

Химическая физика, 2022, T. 41, № 6, стр. 72-79
Моделирование адсорбции водорода и кислорода на наночастицах никеля и платины, расположенных на графитовой подложке с различными дефектами
Н. В. Дохликова 1, *, С. А. Озерин 1, С. В. Доронин 1, Е. И. Руденко 1, М. В. Гришин 1, Б. Р. Шуб 1
1 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова
Российской академии наук
Москва, Россия
* E-mail: dohlikovanv@gmail.com
Поступила в редакцию 06.12.2021
После доработки 13.01.2022
Принята к публикации 20.01.2022
- EDN: WPGOEM
- DOI: 10.31857/S0207401X22060024
Аннотация
С помощью моделирования в рамках теории функционала плотности адсорбции атомарных кислорода и водорода на поверхности наночастиц никеля и платины на подложках графита с различными дефектами были рассчитаны энергии связи адатомов и изменения плотности состояний атомов металла при взаимодействии с адатомами. Установлено, что при адсорбции кислорода на вершине кластера металла плотность состояний снижается больше, чем при его адсорбции на границе кластера металла и подложки графита. Для адсорбции водорода подобных зависимостей не выявлено. Продемонстрировано, что влияние одноатомных дефектов подложки незначительно при адсорбции обоих типов адатомов.
1. ВВЕДЕНИЕ
Квантовохимическое моделирование наноразмерных систем в настоящее время получило широкое распространение благодаря значительно возросшим вычислительным мощностям компьютеров. Для расчета параметров таких систем наиболее оптимально использование теории функционала плотности (ТФП), поскольку ее применение в данный момент дает наилучшую возможность моделирования реальных систем при разумных вычислительных затратах. В частности, в рамках ТФП можно проводить моделирование наноразмерных объектов различных типов – как свободных кластеров, так и структурированных на наноуровне поверхностей. Последние представляют собой распространенный тип наноразмерных систем, использующихся в качестве катализаторов, сенсоров и прочих функциональных наноматериалы. Подробный обзор физико-химических свойств нанесенных наночастиц проведен в работе [1].
Одним из наиболее распространенных и давно известных являются катализаторы на основе платины, однако адсорбционные свойства ее структурированной на наноуровне формы могут отличаться от объемного образца, что открывает возможность создания новых функциональных материалов с управляемыми свойствами. В работе [2] проведено исследование окисления СО на трехатомных кластерах платины (Pt3) и рассчитаны энергетические барьеры возможных вариантов реакций, наиболее вероятный из которых протекает через образование оксидного комплекса платины Pt3O(O). В работе [3] было проведено исследование влияния взаимодействия кластеров Pt размером до 1 нм с подложкой графита на реакцию окисления СО на поверхности кластера с помощью варьирования места адсорбции СО относительно подложки. Авторы этой работы установили, что CO адсорбируется слабее на вершине кластера и сильнее на участках вблизи поверхности, чем на кластере без подложки.
Исследования взаимодействия водорода с малыми кластерами платины Pt1–Pt5, выполненные в работе [4], показали, что водород диссоциирует и хемосорбируется на одноцентровом сайте поверхности кластера. Среди гетерогенных кластеров PtnHm появляются “магические” структуры повышенной стабильности. Моделирование реакции генерирования водорода из воды на кластере платины Pt7 показало, что этот кластер способен отщеплять атом или молекулу Н от молекулы Н2О [5]. В работе [6] проведено исследование кластеров никеля Nin (n = 4–6) с атомами Н, С, О примеси. Показано, что малые кластеры никеля проявляют молекулярные свойства, поскольку не наблюдается единой закономерности в изменении количества изомеров, величин энергии ионизации и сродства к электрону. При этом никель всегда заряжен положительно, а атом примеси – отрицательно. Также никель активно используется в качестве допирующего атома в кластерах благородных и других переходных металлов. Например, в работе [7] показано, что на кластере NiAu6 адсорбция кислорода атомарная.
В наших работах [8, 9] в приближении ТФП было проведено моделирование процесса адсорбции водорода на наночастицах золота и меди, а также влияния подложки графита на систему металл–водород. Указанная компьютерная модель в достаточно хорошем приближении соответствует объектам реального эксперимента по адсорбции водорода на золотых и медных наночастицах, исследованной методами сканирующей туннельной микроскопии/спектроскопии (СТМ/СТС) методик [10, 11]. Совместное использование численного эксперимента с учетом квантовых эффектов и реального эксперимента с атомным разрешением впервые позволило установить, что при взаимодействии атомов водорода с кластерами золота в компьютерной модели изменения энергетического спектра коррелируют с образованием запрещенной зоны у наночастиц золота при адсорбции водорода в реальном эксперименте. Для кластеров меди этот эффект также проявлялся, но в меньшей степени. В данной работе аналогичные исследования по адсорбции водорода и кислорода будут проведены для кластеров платины и никеля. Результаты исследований будут сопоставлены как с экспериментальными данными, так и с результатами работ [8, 9].
2. МЕТОДИКА ИССЛЕДОВАНИЙ
Моделирование наноразмерных систем, имитирующих систему наночастиц Ni и Pt, нанесенных на графитовую подложку с различными дефектами, проводилось в рамках ТФП с использованием программных пакетов Quantum Espresso (QE) [12] и OpenMX (OMX) [13] с открытым исходным кодом. Для расчета электронной структуры использовано приближение LDA в ультрамягком псевдопотенциале с функционалом Пердью–Зингера и радиусом обрезания в 39 Ry. Данные условия расчета были выбраны, исходя из тестовых вычислений, которые показали лучшую сходимость по электронной подсистеме при выбранных параметрах. Наборы атомноцентрированных базисных функций взяты из документации к программному пакету OMX.
Модель подложки представляла собой пластину (slab) графита, состоящую из двух графеновых плоскостей, содержащих по шесть краевых атомов с каждой стороны. Расстояние между плоскостями графена внутри пластины после оптимизации атомной структуры с варьированием параметров ячейки равно 4.34 Å. Величина вакуумного зазора составляла 14.5 Å. Под оптимизацией атомной структуры подразумевается поиск координат атомов, при которых система, состоящая из этих атомов, имеет наименьшее значение энергии.
В данном исследовании рассматривались следующие дефекты подложки: вакансия атома углерода, дефект 5/7, обрывы плоскости графена типа “зигзаг” и “кресло”. Во всех случаях атомная структура верхней плоскости графена была дополнительно оптимизирована без варьирования параметров ячейки для устранения напряжений в структуре при соблюдении единообразия размеров расчетной ячейки.
В качестве исходной модели наночастиц Ni и Pt использовались 13-атомные икосаэдрические изомеры кластеров Ni13 и Pt13. Атомная структура кластеров рассчитывалась при фиксированном положении всех атомов подложки. Поскольку данная работа посвящена исследованию вопросов, касающихся выявления изменений электронной структуры адсорбционного комплекса под влиянием подложки, полученные наноразмерные системы Ni13–графит и Pt13–графит, соответствующие локальному энергетическому минимуму, удовлетворяют поставленным задачам.
Взаимное влияние кластеров друг на друга исследовано с помощью тестовых расчетов с варьированием размеров ячейки, т.е. расстояния между кластерами. Эти расчеты показали, что взаимное влияние кластеров в условиях наших расчетов пренебрежимо мало.
Для каждой системы взаимодействие с адсорбатом было рассчитано для двух случаев. В первом случае положения атомов кластера металла не изменялись при оптимизации, т.е. атомы кластера были фиксированы. Во втором случае положения атомов кластера металла изменялись по ходу оптимизации положения атома адсорбата, т.е. атомы кластера не были фиксированы. Сравнение величин энергий связи в этих двух случаях позволит оценить влияние подвижности атомов металла на энергию связи с адатомами. Для каждой системы проводилось моделирование двух сайтов: в окрестности интерфейса кластер–графит – сайт s2 и на вершине кластера, вдали от интерфейса – сайт s1.
Расчет изменения спроектированной плотности состояний отдельных атомов вблизи сайта адсорбции адатомов является важной частью исследования, поскольку позволяет провести сопоставление результатов СТМ/СТС-экспериментов и моделирования. Для получения более подробной информации изменения s- и d-орбиталей атомов металла исследуются отдельно друг от друга. В предыдущих работах мы показали, что именно состояния в окрестности уровня Ферми вносят основной вклад в изменение туннельного тока в СТС [10]. При небольших значениях напряжения смещения туннельный ток будет определяться только плотностью состояний образца, что позволяется сопоставить изменение вольт-амперных характеристик локальной области образца при адсорбции с изменениями плотности состояний вблизи уровня Ферми при взаимодействии с адатомами.
3. РЕЗУЛЬТАТЫ
Предварительный расчет изменения плотности состояний атомов металла свободных кластеров Ni13 и Pt13 при взаимодействии с адатомами Н и О показал, что в кластерах переходных металлов плотность состояний вблизи уровня Ферми образована в основном d-электронами. Взаимодействие с адатомами вызывает ее незначительное снижение (рис. 1). Величины энергии связи с адатомами Н и О приведены в табл. 1.
Рис. 1.
Спроектированные плотности состояний адсорбционных комплексов Н–Ni13 (а), О–Ni13 (б), Н–Pt13 (г), О–Pt13 (д) и их конфигурационные диаграммы: в – Ni, е – Pt.
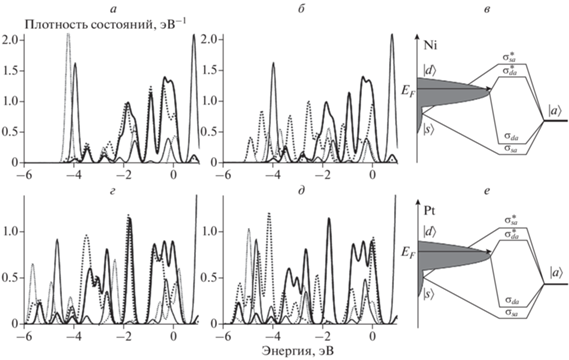
Таблица 1.
Величины энергий связи, адатомов Н и О со свободными кластерами Ni13 и Pt13
| Адсорбат | Энергия связи, эВ | |
|---|---|---|
| Ni13 | Pt13 | |
| H | –3.40 | –3.90 |
| O | –4.58 | –4.34 |
Моделирование влияния графитовой подложки показало, что атомная структура кластера Pt13 даже в системах с подложками с единичными дефектами претерпевает достаточно большую трансформацию, после которой кластер теряет первоначальную икосаэдрическую симметрию. Этот эффект вполне очевиден, так как энергия связи между кластерами и подложкой в этом случае больше, чем в системах с кластерами Au [8] и Cu [9] (табл. 2). Однако, несмотря на то, что величина энергии связи кластера Ni13 с подложками больше, атомная трансформация икосаэдрического кластера Ni13 в системах с единичными дефектами невелика и становится более выраженной в системах с обрывом плоскости графита, особенно при обрыве плоскости типа “зигзаг”, как и для Au. На кластеры Cu и Pt большее влияние оказывал обрыв плоскости типа “кресло”. Согласно данным СТМ/СТС-экспериментов наночастицы платины и никеля обладают тенденцией к концентрации на границах террас графита, что соответствует полученным результатам квантовохимического моделирования [14, 15]. Анализ заселенности по Малликену и построение распределений электронной плотности систем Ni13–графит и Pt13–графит показали накопление небольшого количества отрицательного заряда в подсистеме металла.
Таблица 2.
Величины энергий связи (Eсв) 13-атомных кластеров Cu13, Au13, Ni13, Pt13 с подложками графита без дефекта (1), с вакансией (2), с дефектом Стоуна–Уэльса (3), при обрыве плоскости графена типа “кресло” (4), при обрыве плоскости графена типа “зигзаг” (5)
| Кластер | Eсв, эВ | Заряд | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| Cu | –0.17 | –0.39 | –0.19 | –1.31 | –1.01 | + |
| Au | –0.16 | –0.42 | –0.18 | –1.60 | –1.76 | – |
| Ni | –0.51 | –0.90 | –0.51 | –1.97 | –2.18 | – |
| Pt | –0.36 | –0.53 | –0.44 | –2.78 | –1.94 | – |
Моделирование процесса адсорбции атомов H на поверхности кластеров Ni13 и Pt13 на различных подложках показало, что величины энергий связи вблизи интерфейса и на вершине различаются не слишком сильно (~0.1–0.4 эВ). Более того, нет никакой четкой зависимости энергии сайта от близости и дефектности подложки. Моделирование процесса адсорбции атомов O на поверхности кластеров Ni13 и Pt13 в целом дало сходные результаты, однако различие величин энергий связи сайтов вблизи границы металл–графит и на вершине несколько больше аналогичных значений, полученных для адсорбции атомов Н (табл. 3). Кроме того, в системе Ni13–графит с обрывами плоскости при адсорбции на сайте вблизи интерфейса атом О образует связь с атомом С, а не с атомом Ni. В целом энергия связи адатомов для кластеров на подложке больше энергии связи для свободных кластеров (табл. 1).
Таблица 3.
Величины энергии связи для сайтов адсорбции атомов H и О на кластерах Ni13, Pt13 и подложках графита без дефекта (1), с вакансией (2), с дефектом 5/7 (3), при обрыве плоскости графена типа “кресло” (4), при обрыве плоскости графена типа “зигзаг” (5) при фиксированном (Eсв) и нефиксированном $(Е_{{{\mathbf{св}}}}^{*})$ положениях атомов Ni и Pt (ΔE = Eсв – $Е_{{{\mathbf{св}}}}^{*}$)
| Атом | Сайт | Вид подложки | Eсв | $Е_{{{\text{св}}}}^{*}$ | ΔE | |||
|---|---|---|---|---|---|---|---|---|
| Ni | Pt | Ni | Pt | Ni | Pt | |||
| H | s1 | 1 | –4.35 | –4.37 | –0.02 | –4.28 | –4.32 | –0.03 |
| 2 | –3.81 | –4.41 | –0.60 | –4.38 | –4.42 | –0.03 | ||
| 3 | –4.48 | –4.48 | –0.003 | –4.37 | –4.60 | –0.23 | ||
| 4 | –4.23 | –4.24 | –0.01 | –4.03 | –4.28 | –0.25 | ||
| 5 | –4.13 | –4.15 | –0.03 | –4.02 | –4.18 | –0.15 | ||
| s2 | 1 | –4.21 | –4.24 | –0.03 | –4.14 | –4.21 | –0.07 | |
| 2 | –4.13 | –4.20 | –0.07 | –4.03 | –6.61 | –2.59 | ||
| 3 | –4.24 | –4.26 | –0.03 | –4.15 | –4.19 | –0.04 | ||
| 4 | –4.33 | –4.36 | –0.03 | –4.23 | –4.78 | –0.55 | ||
| 5 | –3.69 | –3.92 | –0.24 | –3.90 | –4.07 | –0.17 | ||
| O | s1 | 1 | –6.39 | –6.59 | –0.20 | –4.86 | –5.29 | –0.43 |
| 2 | –5.08 | –5.16 | –0.08 | –5.45 | –6.48 | –1.03 | ||
| 3 | –6.90 | –6.98 | –0.08 | –5.36 | –5.98 | –0.62 | ||
| 4 | –6.35 | –6.59 | –0.24 | –4.33 | –4.61 | –0.28 | ||
| 5 | –5.92 | –6.21 | –0.28 | –3.98 | –4.65 | –0.67 | ||
| s2 | 1 | –6.39 | –6.59 | –0.20 | –4.37 | –4.75 | –0.38 | |
| 2 | –5.99 | –6.33 | –0.34 | –4.59 | –6.40 | –1.81 | ||
| 3 | –6.38 | –7.06 | –0.68 | –4.89 | –5.15 | –0.26 | ||
| 4 | –6.39 | –6.69 | –0.30 | –3.95 | –4.94 | –0.99 | ||
| 5 | –2.99 | –3.02 | –0.03 | –4.49 | –5.04 | –0.56 | ||
Подвижность атомов Ni влияет на энергию связи с атомами Н и О незначительно, т.е. взаимодействие с адатомами практически не изменяет атомную структуру кластера Ni13. Можно предположить, что в данной системе взаимодействие атомов Ni с подложкой сильнее, чем между атомами Ni в кластере. Влияние подвижности атомов Pt на величину энергии связи с адатомами может приводить к существенной трансформации всего кластера Pt13. Наиболее заметно это проявляется для системы, содержащей подложку с вакансией.
На рис. 2 и 3 приведены рассчитанные спроектированные плотности состояний s- и d-орбиталей атомов Ni и Pt сайтов адсорбции атомов H и О для систем с различными подложками. Расчет показал, что каждая подложка может оказывать достаточно серьезное воздействие на распределение плотности состояний атомов металлов, однако вблизи уровня Ферми по-прежнему расположена “зона” d-орбиталей, как и в свободных 13-атомных кластерах (рис. 1).
Рис. 2.
Спроектированные плотности состояний d-орбиталей (толстая линия), s-орбиталей (тонкая линия) атомов Ni; сплошная линия – до адсорбции, штриховая – после адсорбции атомов Н (а–д), О (е–к); использованные подложки: а, е – бездефектный графит; б, ж – графит с вакансией; в, з – графит с дефектом 5/7; г, и – обрыв плоскости графена типа “кресло”; д, к – обрыв плоскости графена типа “зигзаг”; для каждого адсорбционного комплекса на подложке представлены два сайта: 1 – на вершине, 2 – на интерфейсе.
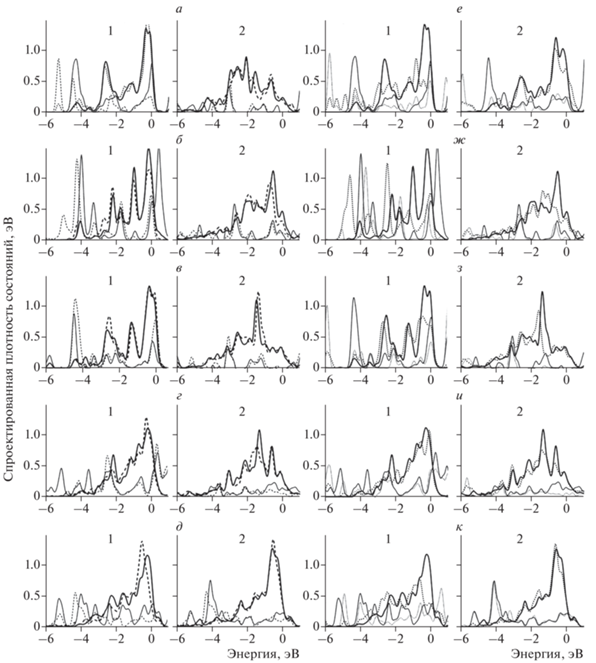

Изменения плотности состояний при адсорбции атомов Н на сайтах на вершине и вблизи интерфейса похожи на изменения при их адсорбции на поверхности свободных кластеров. Можно отметить, что меньшая энергия связи сайта встречается на сайтах с меньшей плотностью d-состояний вблизи уровня Ферми в системах с единичным дефектом подложки, где кластер еще сохраняет упорядоченное строение поверхности. По-видимому, благодаря сильному взаимодействию подложка–кластер явных различий между сайтами на вершине и вблизи интерфейса нет, и активность отдельных сайтов будет обусловлена локальными особенностями поверхности кластера. Полученные результаты соответствуют экспериментальным данным, где экспозиция в атмосфере водорода структурированных на наноуровне покрытий на основе платины и никеля не вызывала существенных изменений вольт-амперных характеристик образца [12, 13].
Изменения плотности состояний при адсорбции атомов О на сайтах на вершине и вблизи границы металл–графит для кластеров, расположенных на подложке, похожи на изменения при адсорбции кислорода на поверхности свободных кластеров. Однако стоит отметить, что для наноразмерных систем с Ni и подложками с единичными дефектами при адсорбции на сайтах на вершине характерно заметное уменьшение плотности состояний вблизи уровня Ферми. При этом для сайтов вблизи интерфейса этого не наблюдается. В меньшей степени этот эффект проявляется для наносистем с Pt и подложками с единичными дефектами. Одной из причин отклонений от такой тенденции предположительно может быть существенная атомная трансформация кластеров Pt. Такие же отклонения встречаются и в системах с подложками с обрывами плоскостей графена, где кластеры металлов также деформированы.
На основании полученных данных можно провести сравнение адсорбционных свойств кластеров переходных металлов Ni13 и Pt13 и ранее исследованных кластеров благородных металлов Cu13 и Au13 [8, 9]. В табл. 2 приведены величины энергии связи исследованных кластеров металлов с подложками графита. Как видно, энергия связи кластеров переходных металлов больше, чем энергия связи кластеров благородных металлов, что, разумеется, оказывает влияние и на адсорбционные свойства этих кластеров.
При слабом взаимодействии с подложкой благородных металлов Cu и Au адсорбция на сайтах вблизи интерфейса становится предпочтительней. Это можно заметить как по величинам изменения связи, так и по изменению плотностей состояний при адсорбции. Cтепень проявления “эффекта интерфейса” для кластеров Au немного больше, чем для кластеров Cu, что может быть обусловлено в том числе и более заметной атомной трансформацией кластеров Au. Также в системах с относительно слабым взаимодействием кластера металла и подложки становится заметным влияние дефектов подложки [8, 9].
При сильном взаимодействии с подложкой переходных металлов Ni и Pt плотность d-состояний вблизи уровня Ферми, в основном определяющая величину энергии связи [16], может изменяться и вблизи интерфейса, и на вершине кластера. Благодаря этому сайты адсорбции атомов Н и О на вершине и вблизи интерфейса становятся равнозначными. Вклад дефектов подложки на фоне ее общего сильного воздействия также становится незначительным. При этом адсорбция атомов О сильнее уменьшает плотность состояний на вершине кластера, если сам кластер трансформировался не слишком сильно.
Можно заметить, что тенденция к большей атомной трансформации и более заметному влиянию подвижности атомов металла на адсорбционные свойства проявляют кластеры более тяжелых металлов, таких как Pt и Au.
4. ЗАКЛЮЧЕНИЕ
В результате ТФП-моделирования адсорбции адатомов Н и О на наночастицах в сстемах Ni13–графит и Pt13–графит установлено, что энергия связи адатомов с кластерами переходных металлов на подложках графита больше, чем в системах свободных кластеров переходных металлов. Одним из факторов увеличения энергии связи может быть появление отрицательного заряда на кластерах металла при взаимодействии с подложкой. Сайты адсорбции атомов Н и О на вершине кластеров переходных металлов и вблизи границы кластер–подложка становятся равнозначными благодаря сильному воздействию подложки. Изменение плотности состояний при адсорбции атомов О на вершине больше, если трансформация кластера невелика. Влияние дефектов подложки на адсорбционные свойства системы кластер–подложка незначительно.
Все расчеты выполнены с использованием вычислительных ресурсов Межведомственного Суперкомпьютерного Центра РАН (МСЦ РАН).
Исследование выполнено за счет гранта Российского научного фонда (проект № 21-73-20010).
Список литературы
Lambert R.M., Pacchioni G. Chemisorption and Reactivity on Supported Clusters and Thin Films. Netherlands, Dordrecht: Springer Science & Business Media, 1997.
Baruah S.D., Gour N.K., Sarma P.J., Deka R.C. // Comput. Theoret. Chem. 2017. V. 1114. P. 1.
Ramos-Sánchez G., Balbuena P.B. // J. Electroanal. Chem. 2014. V. 716. P. 23.
Sebetci A. // Chem. Phys. 2006. V. 331. P. 9.
Xie W.-L., Zhang Z.-H. et al. // Intern. J. Hydrogen Energy. 2017. V. 42. № 7. P. 4032.
Petkov P.St., Vayssilov G.N., Kruger S., Roch N. // Chem. Phys. 2008. V. 348. P. 61.
Nigam S., Sahoo S.K., Sarkar P., Majumder Ch. // Chem. Phys. Lett. 2013. V. 584. P. 108.
Дохликова Н.В., Гатин А.К., Сарвадий С.Ю. и др. // Хим. физика. 2021. Т. 40. № 7. С. 67.
Дохликова Н.В., Гатин А.К., Сарвадий С.Ю. и др. // Хим. физика. 2022. Т. 41. № 4. C. 72.
Дохликова Н.В., Колченко Н.Н., Гришин М.В., Гатин А.К., Шуб Б.Р. // Рос. нанотехнол. 2016. Т. 11. № 11–12. С. 54.
Гатин А.К., Сарвадий С.Ю., Дохликова Н.В., Гришин М.В. // Хим. физика. 2021. Т. 40. № 6. С. 3.
Giannozzi P., Andreussi O., Brumme T. et al. // J. Phys.: Condens. Matter. 2017. V. 29. № 46. P. 30.
Ozaki T., Kino H. // Phys. Rev. B. 2004. V. 69. № 19. P. 19
Гришин М.В., Гатин А.К., Дохликова Н.В. и др. // Хим. физика. 2019. Т. 38. № 1. С. 3.
Гатин А.К., Гришин М.В., Сарвадий С.Ю. и др. // Кинетика и катализ. 2018. Т. 59. № 2. С. 224.
Hammer B., Norskov J.K. // Surf. Sci. 1996. V. 359. № 1–3. P. 306.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика