

Химическая физика, 2022, T. 41, № 2, стр. 51-56
Разработка диагностического подхода, основанного на выявлении посттрансляционных модификаций фибриногена, ассоциированных с окислительным стрессом, методом высокоэффективной жидкостной хроматографии
А. Д. Васильева 1, *, Л. В. Юрина 1, Д. Ю. Азарова 1, В. С. Иванов 1, П. А. Стрельникова 1, А. Е. Бугрова 1, М. И. Индейкина 1, 2, А. С. Кононихин 1, 3, Е. Н. Николаев 3, М. А. Розенфельд 1
1 Институт биохимической физики им. Н.М. Эмануэля, Российской академии наук
Москва, Россия
2 Московский физико-технический институт (национальный исследовательский университет)
Москва, Россия
3 Автономная некоммерческая образовательная организация высшего профессионального образования
“Сколковский институт науки и технологий”
Москва, Россия
* E-mail: alexandra.d.vasilyeva@gmail.com
Поступила в редакцию 27.07.2021
После доработки 14.08.2021
Принята к публикации 20.08.2021
- EDN: IXDKUS
- DOI: 10.31857/S0207401X22020145
Аннотация
В рамках данного исследования был разработан способ упрощенной подготовки проб к анализу методом высокоэффективной жидкостной хроматографии и тандемной масс-спектрометрии (ВЭЖХ–МС/МС) для дальнейшего клинико-диагностического использования, включающий иммунопреципитацию фибриногена на магнитных микрочастицах с последующим ферментативным гидролизом белка. Разработка эффективной методики, которая бы включала выделение целевого белка из исследуемого образца плазмы или любой другой физиологической жидкости с исключением большого количества стадий процесса (например, многочисленных стадий переосаждения, различных видов хроматографии при выделении целевых белков) и использования агрессивных условий (например, при элюировании белков с носителей), а также с уменьшением общей трудоемкости и времени подготовки проб к анализу, является необходимой для использования в протеомных исследованиях.
ВВЕДЕНИЕ
Посттрансляционные модификации (ПТМ) относятся к ковалентной (в том числе ферментативной) модификации белков после биосинтеза. Они могут происходить вскоре после трансляции или на любой стадии жизненного цикла белка, они влияют на укладку белковой молекулы, ее стабильность, клеточную локализацию, активность, взаимодействие с другими белками и биомолекулами. В настоящее время экспериментально обнаружено более 620 типов ПТМ (http://www.uniprot.org/ docs/ptmlist.txt). Накопленные данные показали, что аномальный статус ПТМ какого-либо белка часто указывает на его участие в различных механизмах развития заболеваний, таких как рак, диабет и нейродегенеративные заболевания [1–3] и др. По этой причине посттрансляционные модификации белков выделяются в качестве многообещающего инструмента для прогнозирования заболеваний [4–6].
Однако наблюдается дефицит исследований, которые бы давали представление о разнообразии окислительных ПТМ белков. В основном исследования направлены на ограниченное количественное определение карбонилов или модифицированных ароматических аминокислотных остатков (образование дитирозиновых сшивок, 3-хлортирозина). В настоящее время для количественной оценки ПТМ используются имуноферментный анализ и другие иммунохимические методы, но они более применимы для узких задач в области фундаментальных исследований из-за длительных экспериментальных процедур, необходимости использования специфичных к различным модификациям антител и отсутствием специфичности к конкретному белку, что, в свою очередь, затрудняет исследования механизмов действия окислительного стресса на организм. Масс-спектрометрия является альтернативным подходом, который может обнаруживать широкий спектр модификаций, обеспечивает высокую селективность и специфичность анализа, а также позволяет избежать большинства проблем, связанных с объединением нескольких анализов в одном измерении [7–9]. При всех преимуществах метода масс-спектрометрии и его активном развитии в последние годы, существует ряд нерешенных проблем, которые до сих пор не позволяют применять его в рутинной клинико-лабораторной практике для анализа белковых маркеров, в том числе ПТМ. К ним можно отнести высокую стоимость оборудования и его последующего обслуживания, относительно низкую (по сравнению, например, с иммунохроматографическими методами) пропускную способность, длительную и многостадийную подготовку образцов к анализу, а также сложную обработку и, порой, интерпретацию полученных результатов [10, 11].
Фибрин – это основной белковый компонент тромботического сгустка. Фибриноген – белок-предшественник фибрина, является одним из ключевых факторов свертывания крови [12]. Известно, что фибриноген является белком острой фазы и маркером целого ряда заболеваний. Генетические и фармакологические исследования выявили ключевую роль фибриногена в определении степени местного или системного воспаления [13]. Воспаление является естественным механизмом защиты от патогенов и связано со многими заболеваниями. Многие хронические заболевания, связанные с повышенной продукцией активных форм кислорода, приводят к окислительному стрессу и окислению белков [14]. Фибриноген среди всех белков плазмы проявляет самую высокую чувствительность к воздействию окислителей [15]. Окисление фибриногена ведет к изменению процесса самосборки фибрина и формированию аномальных тромбогенных сгустков. Они характеризуются тонкими индивидуальными фибриновыми фибриллами, малой пористостью, низкой проницаемостью и, как следствие, повышенной устойчивостью к плазминовому гидролизу [16].
Цель данного исследования – разработка метода упрощенной подготовки проб к анализу методом высокоэффективной жидкостной хроматографии и тандемной масс-спектрометрии (ВЭЖХ–МС/МС) для дальнейшего клинико-диагностического использования, включающий иммунопреципитацию фибриногена на магнитных микрочастицах с последующим ферментативным гидролизом белка. Разработка эффективной методики, которая бы включала выделение целевого белка из исследуемого образца плазмы или любой другой физиологической жидкости с последующим гидролизом выделенного белка для дальнейшего анализа методом ВЭЖХ–МС/МС с исключением большого количества стадий процесса (например, многочисленных стадий переосаждения, различных видов хроматографии при выделении целевых белков) и использования агрессивных условий (например, при элюировании белков с носителей), а также с уменьшением общей трудоемкости и времени подготовки проб к анализу, является необходимой для использования в протеомных исследованиях.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для экспериментов была использована пулированная плазма крови доноров, полученная от Московской центральной станции переливания крови. Окисление образцов плазмы проводилось раствором гипохлорита фирмы Sigma-Aldrich (USA) при температуре 37 °С в течение 1 ч при постоянном перемешивании [17–19]. Количество окислителя составляло 300 мкМ. Реакция останавливалась добавлением десятикратного молярного избытка L-метионина.
Иммунопреципитация фибриногена из образцов плазмы проводилась с использованием магнитных микрочастиц с NH2-группой производства компании Sileks (Россия), модифицированных моноклональными (клон 1F3) антителами (МАт) против фибриногена (HyTest, Россия) в течение 1 ч при постоянном перемешивании (соотношение плазма/суспензия магнитных частиц равно 9/1). После окончания инкубации магнитные частицы отделялись от обедненной фибриногеном плазмы с использованием магнитного штатива. Затем частицы пятикратно промывались 1 мл 10 мМ натрий-фосфатного буфера (рН = 7.4) с 0.15 М NaCl также с использованием магнитного штатива. После окончания промывки суспендировали частицы в 1 мл этого же буфера.
Успешность иммунопреципитации молекул фибриногена оценивалась методом электрофореза в полиакриламидном геле (5% – концентрирующий гель, 15% – разделяющий гель) по присутствию полипептидных цепей фибриногена. Электрофорез восстановленных образцов фибриногена, предварительно элюированного с поверхности магнитных частиц 1 М раствором глицина, рН = 2, проводили в присутствии додецилсульфата натрия (SDS) по методике Laemmli. Белок был окрашен красителем кумасси бриллиантовым синим R250 производства компании Pharmacia (Uppsala, Sweden).
Ферментативное расщепление образцов смесью трипсина и Arg-C в соотношении 1 : 1 осуществляли в соответствии с протоколом производителя (Trypsin Gold, mass spectrometry grade, V5280, Arg-C, Sequencing Grade, V1881, Promega, USA). Магнитные частицы с Мат и преципитированным фибриногеном переносились в 50 мМ трис-HCl-буфер с 0.15 М NaCl (рН = 8.0). Гидролиз белков осуществлялся смесью трипсина и Arg-C в соотношении фермент/образец 1/50 в течение 1, 5 и 16 ч при 37 °С. Реакцию останавливали путем добавления муравьиной кислоты до конечной концентрации в 0.1%.
Эффективность гидролиза оценивалась методом электрофореза в полиакриламидном геле (5% – концентрирующий гель, 15% – разделяющий гель). Магнитные частицы после гидролиза были проинкубированы в буфере для образцов с добавлением 5% 2-меркаптоэтанола в течение 5 мин при 90 °С. Электрофорез восстановленных образцов проводили по той же методике, что и для фибриногена (см. выше). Для определения молекулярных масс полипептидных цепей использовали смесь белков-маркеров с молекулярной массой от 10 до 250 кДа: Page Ruler Plus Prestained Protein Ladder, компании Thermo Scientific (USA).
Эксперименты ВЭЖХ–МС/МС выполняли с помощью хроматографа Agilent 1100 производства фирмы “Agilent Technologies Inc.” (USA) c системой автоматического отбора проб и тандемного масс-спектрометра 7T LTQ-FT Ultra фирмы “Thermo Fisher Scientific” (USA). Для хроматографического разделения 1 мкл каждого образца впрыскивали в колонку С18 размером 75 мкм × 12 см производства компании Ammerbuch-Entringen (Germany). Использовали следующую подвижную фазу: растворитель A – 0.1% муравьиная кислота в H2O; растворитель B – ацетонитрил. Хроматографию осуществляли с линейным градиентом путем увеличения относительного содержания растворителя B с 3 до 50% в течение 60 мин. Масс-спектрометрический анализ пептидных фракций проводили с использованием программного обеспечения Xcalibur (Thermo Electron, Бремен, Германия) с автоматическим измерением спектров в двухстадийном режиме [20]. На первом этапе точные массы пептидов были измерены в ячейке ICR в диапазоне m/z = 300–1600 с разрешением R = 50 при m/z = 400 (количество ионов в ячейке ICR было установлено равным 5 · 106). На втором этапе пять наиболее интенсивных пиков первой стадии подвергались столкновительной диссоциации, спектры фрагментов регистрировались в линейной ионной ловушке (число ионов было установлено равным 3 × 104). После фрагментации соответствующие массы родительских пептидов были динамически исключены из рассмотрения в течение следующих 30 с.
Триптические пептиды образцов фибриногена были идентифицированы путем поиска в базе данных UniProtKB (UP000005640-9606 HUMAN, Homo sapiens) с использованием программного обеспечения PEAKS Studio (v.8.5, Bioinformatics Solutions Inc., Canada). Точность определения массы для иона-предшественника была установлена равной 15 ч/млн. Точность массы фрагментов МС/МС – до 0.50 Да.
Фильтрация результатов по средней доле ложных отклонений для пептидов была установлена на уровне <0.1%. Пептиды от шести аминокислотных остатков и содержали максимум три модификации рассматривались для идентификации. Список типов обнаруженных окислительных модификаций приведен в табл. 1.
Таблица 1.
Типы детектированных модификаций
| Моноизотопная масса | Название модификации | Состав | Модифицированные аминокислотные остатки |
|---|---|---|---|
| +15.994915 | окисление | O | Met, Tyr, Trp, Pro, Asn, Asp |
| +31.989828 | диокисление | O (2) | Tyr, Trp |
| +13.979265 | окисление триптофана до оксолактона | H(–2) O | Trp |
| +44.985077 | нитротирозин | H(–1) N O(2) | Tyr |
Идентификация с использованием программы PEAKS достигается путем интеграции результатов поиска в выбранной базе данных и результатов анализа спектров с помощью алгоритма de novo. Алгоритм поиска в базе данных используется для идентификации белков. При поиске в базе данных в качестве вариабельных модификаций указывается только ограниченное число посттрансляционных модификаций для максимизации чувствительности (например, окисление (+15.99) или диокисление (+31.99)). Алгоритм поиска модификаций используется для выявления большего количества ПТМ. На этом этапе анализируются только спектры с высокими показателями достоверности при анализе de novo, но не идентифицированные при поиске в базе данных. Список вариабельных модификаций расширяется и включает более специфические модификации из находящихся в базе данных UNIMOD.
Точные сайты модификации определялись наличием сайт-определяющих фрагментов ионов. Определение достоверных сайтов модификаций было достигнуто с помощью параметра Ascore, включающего доверительную оценку идентификации модификации пептида в конкретной позиции (аминокислотного остатка), рассчитанной с помощью программного обеспечения PEAKS, в качестве –10 log вероятности того, что модификация произошла в сообщенной позиции, сравнительно с другими возможными позициями. В этой работе мы использовали порог фильтрации для параметра Ascore >100.
Все эксперименты повторялись трижды. Аминокислотный остаток, который оказывался модифицированным только в одном из образцов, исключался из дальнейшего анализа. Каждый из аминокислотных остатков, окисленных по меньшей мере на 0.1% (степень модификации >0.1%), считался модифицированным.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Первый этап разрабатываемой методики включает взаимодействие молекул фибриногена с МАт, поэтому крайне важно определить влияние окислителя на эпитопную специфичность МАт к фибриногену. На рис. 1а представлен результат электрофореза смыва фибриногена, преципитированного из нативной и окисленной плазмы, с магнитных частиц, модифицированных МАт. Количество фибриногена, захваченного МАт на поверхности магнитных частиц, не зависит от обработки окислителем образцов плазмы, что свидетельствует о том, что эпитопная специфичность сохраняется.
Рис. 1.
a – электрофореграмма для фибриногена, иммунопреципитированного из нативной плазмы (1), и из плазмы, окисленной 300 мкМ HOCl/–OCl (2) после элюирования с магнитных частиц; б – электрофореграмма белкового элюата с магнитных частиц после 1 (3), 5 (4) и 16 ч (5) ферментативного гидролиза. На дорожке 1 – полипептидные цепи фибриногена, на дорожке 2 – моноклональные антитела против фибриногена; М – маркерные белки.
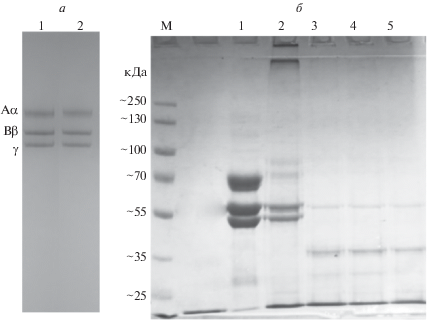
Для верной интерпретации результатов на этапе обработки белковых молекул, иммобилизованных на поверхности магнитных частиц, смесью гидролитических ферментов необходимо понимать насколько полно прошел процесс гидролиза. На рис. 1б представлена электрофореграмма, демонстрирующая, что большая часть белка была успешно гидролизована с поверхности частиц уже через 1 ч обработки ферментами.
По результатам масс-спектрометрии количество детектированных участков молекулы фибриногена, так называемое покрытие, слабо зависит от времени гидролиза (различия внутри группы составляли не более 10%); см. рис. 2а. Изменение покрытия в окисленных образцах может быть вызвано рядом причин, в том числе повреждением участков гидролиза в результате окисления, либо, напротив, экспонированием ранее труднодоступных участков гидролиза в результате гидрофобизации поверхности молекулы белка при окислении.
Рис. 2.
Покрытие аминокислотной последовательности полипептидных цепей фибриногена и детектированные окислительные модификации.
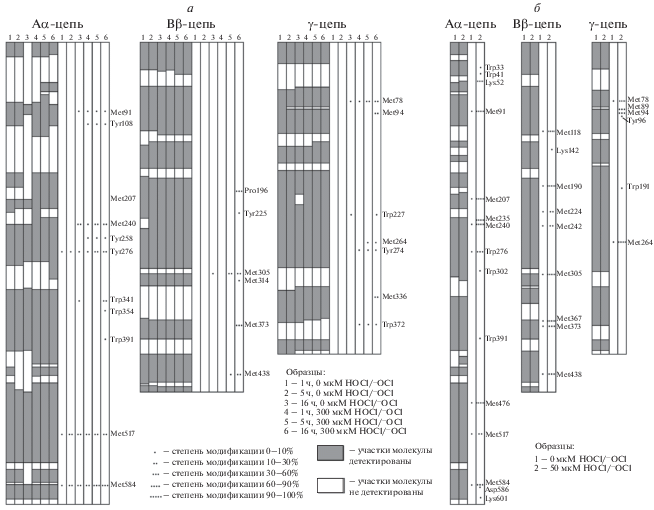
Увеличение содержания (степень модификации) окисленных пептидных форм в молекуле фибриногена наблюдается в окисленном образце относительно нативного, а также степень модификации растет внутри групп образцов с увеличением времени инкубации при обработке гидролитическими ферментами (рис. 2а). Прирост степени модификации внутри групп образцов с увеличением времени обработки может быть объяснен автоокислением белковых молекул, что повышает риск неверной интерпретации результатов. В результате чего, можно предположить, что 1 ч обработки образца гидролитическими ферментами является достаточным для получения достоверных отличий между окисленным и нативным образцами при отсутствии артефактных модификаций.
Методом масс-спектрометрии был детектирован ряд окислительно модифицированных аминокислотных остатков: AαMet91, AαTyr108, AαMet240, AαTyr258, AαTyr276, AαTrp341, AαTrp354, AαTrp391, AαMet517, AαMet584, BβPro196, BβTyr225, BβMet305, BβMet314, BβMet373, BβMet438, γMet78, γMet94, γTrp227, γMet264, γTyr274, γMet336, γTrp372. Ранее опубликованные результаты индуцированного гипохлоритом окисления предварительно очищенного фибриногена [21] коррелируют с данным исследованием (рис. 2б), что свидетельствует о высокой достоверности полученных результатов. Высокая степень модификации аминокислотных остатков при окислении фибриногена в растворе по сравнению с фибриногеном, окисленным в плазме, обусловлена отсутствием буферных белков (прежде всего, альбумина), перехватывающих молекулы окислителя и, как следствие, большую концентрацию гипохлорита относительно исследуемого белка.
ЗАКЛЮЧЕНИЕ
Посттрансляционные модификации (ПТМ) участвуют во многих аспектах биологических процессов, регулируя функции белков. Считается, что аберрантные ПТМ белка часто указывают на его участие в механизмах развития заболеваний. По этой причине посттрансляционные модификации белков выделяются в качестве многообещающего инструмента молекулярной диагностики. Однако, во многом из-за методических сложностей при анализе ПТМ и невозможности проведения глобального скрининга ассоциированных с заболеваниями ПТМ, об их диагностической ценности можно судить только по некоторым, весьма ограниченным и неструктурированным исследованиям. Поэтому разработка высокочувствительного и селективного экспресс-метода для определения типов и уровней ПТМ в белках имеет важное научно-техническое значение.
Разработанный метод подготовки проб для анализа методом ВЭЖХ–МС/МС позволяет сократить общее время анализа до 1 ч гидролиза (при стандартных значениях, составляющих 12–16 ч) и, как следствие, избежать образования ПТМ, характерных для продолжительной подготовки проб или использовании агрессивных условий элюции, и затрудняющих анализ результатов. В перспективе данный метод может быть адаптирован к различным диагностически значимым белковым молекулам и упростить процедуры при работе с большим количеством образцов.
В работе использовали оборудование ЦКП ИБХФ РАН.
Исследование выполнено при поддержке грантом Российского научного фонда № 21-74-00146.
Список литературы
Jin H., Zangar R.C. // Biomarker Insights. 2009. V. 4. P. 191.
Morino C.K., Petersen K.F., Dufour S. et al. // J. Clin. Invest. 2005. V. 115. P. 3587.
Gong C.X., Liu F., Grundkeiqbal I. et al. // J. Neural. Transm. 2005. V. 112. P. 813.
Kumar V., Calamaras T.D., Haeussler D. et al. // Antioxid. Redox. Signal. 2012. V. 17. № 11. P. 1528.
Chung H.S., Wang S.B., Venkatraman V. et al. // Circ. Res. 2013. V. 112. № 2. P. 382.
Xu H., Wang Y., Lin S. et al. // Genomics, Proteomics, Bioinformatics. 2018. V. 16. № 4. P. 244.
Anderson N.L., Anderson N.G., Haineset L.R. et al. // J. Proteome. Res. 2004. V. 3. P. 235.
Bronstrup M. // Expert Rev. Proteomics. 2004. V. 1. P. 503.
Kirkpatrick D.S., Gerber S.A., Gygi S.P. // Methods. 2005. V. 35. P. 265.
Grebe S.K., Singh R.J. // The Clinical biochemist. Reviews. 2011. V. 32. № 1. P. 5.
Pan S., Aebersold R., Chen R. et al. // J. Proteome Res. 2009. V. 8. P. 787.
Medved L., Weisel J.W. // J. Thromb. Haemost. 2009. V. 7. P. 355.
Davalos D., Akassoglou K. // Semin. Immunopathol. 2012. V. 34. P. 43.
Berlett B.S., Stadtman E.R. // J. Biol. Chem. 1997. V. 272. № 33. P. 20313.
Shacter E., Williams J.A., Lim M. et al. // Free Radic. Biol. Med. 1994. V. 17. P. 429.
Weigandt K.M., White N., Chung D. et al. // Biophys. J. 2012. V. 103. P. 2399.
Васильева А.Д., Юрина Л.В., Леонова В.Б. и др. // Хим. физика. 2020. Т. 39. № 6. С. 24.
Щеголихин А.Н., Васильева А.Д., Юрина Л.В. и др. // Хим. физика. 2021. Т. 40. № 2. С. 66.
Вассерман Л.А., Юрина Л.В., Васильева А.Д. и др. // Хим. физика. 2021. Т. 40. № 11. С. 59.
Кононихин А.С., Захарова Н.В., Юсупов А.Э. и др. // Хим. физика. 2019. Т. 38. № 12. С. 59.
Юрина Л.В., Васильева А.Д., Бугрова А.Е. и др. // ДАН. 2019. Т. 484. № 3. С. 367.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика