает значений >99%. Надмолекулярная структура УВ в большой степени зависит от конечной температуры термообработки: чем она выше, тем больше степень графитации и размер кристаллитов (пачки параллельных турбостратных плоскостей) и меньше дефектность структуры.
Пековые УВ делят обычно на два класса: полученные на основе изотропного пека и на основе анизотропного мезофазного пека с упорядоченной структурой. Эти два типа волокон имеют различные структуру, текстуру и физико-химические свойства. Пековые УВ на основе мезофазного пека, благодаря кристаллической графитовой структуре, высокой степени упорядоченности и непрерывной ориентации слоев вдоль оси волокна и, соответственно, высоким значениям модуля Юнга (1000 ГПа), небольшому удельному электрическому сопротивлению (1.0 мкОм · м), хорошей стойкости к высокотемпературной коррозии, широко используются в промышленности [1, 21]. Данные уникальные свойства связаны с идеальной графитоподобной структурой пекового волокна.
Для создания электропроводящего композита важным условием является наличие определенных характеристик у его двух составляющих (матрицы и наполнителя), в первую очередь механических и электрофизических. Для их обеспечения необходимо подобрать правильное сочетание следующих факторов: прочное связывание волокна с полимером, механическая прочность самих волокон, хорошие проводящие свойства. Для повышения адгезии углеродных волокон к полимеру используются различные методы, такие как термохимическая, электрохимическая, плазмохимическая обработка поверхности волокон и т.д. [22–27]. Механические и электрические свойства волокон определяются их структурой, которая, как уже говорилось, зависит от значения температуры термообработки, вида исходного сырья, наличия дефектов и легирующих элементов.
Таким образом, модификация и контроль структуры и свойств УВ на основе пека могут быть достигнуты путем изменения природы исходного пека, температуры термообработки волокна, а также при помощи введения добавок, интеркалянтов, в само волокно. Интеркаляция может проводится с использованием жидких, твердых или газообразных реагентов. Процесс получения интеркалированного соединения из газовой фазы наиболее прост, однако при этом требуется углеродное волокно с высокой степенью структурного порядка. Обычно увеличение проводимости при интеркалировании высокоориентированных графитовых волокон различными интеркалянтами (солями металлов, кислотами) происходит на один порядок [28]. Наибольший эффект в плане получения высокопроводящих волокон был достигнут при использовании в качестве интеркалянта AsF5. При интеркаляции этим соединением волокон на основе бензола с температурой термообработки 3300 °С получено максимальное значение проводимости (σ) при комнатной температуре: σ = 10–3 мОм ⋅ см, что превышает даже проводимость меди σ = 1.7 ⋅ 10–3 мОм ⋅ см [29]. Это значение σ было получено при измерении сопротивления in situ.
Низкие значения электрического сопротивления волокон были получены при бромировании пековых УВ. Так, например, при бромировании УВ в газовой и жидкой фазах электрическое сопротивление уменьшается в несколько раз [5, 30–40]. Авторы работ [32, 41] сообщают об уменьшении сопротивления пековых волокон в 3–5 раз при бромировании в течение 7 дней. В работах [38, 39, 42] было обнаружено, что удельное электрическое сопротивление пековых волокон уменьшается в 7 раз в результате бромирования: с 1.19 до 0.17 мОм · см. Данные значения сопротивления достигаются при 30-часовом бромировании в газовой фазе. При дальнейшем увеличении времени бромировании до 144 ч происходит рост сопротивления до 0.35 мОм · см. Хорошая временнáя и термическая стабильность свойств бромированных волокон, довольно высокая электрическая проводимость и относительная простота их получения делают эти волокна привлекательными для практического использования.
Исследование самого процесса и механизма интеркаляции, в том числе интеркаляции брома в волокно, важно для понимания процессов, происходящих в структуре углеродного материала при его модификации [36, 43–48]. Так, например, изучение кинетики дебромирования [42] показало, что в процессе интеркаляции брома в волокно наряду с разрушением структуры волокна происходит образование химической связи C–Br, причем наиболее интенсивно она образуется только после 96-часового бромирования волокна. При этом с увеличением массы волокна на 55% оно становится более термостойким.
В настоящей работе модификация электрофизических свойств пековых волокон также была проведена в процессе бромирования в газовой фазе. Для изучения структурных преобразований, оказывающих влияние на электрическое сопротивление волокна и происходящих в нем после интеркаляции в него брома, проанализированы данные, полученные с помощью методов комбинационного рассеяния света (КРС) и рентгеновского анализа.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Бромирование в течение 2–144 ч пековых моноволокон было проведено при комнатной температуре в стеклянном сосуде, наполненном газообразным бромом [38]. Диаметр исходных волокон составлял 8–10 мкм. Далее, после удаления избыточного брома в течение 3 сут в вытяжном шкафу образцы выдерживали под вакуумом также в течение 3 сут. В результате бромирования произошло уменьшение электрического сопротивления в 7 раз: с 1.19 до 0.17 мОм · см, в течение первых 30 ч бромирования [38, 39, 42].
Спектры КРС моноволокон регистрировались с помощью спектрометра Т64000 фирмы “Jobin Yvon” (France) c двойным монохроматором, соединенным со спектрографом и охлаждаемым до 140 °С приемником. Источником излучения служил аргоновый лазер мощностью 1.5 мВт с длиной волны λ = 458 нм (размер пятна <2 мкм). Использовали микроскоп с увеличением в 200 раз. Анализ спектров выполнен с помощью разложения формы линий на лоренцевы составляющие.
Структурные исследования исходных и бромированных пековых УВ выполнены с помощью рентгеновского дифрактометра “ДРОН-1,5” с модифицированной коллимацией на Cu(Kα)-излучении. Излучение Cu(Kα) отфильтровывалось никелевой пластиной толщиной 3 мм. Напряжение на трубке составляло 33 кВ, анодный ток – 27 мА. Использовался сцинтилляционный счетчик с амплитудным дискриминатором. Образцы УВ для обеспечения одинаковых условий проведения эксперимента и уменьшения влияния анизотропии измельчали и набивали в металлические кюветы с рабочим объемом 17 × 17 × 1.5 мм3. При этом величина облучаемой поверхности образца была достаточной, чтобы получать истинную интенсивность рефлексов на медном излучении, начиная со значения брэгговского угла θ = 2°. Запись образцов проводили в интервале углов θ от 1° до 45°. Измерение углового положения рефлексов осуществляли с точностью до 0.01°. Все образцы исследовались как в отраженном, так и в прошедшем свете, что позволило получить дополнительные рентгенографические характеристики.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Спектры микро-КРС для исходных (а) и бромированных в течение 48 ч (б) пековых моноволокон приведены на рис. 1. Для исходного волокна (рис. 1а) характерны две интенсивные линии: линия вблизи ~1582 см–1, приписываемая активной КР-моде ${{Е}_{{2{{g}_{2}}}}}$ и связанная с С=С-связью графитовой сетки, и линия вблизи ~2740 см–1 с плечом в области ~2480 см-1, соответствующая пику при ~2730 см–1 в спектре второго порядка чистого графита, а также малоинтенсивная линия в области ~1360–1400 см–1, индуцированная процессом разупорядочения графитовой структуры и связанная с мелкокристаллической структурой волокна. Малая величина отношений интегральных интенсивностей линий: S1360/S1582 и S2480/S2740 (табл. 1), характеризует исследуемое волокно как высокоупорядоченную систему. Ширина линий при ~1582 и ~1360 см–1 на полувысоте характеризует порядок в графитовых плоскостях [49]. Для наших исходных волокон ширина основной линии ${{Е}_{{2{{g}_{2}}}}}$ на полувысоте (1/2)ν1582 аналогична ширине основной линии на полувысоте у высокочистого мелкокристаллического графита [49]. При бромировании волокна происходят изменения в спектре КРС (рис. 1б): уширение основных линий при ~1582 и ~2740 см–1, возрастание интенсивностей линий при ~1360–1400 и ~2480 см–1, появление новых линий в областях ~242, ~500, ~700, ~1600–2000, ~3000 см–1. Положение линий вблизи ~1582 и ~2740 см–1 практически не зависит от времени бромирования (табл. 2), что свидетельствует о наличии графитовой составляющей во всех образцах волокон. Положение линии при ~1360 см–1 не меняется при увеличении времени бромирования до 96 ч, а затем наблюдается ее смещение в сторону высоких частот, которое составляет ∆ν1360 = 19 см–1. С увеличением времени бромирования (табл. 1) также происходит возрастание отношения интегральных интенсивностей всех линий спектра первого порядка, а также увеличение ширины на полувысоте линии в области ~1582 см–1 спектра первого порядка. Ширина на полувысоте линии при ~1360 см–1 резко возрастает после двухчасового бромирования и остается практически неизменной при увеличении времени последнего до 120 часов бромирования. Это свидетельствует о разупорядочении графитовой структуры, появлении беспорядка и аморфной фазы в структуре волокна при бромировании. Увеличение отношения интегральных интенсивностей линий S1360/S1582 при бромировании в течение 96 ч при постоянном значении интегральной интенсивности S1582 указывает на интенсивное разрушение больших кристаллитов, рост мелкокристаллической фазы в волокне, а также на разрушение самой графитовой структуры и появление и рост аморфной фазы. При времени бромирования до 24 ч это отношение изменяется всего в 3 раза, при 24-часовом бромировании оно возрастает уже в 16 раз, при 48-часовом – в 33 раза, а после бромирования в течение 96 ч величина отношения S1360/S1582 возрастает в 100 раз по сравнению с начальным значением. Об аморфной фазе в волокне при бромировании в течение ≥96 ч свидетельствует появление новой линии в спектре первого порядка в области ~1530 cм–1, характерной для аморфного углерода.
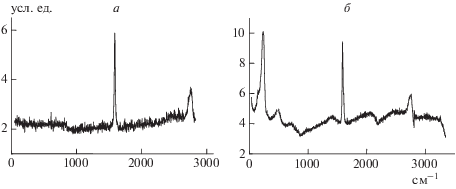
Таблица 1.
Отношения интегральных интенсивностей и ширин на полувысоте линий первого и второго порядка (вычисленных на основе разложения на лоренцианы) спектров КРС пековых моноволокон, бромированных в течение 0–120 ч
| tBr, ч | S1360/S1582 | S1600/S1582 | S1530/S1582 | (1/2)ν1582 | (1/2)ν1360 | S2480/S2740 | S3000/S2740 | (1/2)ν2740 | (1/2)ν2480 |
|---|---|---|---|---|---|---|---|---|---|
| первый порядок | второй порядок | ||||||||
| 0 | 0.03 | – | – | 17.5 | 13.2 | 0.079 | – | 60 | 78 |
| 2 | 0.08 | 0.1 | – | 18.1 | 15.7 | 0.44 | 0.1 | 63 | 100 |
| 24 | 0.5 | 0.17 | – | 20.0 | 237 | 0.7 | 0.16 | 90 | 172 |
| 48 | 0.99 | 0.3 | – | 23.0 | 245 | 0.9 | 0.21 | 95 | 210 |
| 72 | 1.8 | 0.7 | – | 25.0 | 245 | 1.0 | 0.26 | 100 | 250 |
| 96 | 2.9 | 0.94 | 0.2 | 29.6 | 240 | 1.12 | 0.65 | 110 | 353 |
| 120 | 3.4 | 1.23 | 0.68 | 62.3 | 242 | 1.22 | 0.74 | 104 | 370 |
Таблица 2.
Положение линий спектра КРС и рентгенографические характеристики пековых моноволокон, бромированных в течение 0–120 ч
| tBr, ч | ν1360, см–1 | ν1582, см–1 | ν2740, см–1 | d002, Å | Lс, Å | Lа, Å | Сг |
|---|---|---|---|---|---|---|---|
| 0 | 1361 | 1582.4 | 2740 | 3.370 | 200 | 610 | 100.0 |
| 2 | 1361 | 1582.4 | 2740 | 3.371 | 210 | 610 | 100.5 |
| 24 | 1362 | 1582.4 | 2741 | 3.373 | 235 | 610 | 101.0 |
| 48 | 1363 | 1582.5 | 2742 | 3.375 | 255 | 610 | 103.0 |
| 72 | 1363.5 | 1582.3 | 2734 | 3.381 | 245 | 610 | 79.0 |
| 96 | 1380 | 1581.7 | 2743 | 3.381 | 200 | 610 | 64.5 |
| 120 | 1376 | 1581.8 | 2740 | 3.380 | 205 | 610 | 68.5 |
Появление новых линий в низкочастотной области спектра (интенсивной линии при ~242 см–1 с плечом в области ~160–170 см–1 и менее интенсивных линий при ~500 и ~750 см–1) связано с присутствием брома в образце. Отношение площадей составляющей ~160 см–1 к линии ~242 см–1S160/S242, равное 0.4, и отношение S500/S242, равное в среднем 0.4, практически не зависят от времени бромирования. Колебания в области ~242 см–1 связаны с валентными колебаниями анионов брома ${\text{Br}}_{3}^{ - }$ и ${\text{Br}}_{5}^{ - }$ появившимися в графитовой структуре из-за переноса заряда (электрона) с графитовых слоев, граничащих с бромом, и являющимися свидетельством интеркалированного в графит брома [38]. Колебания в областях ~500 и ~750 см–1 в спектрах КРС связаны с возникновением связи С–Br в процессе бромирования [38, 50, 51]. Следует, однако, заметить, что в работе [52] существовала отличная от нашей интерпретация колебаний в областях ~500 и ~750 см–1. Авторы работы [52] эти частоты рассматривают как удвоенное и утроенное значения частоты ω0 ~ 242 см–1. Линия при ~323 см–1, соответствующая частоте колебаний свободной молекулы Br2, не наблюдалась ни в одном из представленных образцов, что свидетельствует об отсутствии свободного брома во всех образцах волокон. Однако во всех бромированных образцах отчетливо наблюдается высокочастотная, неразрешенная линия при ~1600 см–1 в виде симметричного плеча основной полосы, связанная, возможно, с колебаниями связи С=С в приграничных к брому слоях волокон; причем интенсивность ее растет с увеличением времени бромирования. Появление подобных слоев облегчает перенос зарядов и приводит к снижению сопротивления бромированных волокон по отношению к исходному волокну в 7 раз.
Из табл. 1 также видно, что линии спекров КРС второго порядка шире, чем линии спектра первого порядка как в исходных, так и в бромированных волокнах; кроме того, спектр второго порядка плохо разрешен (рис. 1б). Тем не менее отношения интегральных интенсивностей линий спектра второго порядка аналогичны отношениям интегральных интенсивностей линий спектра первого порядка.
Доминирующей линией спектра второго порядка является линия при ~2740 см–1, которая может быть представлена как 2 · 1365 = 2730 см–1 [51]. Широкая линия в области ~2480 см–1 связана с комбинацией мод высокой плотности фононных состояний с различными волновыми векторами (860 + 1620 = 2480 см–1) [53]. Линия при ~3000 см–1 появляется только в бромированных образцах. Эта линия, индуцированная процессом разупорядочения графитовой решетки, характеризуется комбинацией мод (~1365 + 1620 = 2985 см–1) [53]. Отношение S3000/S2740 незначительно меняется при увеличении времени бромирования до 72 ч (табл. 1). Большие изменения происходят при времени бромирования >96 ч, что также подтверждает факт разрушения графитовой структуры волокна и создание мелкокристаллической фазы при больших временах бромирования.
Для более точной характеристики разрушений в структуре волокна, происходящих при бромировании, были использованы данные рентгенографического анализа [54], который является прямым методом исследования фазового состава вещества, с помощью которого можно идентифицировать начальные и конечные продукты процесса и проследить последовательность изменений, происходящих в материале. Из рентгеновских дифракционных спектров проб графитированного волокна можно определить следующие важнейшие структурные характеристики [55–58]: d002 – расстояние между графитовыми плоскостями (002), Lc – размер кристаллитов вдоль оси “с”, La – размер кристаллитов перпендикулярно оси “с”, Сг – степень графитации, а также получить информацию о кристаллической структуре волокна и выявить преимущественную ориентацию кристаллитов относительно оси волокна. Значения Lc, La, d002 были определены с использованием формулы Брэгга и Шеррера [55, 56], а также формулы Селякова [59]: nλ = 2dsinѲ, L = Kλ/(βcosΘ), где Θ – угол рассеяния, d – внутриплоскостное расстояние, λ – длина волны рентгеновских лучей, β – полуширина максимума интенсивности линии в радианах. Значение K для Lc составляло 0.9, для La – 1.84 [60]. Степень графитации волокна рассчитывают по формуле Сг = (3.440 – d002)/(3.440 – 3.354) [41, 61].
Однако в этой формуле используется лишь величина d002 без учета угловой ширины рефлекса. В работе [62] была предложена формула, учитывающая обе характеристики: Сг = Lc/(d002 – 3.22). Однако в данном случае полуширину отражения d002 было предложено определять не прямым измерением на дифрактометре, а графическим методом, что усложняло процесс обработки спектра и служило источником дополнительных ошибок. Кроме того, для графитовых объектов значение поправочного коэффициента, равное 3.22 Å, кристаллохимически не оправдано. Поэтому для определении степени была использована скорректированная формула [54, 63], в которой величина 3.22 Å заменена на 3.35 Å, что соответствует величине d002 идеально окристаллизованного графита: Сг = (Lc · 10–2)/(d002 – – 3.35). Рентгенографические характеристики d002, Lс, Lа и Сг приведены в табл. 2. Значения Сг и Lс рассчитаны по спектрам, полученным в отраженном свете, а значения Lа – по спектрам, полученным в прошедшем свете, по рефлексу d110.
Согласно данным рентгенографического анализа (увеличение d002, уменьшение Сг; см. табл. 1), в результате бромирования происходит некоторое разуплотнение графитовой структуры волокна. Размеры кристаллитов изменяются таким образом: при постоянном значении Lа значение Lс сначала увеличивается, затем уменьшается. Это связано с тем, что наиболее интенсивно интеркалированное соединение образуется в местах наличия дефектов структуры. Сначала происходит кажущееся увеличение доли графитовой компоненты и своего рода “обогащение” УВ высокоориентированными кристаллами графита в связи с сокращением дефектов, что приводит к уменьшению электрического сопротивления волокна в 7 раз. Однако при увеличении времени бромирования от 48 до 96 ч из-за того, что количество дефектов в волокне за счет его контакта с бромом резко уменьшилось, также в небольшом количестве начинают образовываться связи С–Br. Происходит некоторое измельчение графитовых частиц в направлении оси “с”, что приводит к уменьшению среднего размера Lс. При времени бромирования >96 ч бром начинает разрушать саму графитоподобную структуру волокна, интенсивно вступая в химическую связь с атомами углерода графитовой сетки. В результате этого наблюдается рост электрического сопротивления волокна (с 0.17 до 0.35 мОм · см) [38] и появляется линия при ~1530 cм–1 в спектре КРС, соответствующая аморфной фазе. Рентгенографические характеристики при времени бромирования >96 ч остаются практически неизменными (табл. 2). Наблюдается лишь незначительное кажущееся увеличение значения Сг, имеющее место благодаря уменьшению количества дефектов структуры, по которым происходит бромирование.
Таким образом, данные рентгеноструктурного анализа (усиление фона некогерентного рассеяния, уменьшение степени графитации, разуплотнение графитовой структуры волокна) и данные спектроскопии КРС (уширение основных линий при ~1582 и ~2740 см–1, отвечающих за порядок в структуре волокна, возрастание интенсивностей линий при ~1360–1400 и ~2480 см–1, отвечающих за разупорядочение в графитоподобной структуре волокна, появление новых линий, связанных с присутствием брома) подтверждают факт разрушения графитоподобной структуры волокна в процессе бромирования. Данные изменения в структуре, происходящие на начальных стадиях бромирования и связанные с уменьшением количества дефектов на поверхности волокна благодаря воздействию газообразного брома, приводят к уменьшению электрического сопротивления пекового УВ в 7 раз. При временах бромирования >96 ч происходят значительные разрушения уже самой графитоподобной структуры волокна, что приводит к появлению аморфной фазы и, соответственно, к некоторому увеличению электрического сопротивления. Соответственно, дальнейшее бромирование волокна является нецелесообразным, так как оно может привести к полному разрушению графитоподобной структуры волокна и, соответственно, к значительному увеличению электрического сопротивления.
Следует заметить, что некоторое расхождение в величинах времени бромирования (для КРС – 48 ч, для рентгенографического анализа – 72 ч), определяющего начало значительных разрушений в графитовой структуре, в двух методах исследования связано с их спецификой. Спектроскопия КРС характеризует тонкий поверхностный слой толщиной ≈0.1 мкм и дает информацию о структуре волокна, усредненную по площади пучка лазера, в то время как рентгеновские методы из-за различных деформаций, изогнутости и возможных дефектов, присущих графитоподобным слоям волокна, позволяют определить не реальные физические размеры слоя, а некие эффективные параметры, относящиеся к областям когерентного рассеяния. Тем не менее оба метода подтверждают значительные изменения в структуре волокна, происходящие при бромировании.
ЗАКЛЮЧЕНИЕ
Результаты исследований показали эффективность применения газообразного брома для модификации поверхности пекового углеродного волокна, которая приводит к снижению его электрического сопротивления в 7 раз. Процессы, происходящие при интеркалировании бромом пекового УВ, носят общий характер. Данные, представленные в настоящей статье, будут полезны для понимания процесса бромирования и при решении задач получения углеродных волокон, обладающих высокой проводимостью.
Работа выполнена в рамках госзадания ИБХФ РАН (регистрационный номер 01201253304).
Список литературы
Yuan G., Xue Z., Cui Z. et al. // ACS Omega. 2020. V. 5. № 34. P. 21948.
Анпилова А.Ю., Масталыгина Е.Е., Храмеева Н.П. и др. // Хим. физика. 2020. Т. 39. № 1. С. 66; https://doi.org/10.31857/S0207401X20010021
Куперман А.М., Горбаткина Ю.А., Турусов Р.А. // Хим. физика. 2012. Т. 31. № 8. С. 50.
Guimarães C.J.B., Aguiar A.P., de Castro A.T. // Polímeros. 2021. V. 31. № 1. P. e2021011; https://doi.org/10.1590/0104-1428.08720
Kim B-.J., Kotegawa T., Eom Y. et al. // Carbon. 2016. V. 99. P. 649; https://doi.org/10.1016/j.carbon.2015.12.082
Shimanoe H., Ko S., Jeon Y.-P. et al. // Polymers. 2019. V. 11. № 12. P. 1911; https://doi.org/10.3390/polym11121911
Özsin G., Pütün A.E. // J. Fac. Eng. Architect. Gazi Univ. 2018. V. 33. №. 4. P. 1433; https://doi.org/10.17341/gazimmfd.4164440
Kim B.-J., Kil H., Watanabe N. et al. // Current Org. Chem. 2013. V. 17. № 13. P. 1463; https://doi.org/10.2174/1385272811317130013
Ni G., Jiang W., Shen W. // Chem. Select. 2019. V. 4. № 13. P. 3690; https://doi.org/10.1002/slct.201803764
Liu J., Chen X., Liang D. et al. // Energy Sources, Part A: Recovery, Utilizat., Environ. Effects. 2020; https://doi.org/10.1080/15567036.2020.1806952
Arai Y. // High-Performance and Specialty Fibers. The Soc. Fiber Sci. Technol. Tokyo, Jap. (ed.): Springer, 2016. Ch. 21. P. 343; https://doi.org/10.1007/978-4-431-55203-1_21
Lim T.H., Yeo S.Y. // Sci Rep. 2017. V. 7. P. 4733; https://doi.org/10.1038/s41598-017-05192-5
Kим C. // The Chemical J. 2014. № 10. C. 64; http://tcj.ru/wp-content/uploads/2014/11/2014_10_63-73_PLAST-Syre.pdf
Корнеева Н.В., Кудинов В.В., Крылов И.К. и др. // Хим. физика. 2019. Т. 38. № 9. С. 67; https://doi.org/10.1134/S0207401X19090036
Morgan P. Carbon fibers and their composites. Boca Raton: Taylor & Francis Group, CRC Press. 2005.
Мухамерзянов А.Т., Мухамерзянова А.А., Гимаев Р.Н. и др. // Вестн. Башк. Ун-та. 2015. Т. 20. № 4. С. 1218.
Дошлов О.И., Кондратьев В.В., Угапьев А.А. и др. // Изв. вузов. Прикл. химия и биотехн. 2014. № 2(7). С. 31.
Сидоров О.Ф., Селезнев А.Н. // Рос. хим. журн. 2006. Т. L. № 1. С. 16.
Matsumoto T. // Pure Appl. Chem. 1985. V. 57. P. 1553.
Riggs D.M., Shuford R.J., Lewis R.W. // Graphite fibers and composites / Ed. Lubin G. Handbook of Composites. N.Y.: Van Nostrand Reinhold Co., 1982. P. 196.
Yuan G., Li X., Xiong X. et al. // Carbon. 2016. V. 115. P. 59; https://doi.org/10.1016/j.carbon.2016.12.040
Alexander M.R., Jones F.R. // Carbon. 1994. V. 32. № 5. P.785.
Severini F., Formaro L., Pegoraro M. et al. // Carbon. 2002. № 40. P. 735.
Lee S., Kim T.R., Ogale A.A. et al. // Synth. Met. 2007. V. 157. P. 644.
Bing X., Wang X., Lu Y. // Appl. Surf. Sci. 2006. V. 253. Issue. 5. P. 2695.
Ma Y.J., Wang J.L., Cai X.P. // Intern. J. Electrochem. Sci. 2013. V. 8. P. 2806.
Alway-Cooper R.M., Anderson D.P., Ogale A.A. // Carbon. 2013. V.59. P. 40; https://doi.org/10.1016/j.carbon.2013.02.048
Dresselhaus M.S., Endo M. // Graphite Intercalation Compounds II: Transport and Electronic Properties / Eds. Zabel H., Solin S.A. Springer Ser. Mater. Sci. V. 18. Berlin: Springer–Verlag, 1992. P. 347.
Shioy J., Matsubara H., Murakami S. // Synth. Met. 1986. V. 14. P. 113.
Hooley J.G., Deitz V.R. // Carbon. 1978. V. 16. № 4. P. 251.
Dresslhaus M.S., Dresslhaus G. //Adv. Phys. 1981. V. 36. № 2. P. 139; https://doi.org/10.1080/00018738100101367
Ho C.T., Chung D.D.L. /// Carbon. 1990. V. 28. Issue 6. P. 831; https://doi.org/10.1016/0008-6223(90)90331-R
Klimenko I.V., Zhuravleva T.S., Jawhari T. // Synth. Met. 1997. V. 86. Issue 1–3. P. 2337; https://doi.org/10.1016/S0379-6779(97)81150-2
Kim B.J., Eom Y., Kato O. et al. // Carbon. 2014. V. 77. P. 747; https://doi.org/10.1016/j.carbon.2014.05.079
Choi Y.O., Yang. K.S. // Fibers Polym. 2001. V. 2. P. 178; https://doi.org/10.1007/BF02875342
Liang D., Liu D., Yang S. et al. // Polymers. 2020. V. 12. № 12. P. 3059; https://doi.org/10.3390/polym12123059
Mathur R.B., Bahl I.P., Kannan A. et al. // Carbon. 1996. V. 34. Issue 10. P. 1215; https://doi.org/10.1016/0008-6223(96)00089-9
Klimenko I.V., Zhuravleva T.S. // Mater. Today: Proceedings. 2018. V. 5. Issue 12. P. 25987; https://doi.org/10.1016/j.matpr.2018.08.017P. 25987
Klimenko I.V., Shchegolikhin A.N., Zhuravleva T.S. // Synth. Met. 1995. V. 71. P. 1773
Gaier J., Ditmars N.F., Dillon A.R. // Carbon. 2005. V. 43 Issue 1. P. 189; https://doi.org/10.1016/j.carbon.2004.09.005
Jaworske D.A., Gaier J.R., Maciag C. et al. // Carbon. 1987. V. 25. Issue 6. P. 779.
Клименко И.В., Журавлева Т.С., Бибиков С.Б. // Высокомолекуляр. соединения. А. 2000. Т. 42. № 2. С. 320.
Jacquesa E., Kjell M.H., Zenkerta D. et al. // Carbon. 2014. V. 59. P. 246; https://doi.org/10.1016/j.carbon.2013.03.015
Ouchi Y., Takenaka A., Kinumoto T. et al. // Carbon. 2013. V. 55. P. 372; https://doi.org/10.1016/j.carbon.2012.12.040
Johannisson W., Harnden R., Zenkert D. // Proc. Nation. Acad. Sci. 2020. V. 117. № 14. P. 7658; https://doi.org/10.1073/pnas.1921132117
Ghosh S., Bhattacharjee U., Patchaiyappan S. et al. // Adv. Energy Mater. 2021. V. 11. P. 2100135; https://doi.org/10.1002/aenm.202100135
Fredi G., Jeschke S., Boulaoued A. et al. // Multifunction. Mater. 2018. V. 1. № 1. P. 015003; https://doi.org/10.1088/2399-7532/aab707
Гришин М.В., Гатин А.К., Сарвадий С.Ю. и др. // Хим. физика. 2020. Т. 39. № 7. С. 63; https://doi.org/10.31857/S0207401X20070067
Knight D.S., White W.B. // J. Mater. Res. 1989. V. 4. № 2. P. 385.
Huong P.V. // Sol. State Comm. 1993. V. 88. № 1. P. 23.
Tebbe F., Harlow R., Chase D. et al. // Science. 1992. V. 256. P. 822.
Dresselhaus M.S., Dresselhaus G. Light Scattering in Solids III / Cardona M., Guntherodt G. V. 51. Topics in Appl. Phys. Berlin: Springer-Verlag, 1982.
Dresselhaus M.S., Dresselhaus G., Sugihara R. et al. Graphite Fibers and Filaments. Springer Proc. Mater. Sci. V. 5. Berlin: Springer, 1988.
Клименко И.В., Королев Ю.М., Журавлева Т.С. // Высокомолекуляр. соединения. А. 2001. Т. 43. № 2. С. 357.
Mohindar S.S., Pavlovic A.S. // Carbon. 1993. V. 31. Issue 4. P. 557.
Endo M., Kim C., Karaki T. et al. // Ibid. 1998. V. 36. Issue 11. P. 1633
Rezende L., Oliveira Chaves A., Moraes S.L.L. // Braz. J. Geol. 2021. V. 51. № 1. P. e20200083; https://doi.org/10.1590/2317-4889202120200083
Tagiri M., Yago Y., Tanaka A. // The Island Arc. 2000. V. 9. № 2. P. 188; https://doi.org/10.1046/j.1440-1738.2000.00272.x
Selyakow N.Zs. // Phys. 1925. V. 31. P. 439.
Warren B.E. // Phys. Rev. 1941 V. 59. P. 693.
Pacault A. Chemistry and Physics of Carbon. / Ed. Walker L., Jr. V. 7. N.Y.: Marcel Dekker, 1971.
Tagiri M. // J. Japan. Assoc. Min. Petr. Econ. Geol. 1981. V. 76. № 11. P. 345; https://doi.org/10.2465/ganko1941.76.345
Луковников А.Ф., Королев Ю.М., Голован Г.С. // ХТТ. 1996. № 5. С. 3.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика