

Химическая физика, 2022, T. 41, № 2, стр. 78-85
Взаимодействие диоксида азота с терлоном – поли-п-фенилентерефталамидом
Т. В. Похолок 1, *, С. М. Ломакин 1, И. Г. Калинина 2
1 Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
Москва, Россия
2 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова
Российской академии наук
Москва, Россия
* E-mail: pgb@sky.chph.ras.ru
Поступила в редакцию 27.07.2021
После доработки 16.08.2021
Принята к публикации 20.08.2021
- EDN: DRMGDA
- DOI: 10.31857/S0207401X2202008X
Аннотация
Изучение свойств полимерных волокон представляет собой актуальную задачу, важную для создания новых перспективных материалов и модификации их свойств в условиях эксплуатации. С помощью методов ЭПР- и ИК-спектроскопии, термоаналитических методов, электронной сканирующей микроскопии проведено комплексное исследование низкотемпературного окисления на воздухе волокон поли-п-фенилентерефталамида (терлона), предварительно нитрованного окислами азота, и представлен механизм этого процесса. В качестве инициатора окисления установлены образующиеся при нитровании фенилнитритные фрагменты макромолекул. Деструкция полимера происходит в результате распада N-нитрозоамидов, N-нитросоединений и N-нитритов – продуктов нитрования терлона. Методом ИК-спектроскопии определены основные продукты нитрования терлона – сложные эфиры и карбоновые кислоты.
ВВЕДЕНИЕ
Терлон относится к классу “арамидных волокон” и входит как сополимер в состав высокомодульных, термо- и огнестойких волокон в процентном соотношении, превышающем 70%. Благодаря этим качествам “арамидные волокона” широко используются в авиационной технике и в изделиях специального назначения. Для “арамидов” характерна высокая устойчивость к воздействию многих внешних факторов: кислорода, света, радиации. Это связано с высокой прочностью связей N–H и ароматических связей C–H, препятствующих образованию в них свободных радикалов при температурах ниже 300 °С. Однако в атмосфере, содержащей оксиды азота, их эксплуатационные свойства значительно ухудшаются. Резко падают как прочностные свойства, так и термическая стабильность.
Ранее нами было показано [1–6], что по отношению к диоксиду азота (NO2) эти полимеры (ароматические полиамиды, полиамидимиды и полиимиды) весьма реакционноспособны уже при комнатной температуре. Специфическое ион-радикальное инициирование превращает химически инертные ароматические кольца в более активные циклогексадиеновые структуры [7]. В качестве инициатора выступает нитрозилнитратная (НН) форма ${\text{N}}{{{\text{O}}}^{ + }}{\text{NO}}_{3}^{ - },$ представленная в реакции диоксида азота с ароматическим полиамидом фенилоном. При этом осуществляется перенос электрона с NH-группы мономерного звена на катион нитрозилнитрата ${\text{N}}{{{\text{O}}}^{ + }}{\text{NO}}_{3}^{ - }$ [7]. В полимере накапливаются азотсодержащие макрорадикалы, служащие источником большого числа разнообразных валентно-насыщенных продуктов нитрования фенилона [8], первичными из которых являются N-нитрозо- и N-нитрито-производные. Они легко распадаются, и в арамиде возникает много относительно стабильных при комнатной температуре соединений, чьи полимерные звенья содержат фенильные кольца и присоединенные нитро-, нитрит- и нитрозогруппы, вызывая резкое ухудшение термостойкости и ударной прочности полимера [8, 9].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали волокна терлона (бледно-желтого цвета) промышленного поли-п-фенилентерефталамида – аналога кевлара, полученного посредством поликонденсации дихлорангидрида терефталевой кислоты и п-фенилендиамина. Диоксид азота получали путем термического разложения Pb(NO3)4 в вакууме (продукты: Pb(NO3)2 + + 2NO2 + O2). Смесь 2NO2 + O2 пропускали через заполненную P2O5 трубку для удаления воды с последующей конденсацией NO2 в ловушку, охлажденную до –35 °С смесью ацетона с сухим льдом. Полученный NO2 очищали от следов кислорода трехкратным повторением цикла “замораживание газа до 77 К и откачка O2 до ≈10–3 Торр – размораживание до комнатной температуры” [9]. Очищенный NO2 хранили в специальной емкости, присоединенной к вакуумной установке. Для проведения опыта образец волокна терлона массой 0.06 г помещали в цилиндрическую кварцевую ампулу для ЭПР-измерений, соединенную с шарообразной колбой емкостью 500 см3, которая была снабжена встроенными кранами, обеспечивающими вакуумирование и напуск газов в ампулу и спектроскопическую кварцевую кювету для определения концентрации NO2. После откачки системы до давления в 1.3 Па колбу с ампулой отключали краном от вакуумной установки и заполняли очищенным NO2. Параллельно этот же газ заполнял и снабженную краном кварцевую кювету. После заполнения системы газом краны на колбе и кювете закрывали. Концентрацию NO2 определяли по оптической плотности при длине волны λ = 410 нм [9]. Регистрацию спектров осуществляли на спектрофотометре компании Shimadzu UV mini-1240 (Japan). Ампулу с образцом заполняли газом NO2 концентрацией 2 · 10–3 моль/л и при 98 °С прогревали в течение 8 ч. Затем образец хранили при комнатной температуре 3 мес в атмосфере NO2. Для исследования ИК-спектров NO2 скачивали, волокна терлона распушали и помещали в специальную кювету. Оставшиеся волокна хранили на воздухе в темном месте в ампуле. Для ЭПР-измерений ампулу с образцом, заполненную газом ([NO2] = 2 · 10–3 моль/л), помещали непосредственно в резонаторе ЭПР-спектрометра. Использовали спектрометры РЭ-1306 и ЭПР-10 МИНИ (ООО “Резонанс-М”, Санкт-Петербург) со следующими характеристиками: частота ВЧ-модуляции – 100 кГц, амплитуда ВЧ-модуляции – 1–3 Гс, мощность СВЧ – 1–3 мВт.
Значения g-факторов и констант сверхтонкого взаимодействия $A_{{\parallel , \bot }}^{{\text{N}}},$ определяли при одновременной регистрации сигналов ЭПР образца и ионов Mn2+ в решетке MgO. ИК-спектры исходных и экспонированных в NO2 образцов регистрировали методом многократного нарушенного полного внутреннего отражения на ИК-спектрометре с фурье-преобразователем марки Tensor 27 (Bruker) со стандартной АТР−ячейкой Pike Miracle (ZnSe). Воспроизводимость измерений составляла 1–2%.
Термогравиметрический и дифференциальный термический анализы (далее – ТГА и ДТА) образцов исходного и нитрованного терлона проводили на синхронном термоанализаторе STA 449 F3 компании NETTZCH (Germany). Навески образцов составляли 10–12 мг. Процесс деструкции полимеров проводили на воздухе при скорости потока газа 30 мл/мин и линейной скорости нагрева 10 °С/мин в диапазоне температур 30–900 °С. Изменения потери массы регистрировались с точностью до 1–3 мг, относительная погрешность измерения температуры составляла ±1.5 °С, тепловых эффектов – ±3%. Процесс описывали зависимостями потери массы (ТГА), скорости потери массы (ДТГ) и тепловых эффектов (ДТА) от температуры. В качестве параметров, характеризующих деструкцию, были выбраны: по кривым ТГА – температуры стадийности процесса потери массы (Т) и по кривым ДТГ – максимальная скорость потери массы (Wmax), по кривым ДТА – величины тепловых потоков, сопровождающих стадии потери массы (Q, кДж/г). Термомеханические свойства исследовали на приборе ТМА-402 фирмы Netzsch (Germany) со сферическим индектором диаметром 3 мм при нагрузке 50 г и скорости нагрева 10 град/мин в диапазоне температур 25–600 °С. Измерения проводили на воздухе.
Испытание механической прочности нитей нитрованного терлона проводились на разрывной машине Instron 3365 (USA). Микроструктура волокон изучена методом сканирующей электронной микроскопии на приборе Tescan MiraL MU компании Tescan (Czech) при низких значениях тока и ускоряющего напряжения 1 кВ в режиме высокого вакуума без напыления.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Методом ЭПР-спектроскопии изучено действие диоксида азота на термостойкий ароматический полиамид (поли-п-фенилентерефталамид) терлон. Показано, что при длительном экспонировании терлона в диоксиде азота (до 8 ч) и последующем прогреве при 98 °С в течение 15 ч и более в спектре ЭПР наблюдается сигнал, представляющий собой анизотропный триплет иминоксильных радикалов (Rim) со следующими параметрами:
Рассмотрим возможный механизм взаимодействия терлона с диоксидом азота. Реакция терлона с окислами азота инициируется переносом электрона с NH-группы на катион нитрозилнитрата ${\text{N}}{{{\text{O}}}^{ + }}{\text{NO}}_{3}^{ - },$ находящегося в химическом равновесии с неполярной формой димера N2O4 [6, 7]: O2N–NO2 ↔ 2NO2 ↔ ONONO2.
Нитрозилнитрат проявляет сильные окислительные свойства и способен инициировать радикальные реакции в первичном процессе переноса электрона от амидных групп арамида на НН с образованием промежуточных катион-макрорадикалов и оксида азота [6, 7]. В роли доноров электрона могут выступать амидные группы и фениленовые кольца макромолекул, и происходит окислительное взаимодействие амидных групп мономерного звена полимера с нитрозилнитратной формой димера NO2 (Схема 1 ).
Схема 1
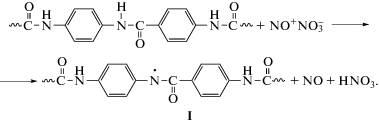
На первой стадии образуется радикальная пара, включающая NO и амидильный радикал I. Изомерными формами радикала I являются радикалы циклогексадиенильного типа, существующие в нескольких электронно-таутомерных формах [6, 7]. Присоединяя оксид азота, они превращаются в нитрозосоединения и легко изомеризуются в оксимы, которые в реакции с NO2 генерируют стабильные иминоксильные радикалы, как описано нами [6, 7] на примере нитрования фенилона.
Инициированные амидильные радикалы I реагируют с NO2 и NO, образуя широкий набор стабильных N-нитро-, N-нитрит- и N-нитрозобензольных соединений, которые термически не стабильны и разрушаются при нагревании и хранении терлона с разрывом полимерной цепи и накоплением сложных эфиров и карбоновых кислотных групп, вызывая резкое ухудшение термостойкости и ударной прочности полимера [10–12]. К первичным валентно-насыщенным продуктам реакции относятся распадающиеся при нагревании N-нитрозо- (II) и N-нитрит-производные (III) [6, 7] (Схема 2 ).
Схема 2
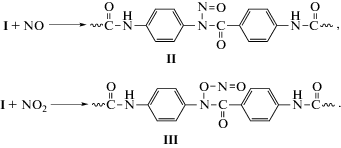
Среди вторичных продуктов образуются свободные иминоксильные радикалы $R_{{im}}^{\centerdot },$ из которых наиболее устойчивыми являются радикалы, обладающие орто-хиноидным строением [6, 7]. Наличие подобных структур обуславливает сильное окрашивание нитрованных образцов (волокон) по типу представленного в нашей статье [8].
В соответствии с работами [6, 7] иминоксильные радикалы (Rim) образуются с участием промежуточного электронного таутомера Ia путем “внутриклеточных” реакций. Основные стадии процесса нитрования терлона приведены на Схеме 3 .
Схема 3
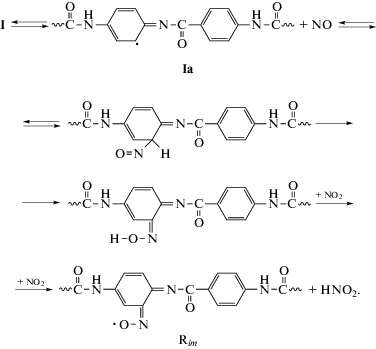
Термическая нестабильность соединений II и III обусловлена ослаблением связи N–N и N–O вследствие отталкивания несвязывающих неподеленных пар электронов на атомах N и O.
Инфракрасные спектры нитрованного терлона
В ИК-спектрах образцов терлона, обработанных в разных режимах нитрования диоксидом азота NO2 в концентрации 2 · 10–3 моль/л и продолжительности экспозиции их на воздухе, было проведено исследование трех групп образцов: 1-я группа – 3 мес нитрования; 2-я – 7 лет хранения на воздухе; 3-я – 10 лет хранения. Сравнивая ИК-спектры исходных и нитрованных волокон (рис. 1), можно выделить группы полос с интенсивностью, уменьшающейся в интервале частот ν = 3600–2500, проходящей через максимум при ν = 3322 см–1, 1743–1000 см–1 и возрастающей при ν = 1442–1000 см–1. Первая группа полос относится к полосам исходного терлона и расходованию связей N–H (ν = 3340 см–1) и водородных связей (ν = = 3500–2500 см–1) между амидными группами, обеспечивающими прочность полимера и разрушающимися при нитровании терлона. К полосам с возрастающей интенсивностью относятся частоты функциональных групп продуктов нитрования. Усредненные значения частот пиков: 1610, 1510, 1407, 1307, 1250, 1113, 1016, 959, 859–890 см–1, в дальнейшем позволяют отнести их к концевым фрагментам макромолекул – сложным эфирам ароматических и карбоновых кислот, идентичных бензальдегиду и бензойной кислоте, являющихся фрагментами концевых групп молекулы терлона), а к продуктам нитрования полимерной молекулы – ароматические нитросоединения, нитриты, нитраты, азотную кислоту [13–16]. Полосы, соответствующие этим соединениям, появляются в ИК-спектрах нитрованного терлона, один из которых представлен на рис. 1. Принципиальным отличием представленного на этом рисунке ИК-спектра является наличие полосы с максимумом ν = 1442 см–1, соответствующей N-нитрозосоединениям, которая отсутствует в спектрах при больших временах экспозиции на воздухе.
Рис. 1.
ИК-спектры волокон терлона до (1) и после нитрования (2) в течение 6 ч при [NO2] = 2 · 10–3 моль/л при Т = 98 °С и хранения их в течение 3 мес при 25 °С.
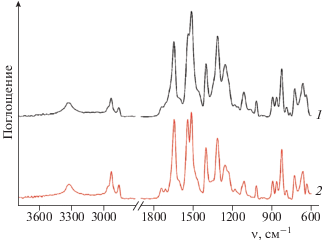
Как будет показано ниже, распад N-нитрозосоединений приводит к разрыву молекулы терлона и последующей деструкции полимера. Следует отметить, что пики полосы в диапазоне частот 3600–2500 см–1 смещаются и расплываются за счет аморфизации кристаллической фазы при действии диоксида азота на терлон. По этой же причине увеличивается полоса с максимумом ν = 1018 см–1. Химическая структура волокнá отличается высокой степенью ориентированности и жесткости. Эти характеристики, в частности, обусловлены наличием в структуре большого количества ароматических бензольных колец. По своей структуре волокно может быть отнесено к сетчатым полимерам. Жесткие полимерные цепи находятся в распрямленном состоянии и образуют очень плотную упаковку в объеме волокнá, что определяет его высокие механические свойства. Кристаллическая природа полимера обеспечивает высокую термическую стойкость волокон, а наличие ароматических колец в структуре макромолекулы обуславлавает стабильность волокон терлона [17].
Механическая прочность волокон терлона
Проведенные нами измерения механической прочности нитрованных волокон терлона методом разрывного напряжения показали, что прочность волокон уменьшается на 30–35% после трехсуточной экспозиции нитей при комнатной температуре, предварительно нитрованных в NO2 с концентрацией 1.5 · 10–3 моль/л в течение 6 ч при Т = 98 °С. Относительная разрушающая нагрузка нити терлона (P/P0) для образцов, нитрованных в NO2 с концентрацией 2 · 10–3 моль/л при Т= 98 °С и хранившихся разное время при 25 °С в атмосфере NO2 (1, 3, 7, 14 сут и 3 мес) составила 0.855, 0.785, 0.584, 0.54–0.58, 0.48–0.51, соответственно, при P0 = (8.35 ± 0.45) кГс.
В работах [11, 12] предложен механизм распада соединений типа N-нитрозо- и N-нитритпроизводных II и III, а также N-нитроамидов, образующихся при нитровании амидов и полиамидов. Показано, что продуктами распада являются сложные эфиры с выходом 81–84%, органические кислоты с выходом 16–18% и азот. В работе [12] на примере нитрования диоксидом азота нитей нейлона по объему выделившегося азота были определены кинетические параметры деструкции поликапрамида (ПКА) в трех средах: в ксилоле, керосине и воде в интервале температур 80–110 °С, и показано, что процесс описывается кинетическим уравнением реакции первого порядка. Были определены значения энергии активации распада Ea = 26 ккал/моль, констант скорости распада kd в указанном выше интервале температур, времен полураспада τ0.5. Так, при T = 80 °С τ0.5 = 33 мин, kd = 0.021 мин–1. Величина kd в воде в 2 раза больше, чем в ксилоле и керосине. Молекулярный вес нейлона уменьшался от 10.000–12.000 ед. до 300–500 ед. за 3 мес. Волокно приобретало бледно-кремовый цвет и рыхлую поверхность за счет деструктивных процессов в полимере, связанных с выделением азота и разрывами макромолекул при распаде N-нитрозоамидов. На лицо разрушение полимера при действии диоксида азота на алифатический полиамид.
Эффективный процесс деструкции пленок ПКА наблюдали в работе [6]. Нитрозирование как алифатических, так и ароматических полиамидов диоксидом азота приводит к разрушению системы водородных связей между соседними макромолекулами и ухудшению механических свойств полимера. Экспонированные в NO2 при [NO2] = (4–8.5) · 10–5 моль/л тонкие (1–5 мкм) пленки ПКА разрушались при минимальных механических воздействиях и после более чем 10 ч нитрования превращались в желтую, очень вязкую жидкость. Для ароматического полиамидимида в присутствии NO2 было установлено, что в результате протекания последовательности реакций значение начальной скорости накопления иминоксильных макрорадикалов является нижней границей для скорости деструкции макромолекул, (Wd)0 [6]. Проведенная оценка показала, что в условиях экспериментов при 25 °С (Wd)0 = = 10–8 моль/кг · с.
Используя эти значения и приведя их к реальным условиям хранения и эксплуатации, можно спрогнозировать техническую пригодность волокна и определить его конкретный ресурс [18–21]. Так, для ПКА рассчитанная величина kd = 1.06 · · 10–3 мин–1 при 25 °С в 20 раз меньше значения kd, полученного при 80 °С, а время полураспада N-нитрозоамида нейлона составляет 26 ч. Приблизительная оценка величины kd при 25 °С для нитрованного терлона с учетом времени полураспада (разрушения) волокна (4 года) равна 3.3 · 10–5 мин–1. Величина kd для терлона до 300 и более раз меньше kd для нитрованного ПКА.
Термическая устойчивость и изменения структуры нитрованного терлона
Для установления влияния диоксида азота на термическую устойчивость волокна терлона было проведено исследование образцов терлона, обработанных в разных режимах нитрования и продолжительности экспозиции их на воздухе, термоаналитическими методами ТГА, ДТА и термомеханического анализа (ТМА). На рис. 2, 3 приведены соответствующие кривые ТГА, ДТА, ТМА, из которых видно что, свойства и параметры нитрованных образцов существенно отличаются от исходных, не нитрованных волокон. Так, из сравнения параметров, приведенных на рис. 2 видно, что термическая деструкция нитрованного волокна протекает при меньших значениях температур и потери массы (∆), чем деструкция исходного терлона. Температура начала интенсивной потери массы – Тн1 = 488.43 °С при 7%-ной потере массы для исходного терлона и Тн2 = 475.31 °С при 12%-ной потере массы для нитрованного терлона; Тmax 1 при максимальной скорости деструкции равна 497.75 °С для исходного терлона и Тmax 2 равна 482.17 °С для нитрованного терлона. Температура окончания деструкции Тк 1 = 572.47 °С и Тк 2 = = 510.80 °С при потере массы около 99.7% (т.е. оба полимера разлагаются без твердого остатка). Из анализа кривых ДТА рис. 2 следует, что величины площадей эндопиков исходного, A1 = = ‒241 388.861 мкВ ∙ с. и нитрованного терлона, A2 = –122 680.179 мкВ ∙ с, различаются в два раза, причем площадь пика для нитрованного терлона мешьше, чем площадь эндопика для исходного терлона, что свидетельствует о меньших энергетических затратах и указывает на более интенсивный процесс деструкции нитрованного полимера.
Рис. 2.
Потеря массы волокон терлона по данным ТГА и ДТА до (а) и после нитрования (б) в течение 6 ч при [NO2] = = 2 · 10–3 моль/л при Т = 98 °С и 3 мес хранения при 25 °С.
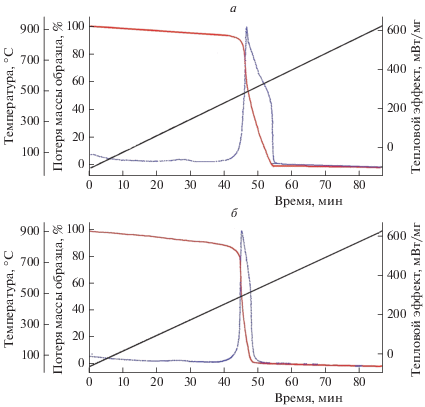
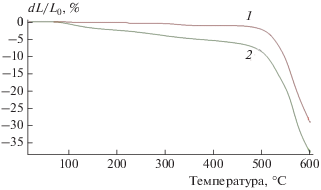
Рис. 3.
Температурная зависимость потери прочности волокон терлона до (1) и после нитрования (2) в течение 6 ч при в [NO2] = 2 · 10–3 моль/л при Т = 98 °С и 3 мес хранения при 25 °С по данным ТМА.
Как показано в работах [22–25], основными продуктами деструкции в начальной стадии процесса (до 10% потери массы) являются газообразные СО, СО2 и H2O. При Т ≥ 500 °С в состав продуктов распада входят фрагменты макромолекулы терлона, включая бензойную кислоту, п-фенилендиамин, бензонитрил, анилин, терефталонитрил. Предполагается, что деструкция терлона происходит по амидным группам мономерного звена полимера. Энергия активации термической деструкции на воздухе и в атмосфере азота равна 207 и 219 кДж ∙ моль–1 соответственно.
Основные продукты термической деструкции терлона идентичны продуктам нитрования волокон, идентифицированным выше по ИК-спектрам: сложным эфирам ароматических и карбоновых кислот – бензальдегиду и бензойной кислоте, являющимся фрагментами концевых групп молекулы терлона.
Изменения структуры волокон терлона при нитровании
Разрушающее действие диоксида азота на структуру волокна наблюдается из снимков образцов, сделанных с помощью электронного сканирующего микроскопа (ЭСМ) при одинаковых параметрах съемки. Степень разрушения волокна терлона можно визуально оценить по рис. 4. Сравнивая исходное и нитрованное волокно, хранившееся на воздухе более двух лет, отметим, что различий почти нет. Упакованные в ровные прямолинейные “снопы” волокнá в основном однородны по внешнему виду. На некоторых участках появляются первые дефекты в виде заусенцев и разломов, похожих на разрезы. С увеличением времени экспозиции (хранения) до трех и более лет дефектность волокон усиливается, на поверхности появляются изломы, каверны, сколы, трещины. Прямолинейность волокон исчезает, они теряют прозрачность, становятся хрупкими, чешуйчатыми и превращаются в стружку, а затем – в опилки (почти в труху). Все повреждения наглядно видны на рис. 4б. Кроме этого, наблюдается изменение окраски волокон от бледно-желтого до насыщенного желтого цвета.

ЗАКЛЮЧЕНИЕ
С помощью методов ЭПР и ИК-спектроскопии, ТГА, ДТА, ТМА и ЭСМ проведено комплексное исследование взаимодействия диоксида азота с терлоном и представлен универсальный механизм низкотемпературного окисления на воздухе волокон термостойкого ударопрочного ароматического полиамида поли-п-фенилентерефталамида (терлона). Димер NO2 в форме нитрозилнитрата инициирует ион-радикальные реакции с образованием продуктов нитрозирования за счет переноса электрона c NH-группы мономерного звена на катион нитрозилнитрата. Разнообразные превращения возникающего катион-радикала приводят к синтезу широкого набора относительно стабильных N-нитро-, N-нитрит-, N-нитрозобензольных соединений и стабильных азотсодержащих радикалов, что приводит к резкому ухудшению эксплуатационных свойств терлона. Показано, что деструкция нитрованного терлона на воздухе происходит и при комнатной в результате распада основных продуктов нитрования N-нитрозоамида и N-нитритамида. При этом образуются сложные эфиры ароматических кислот (фенилбензоатные фрагменты) и кислотные группы бензойной кислоты в качестве концевых групп разрывов полимерной цепи. Прочность нитрованных волокон терлона уменьшается на 30% за одни сутки экспозиции в диоксиде азота при [NO2] = 1.5 · 10–3 моль/л за счет распада водородных связей, а дальнейшее хранение этих волокон на воздухе в течение 3–4 лет приводит к полному их разрушению. Эти результаты приводят к выводу о высокой чувствительности терлона – ароматического полиамида к диоксиду азота и требованию особой чистоты атмосферы в помещениях, в которых хранятся или функционируют изделия из ароматических полиамидов (даже в следовых количествах).
Авторы выражают огромную благодарность А.Е. Флатовой за измерения микроструктуры волокон и А.А. Далинкевич за измерения разрывной прочности волокон.
Работа выполнена за счет субсидии, выделенной на выполнение госзадания (регистрационный номер 0120125305), а также в рамках финансирования госзадания ФИЦ ХФ им. Н.Н. Семёнова РАН № ГЗ-0082-2019-0008 (регистрационный номер АААА-А20-120030590042-8).
Список литературы
Ломакин С.М., Хватов А.В., Сахаров П.А. и др. // Хим. физика. 2020. Т. 39. № 11. С. 58.
Ломакин С.М., Шаулов А.Ю., Коверзанова Е.В. и др. // Хим. физика. 2019. Т. 38. № 4. С. 74.
Коверзанова Е.В., Усачев С.В., Ломакин С.М. и др. // Хим. физика. 2019. Т. 38. № 6. С. 53.
Хватов А.В., Бревнов П.Н., Шилкина Н.Г., Ломакин С.М. // Хим. физика. 2019. Т. 38. № 6. С. 71.
Бревнов П.Н., Новокшонова Л.А., Крашенинников В.Г. и др. // Хим. физика. 2019. Т. 38. № 9. С. 54.
Zaikov G., Davydov E., Gaponova I., Pokholok T., Pariiskii G. Interaction of Polymers with Polluted Atmosphere. Shawbury, SY4 4NR. UK: iSmithers – A Smithers Group Company, 2009. P. 264.
Pokholok T.V., Gaponova I.S., Davydov E.Ya., Parii-skii G.B. // Polym. Degrad. Stab. 2006. V. 91. № 10. P. 2423.
Похолок Т.В., Гапонова И.С., Парийский Г.Б. и др. // Вестн. технологич. ун-та. 2015. Т. 18. № 5. С. 51.
Похолок Т.В., Гапонова И.С., Парийский Г.Б., Ломакин С.М., Михеев Ю.А. // Хим. физика 2017. Т. 36. № 9. С. 11.
White E.H. // J. Amer. Chem. Soc. 1955. № 20 . V. 77. P. 6011.
White E.H. // Ibid. P. 6014.
Porter M.R. // J. Polym. Sci. 1958. № 126. V. 33. P. 447.
Беллами Л. ИК-спектры сложных молекул. М.: Мир, 1963.
Тарасевич Б.Н. ИК-спектры основных классов органических соединений. Справочные материалы. М.: Изд-во МГУ, 2012.
George W.O., McIntyre P.S. Infrared Spectroscopy Analytical Chemistry by Open Learning / Ed. Mowthorpe D.J. Publ. on behalf of ACOL, Thames Polytechnic. London: By John Wiley and Sons, 1987.
Васильев А.В., Гриненко Е.В., Щукин А.О., Федулина Т.Г. Инфракрасная спектроскопия органических и природных соединений. Учебное пособие. СПб.: СПбГЛТА, 2007.
Перепелкин К.Е. Структура и свойства волокон. М.: Химия, 1985.
Бенсон С. Термохимическя кинетика. М.: Мир, 1971.
Денисов Е.Т., Саркисов О.М., Лихтенштейн Г.И. Химическая кинетика. М.: Химия, 2000.
Эмануэль Н.М., Бучаченко А.Л. Химическая физика старения и стабилизации полимеров. М.: Наука, 1982.
Эмануэль Н.М., Кнорре Д.Г. Курс химической кинетики. М.: Высш. шк., 1984.
Pramoda K.P., Chung T.S., Liu S.L., Oikawa H., Yamaguchi A. // Polym. Degrad. Stab. 2000. V. 67. P. 365.
Gowenlock Brian G., Pfab Josef, Young Victor M. // J. Chem. Soc. Perkin Trans. 1997. V. 2. P. 1793.
Brown James R., Holgeman Daryl K.C. // Polymer. 1982. V. № 3. P. 365.
Калашник А.Т., Паникарова Н.П., Довбий Е.В. и др. // Высокомолекуляр. соединения. А. 1977. Т. 19. № 12. С. 2747.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика