

Химическая физика, 2022, T. 41, № 6, стр. 65-71
Молекулярная динамика фермент-субстратных комплексов в гуанозинтрифосфат-связывающих белках
М. Г. Хренова 1, 2, *, И. В. Поляков 1, 3, А. В. Немухин 1, 3
1 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
2 Институт биохимии им. А.Н. Баха Федерального исследовательского центра
“Фундаментальные основы биотехнологии” Российской академии наук
Москва, Россия
3 Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
Москва, Россия
* E-mail: khrenova.maria@gmail.com
Поступила в редакцию 10.01.2022
После доработки 15.02.2022
Принята к публикации 20.02.2022
- EDN: CZJHOA
- DOI: 10.31857/S0207401X22060061
Аннотация
Методами молекулярной динамики с потенциалами, построенными по теории квантовой механики/молекулярной механики, сопоставлены динамические свойства фермент-субстратных комплексов в реакции гидролиза гуанозинтрифосфата (GTP) нативным ферментом Ras и его вариантом G12VRas с онкогенной точечной заменой. Молекулярные модели систем, включающих гуанозинтрифосфат-связывающий белок Ras, белок-ускоритель GAP, субстрат GTP и каталитическую молекулу воды, построены на основе атомных координат комплекса из базы данных белковых структур. Анализ полученных распределений вдоль молекулярно-динамических траекторий для расстояний между атомом кислорода каталитической молекулы воды в активном центре комплекса и атомом фосфора γ-фосфатной группы GTP показал, что замена G12V приводит к росту популяций с большими расстояниями между реактантами, снижая таким образом долю реакционных конформаций для реакции гидролиза.
ВВЕДЕНИЕ
Методы квантовой механики/молекулярной механики (КM/MM) активно используются в настоящее время для расчетов поверхностей потенциальной энергии при моделировании ферментативных реакций. Существует потребность в вычислении фрагментов поверхностей энергии Гиббса для анализа химических реакций в белковых макромолекулах, поэтому не следует ограничиваться рассмотрением поверхностей потенциальной энергии, которые могут быть построены в рамках “статического” подхода КM/MM или в рамках моделей молекулярных кластеров. Именно поэтому в последние годы активно развивается метод молекулярной динамики (МД) с потенциалами КM/MM. Как и во всех приложениях теории КM/MM [1], вся молекулярная система, описывающая белковый комплекс, подразделяется на квантовую подсистему, в которой происходят разрывы и образования химических связей, и молекулярно-механическую подсистему, существенно большего размера, окружающую активный центр. При этом для оценок энергий и сил, действующих на ядра атомов в квантовой части, целесообразно использовать хорошо зарекомендовавшие себя подходы квантовой химии. В качестве таковых рационально применять методы теории функционала электронной плотности (ТФП) с гибридными функционалами, в частности использовать функционал PBE0 [2], атомно-центрированные базисные наборы типа 6-31G** и учитывать дисперсионные вклады D3 [3]. Расчеты МД-траекторий с потенциалами КM(ТФП)/MM стали практически возможными для систем с числом атомов до 150 в квантовой части с применением интерфейсов [4] программы молекулярной динамики NAMD [5] и программы квантовой химии TeraChem [6], в которых эффективно используются возможности графических процессоров.
В настоящей работе метод КM(PBE0-D3/6-31G**)/MM(CHARMM) MД применен для сравнения динамических свойств фермент-субстратных (ES) комплексов для нативного варианта гуанозинтрифосфат-связывающего белка Ras и его варианта с точечной заменой аминокислотного остатка глицина на валин в позиции 12 (G12VRas) в комплексе с белком-ускорителем GAP. Ферментативная система Ras–GAP отвечает за гидролиз гуанозинтрифосфата (GTP), результатом которого является перевод активного состояния сигнального белка Ras в неактивное. Моделирование свойств комплекса Ras–GAP представляет важнейшую задачу для биомедицинских исследований, поскольку точечные замены аминокислотных остатков оказывают серьезное влияние на функционирование этого комплекса в организме человека. Замена G12V в белке Ras является одной из наиболее распространенных у онкологических пациентов [7]. Результаты кинетических исследований [8] показывают, что каталитическая константа скорости гидролиза GTP снижается с 18 до 0.01 с–1 при переходе от нативного комплекса Ras–GAP к варианту G12VRas–GAP. Таким образом, молекулярное моделирование должно объяснить причины столь заметного снижения каталитической активности фермента при точечной замене G12V, несмотря на то, что по данным рентгеноструктурного анализа комплекса Ras–GAP [9] аминокислотный остаток в позиции 12 находится несколько в стороне от активного центра.
Предшествующие работы [10, 11] по моделированию методом КM/MM MД свойств фермент-субстратных комплексов в системе Ras(GTP)–GAP с нативным белком Ras и с мутантами G12VRas, G13VRas были выполнены с использованием функционала BLYP и смешанного базиса плоских волн и гауссовых функций для расчетов энергий и сил в квантовой подсистеме в программе CP2K [12]. Поскольку применение простых обобщенно-градиентных функционалов для моделирования ферментативных систем может приводить к значительным ошибкам [13], в настоящей работе анализируются причины замедления каталитической активности комплекса G12VRas-GAP по сравнению с нативным вариантом белка Ras с использованием более надежных методов.
ИНТЕРФЕЙС ПРОГРАММ МОЛЕКУЛЯРНОЙ ДИНАМИКИ И КВАНТОВОЙ ХИМИИ
Использование КM/MM-интерфейса в процедурах NAMD [4] может быть реализовано как с помощью встроенного в программу вызова предопределенных программ квантовой химии (ORCA, MOPAC), так и методом вызова любой отдельной программы, осуществляющей функции интерфейса для любого квантовохимического программного обеспечения (ПО). То есть NAMD напрямую или же через вызов отдельной программы формирует файл задачи для квантового расчета, задавая все необходимые параметры для КM-подсистемы, а также координаты и величины точечных зарядов ММ-подсистемы в нужном формате. По окончании квантовохимического цикла работы ПО считывается полная энергия квантовой системы в поле зарядов, силы в квантовой части и частичные заряды на атомах квантовой подсистемы по результатам выбранной процедуры, например по малликеновскому анализу заселенностей или же методом подгонки под электростатический потенциал КM-подсистемы (RESP). Учет сил, действующих от КM-подсистемы на точечные заряды в ММ-части, обычно проводится в пределах 12–14 Å (так называемого радиуса отсекания). Полученные заряды могут быть использованы и для расчета сил, действующих со стороны атомов квантовой части на ближайшее окружение точечных зарядов (в пределах радиуса отсекания), что является приближением по сравнению со считыванием этих сил из файла выдачи квантовохимического ПО. Такое приближение будет негативно влиять на сохранение полной энергии в рамках расчетов в микроканоническом ансамбле (NVE) за счет наличия нескомпенсированной силы, совершающей работу. В случае изобарно-изотермического ансамбля (NPT) это обстоятельство не является критичным, поскольку влияние параметров термостата и баростата оказывается более существенным, чем описываемые действия с зарядами. Кроме того, при расчетах методом КM/MM MД профилей свободной энергии химических реакций полная энергия системы не используется.
Очевидно, наилучшим вариантом работы КM/MM-интерфейса является считывание сил, действующих со стороны атомов квантовой части на MM-заряды, полученные непосредственно в рамках квантовохимического расчета, как, например, реализовано в оригинальном интерфейсе ORCA–NAMD. По этому же принципу мы модифицировали стандартную программу интерфейса TeraСhem–NAMD. Принципиальной разницей в файлах выдачи квантовохимических программ ORCA и TeraСhem является то, что в файлах выдачи программы ORCA содержатся силы, действующие на каждый точечный заряд со стороны квантовой плотности, которые программа NAMD может считать напрямую, тогда как в случае TeraСhem предусмотрена лишь выдача суммарной силы, действующей на каждый точечный заряд, т.е. суммарного взаимодействия как с электронной плотностью КM-атомов, так и со всеми другими точечными зарядами. Таким образом, чтобы получить значения, пригодные для считывания программой NAMD, необходимо вычесть из полной силы, действующей на каждый точечный заряд, силы, действующие со стороны всех остальных зарядов. Это несложная процедура, которая требует расчета большого количества вкладов типа приведенных ниже для всех координат x, y и z и всех точечных зарядов, используемых в квантовом расчете:
(1)
$\sum\limits_{i,j}^N {{{F}_{{ij}}}\left( x \right)} = \frac{{{\text{P}}{{{\text{C}}}_{i}}{\text{P}}{{{\text{C}}}_{j}}\left( {{{x}_{i}} - {{x}_{j}}} \right)}}{{R_{{ij}}^{3}}},$Поскольку программа интерфейса изначально написана на интерпретируемом языке программирования Python, расчет вкладов вида (1) может занимать около минуты процессорного времени в случае ∼3000 точечных зарядов в расчетах модельных белковых систем, что абсолютно неприемлемо при вычислении длительных КM/MM MД-траекторий. Используя компилятор функций Numba для Python, удается снизить время счета до ~1 с, что делает модифицированный интерфейс пригодным для применения в реальных задачах.
Важным обстоятельством при практическом использовании метода КM/MM MД для оценок профилей свободной энергии ферментативных реакций является скорость расчета энергии и градиентов энергии для квантовой подсистемы в поле зарядов в рамках адекватного приближения, например с гибридными функционалами и двухэкспонентными базисами с учетом поляризационных функций для всех атомов. И хотя на данный момент существует ряд программных пакетов квантовой химии, которые выполняют именно такую задачу, но скорость счета может отличаться в десятки раз в рамках как одного и того же центрального процессора (CPU), так и между CPU и графическим ускорителем (GPU). Именно поэтому был выбран программный пакет TeraСhem [6], который обеспечил наибольшую скорость расчета во всех предварительных тестах квантовохимических программ как на CPU, так и на GPU.
ПОСТРОЕНИЕ МОДЕЛЬНЫХ СИСТЕМ
Для построения полноатомных молекулярных моделей комплексов Ras(GTP)–GAP в качестве источника начальных координат атомов была использована структура PDB 1WQ1 [9] из базы данных белковых структур (PDB), полученная методами рентгеноструктурного анализа. Вместо молекулы гуанозинтрифосфата в структуре PDB 1WQ1 располагается нереакционный аналог субстрата. Атомы водорода были добавлены в систему в соответствии с представлениями о валентности, а также с учетом протонированных состояний ионогенных групп при нейтральных значениях pH в комплексах Ras(GTP)–GAP и G12VRas(GTP)–GAP. Полученные модельные системы были полностью сольватированы молекулами воды.
Предварительная релаксация молекулярных систем с общим числом атомов около 57500 была проведена методом классической молекулярной динамики с силовым полем CHARMM36 [14] и использованием программы NAMD [5]. На рис. 1 показан фрагмент структуры активного центра фермента в реакции гидролиза GTP в активном центре комплекса Ras(GTP)–GAP. Механизм этой реакции активно обсуждался в литературе [15–19]. Согласно результатам недавних работ по моделированию методом КM/MM [18, 19], на первых стадиях реакции происходит нуклеофильная атака молекулой воды γ-фосфатной группы GTP с последующим разрывом химической связи фосфор–кислород PG–O3B, причем активную роль в перераспределении химических связей играет боковая цепь аминокислотного остатка глутамина Gln61. При анализе динамического поведения ES-комплексов для двух исследуемых систем в качестве критериев выбирались следующие межатомные расстояния: PG…OW, O2G…H1w, PG–O3B, O3G…H1E (рис. 1).
Рис. 1.
Фрагмент активного центра в комплексе Ras(GTP)–GAP. Прозрачными лентами показаны белки GAP и Ras, лентами с заливкой – фрагмент P-петли, содержащей остаток Gly12. Стержнями показаны молекула гуанозинтрифосфата GTP, каталитическая молекула воды Wcat и остаток Gln61. Стрелка – направление нуклеофильной атаки атомом кислорода Ow каталитической молекулы воды Wcat атома фосфора γ-фосфатной группы PG. Штриховые линии – водородные связи, способствующие эффективной реакции; штрих-пунктирная линия – водородная связь, образующаяся в нереакционной конформации.
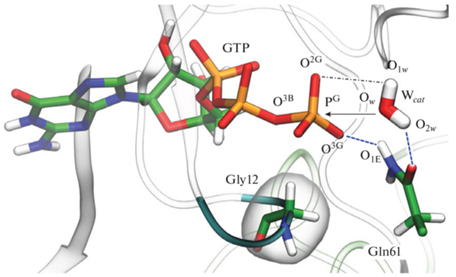
При расчетах методом КM(PBE0-D3/6-31G**)/MM(CHARMM) MД к квантовой подсистеме были отнесены фосфатные группы GTP, аминокислотные остатки Gly/Val12, Gly13, Lys16, Ser17, Thr35, Ile36, Ala59, Gly60, Gln61 и катион магния от Ras, три молекулы воды и боковая цепь Arg789 от белка-ускорителя GAP (всего 112 атомов, не считая атомов водорода, добавляемых на границе квантовой и молекулярно-механической подсистем).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
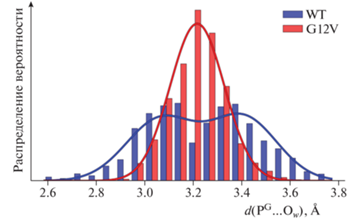
Для определения влияния аминокислотной замены на каталитические свойства мы проанализируем распределения ключевых межатомных расстояний в структуре ES-комплексов Ras(GTP)–GAP в нативной форме или с точечной заменой G12V в белке Ras. На рис. 2 представлены распределения расстояний между атомом кислорода каталитической молекулы воды и атомом фосфора γ-фосфатной группы GTP. Для системы с G12VRas наблюдается нормальное распределение, характеризующееся значениями расстояний нуклеофильной атаки (3.22 ± 0.11) Å. Для нативной формы распределение содержит две равно представленные популяции, характеризующиеся следующими параметрами: (3.07 ± 0.02) Å и (3.41 ± 0.02) Å. Очевидно, чем короче путь нуклеофильной атаки, т.е. чем меньше расстояние между атомом кислорода молекулы воды и атомом фосфора, тем легче проходит реакция. Следовательно, в первой популяции представлены состояния с большей реакционной способностью.
Рис. 2.
Распределения расстояний нуклеофильной атаки в структуре фермент-субстратного комплекса для нативной формы Ras (WT) и варианта G12VRas (G12V).
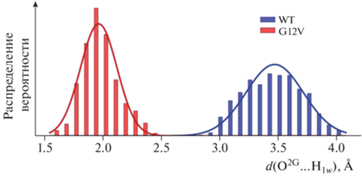
Однако описание систем только лишь на основании величин расстояний нуклеофильной атаки не дает полной картины. Это связано с тем, что для прохождения реакции должна быть правильно ориентирована молекула воды, т.е. неподеленная электронная пара кислорода должна быть ориентирована на электрофильный сайт фосфора [20, 21]. Поэтому мы также проводим анализ других геометрических параметров системы. В частности, при правильной ориентации каталитической молекулы воды атом водорода H1w образует водородную связь с основной цепью карбонильной группы Thr35. Однако возможна и другая ориентация, в которой водородная связь образуется с атомом O2G γ-фосфатной группы субстрата, что блокирует протекание реакции. Таким образом, контролируя длину водородной связи O2G…H1w, можно судить о правильности ориентации молекулы воды (рис. 3). Распределения для нативного фермента и для варианта с заменой G12V заметно различаются; для последнего характерно наличие водородной связи между каталитической молекулой воды и атомом кислорода γ-фосфатной группы, в то время как в нативном ферменте расстояние O2G…H1w превышает 3 Å.
Рис. 3.
Распределения длин водородной связи O2G…H1w для нуклеофильной атаки в структуре фермент-субстратного комплекса для нативной формы Ras (WT) и варианта G12VRas (G12V).
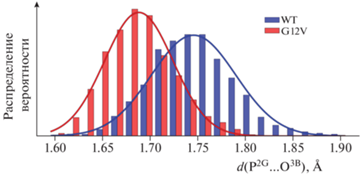
Правильная ориентация молекулы воды и образование предреакционного комплекса сопровождаются удлинением разрываемой связи PG–O3B (рис. 4). Распределение длины связи для нативной формы смещено в сторону больших значений на 0.06 Å: (1.75 ± 0.04) Å для нативной формы и (1.69 ± 0.04) Å для варианта G12VRas.
Рис. 4.
Распределения длин связи PG…O3B нуклеофильной атаки в структуре фермент-субстратного комплекса для нативной формы Ras (WT) и варианта G12VRas (G12V).
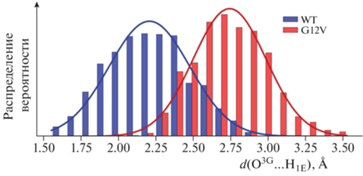
На второй стадии реакции осуществляется концертный перенос протона H2w с молекулы воды на атом кислорода Gln61 и перенос протона H1E на атом кислорода γ-фосфатной группы O3G. Более короткий путь переноса протона вдоль ориентированных цепей водородных связей должен способствовать более эффективному протеканию реакции. Из рис. 5 видно, что для варианта G12VRas лишь небольшая популяция образует водородную связь O3G…H1E, притом что даже эта часть характеризуется слабыми связями, в то время как для нативной формы распределение показывает наличие стабильной водородной связи, характеризующейся длиной (2.21 ± 0.27) Å.
Рис. 5.
Распределения длин связи O3G…H1E нуклеофильной атаки в структуре фермент-субстратного комплекса для нативной формы Ras (WT) и варианта G12VRas (G12V).
В работе [10] проводилось аналогичное моделирование динамического поведения ES-комплексов, однако уровень описания квантовой подсистемы был ниже того, который использован в данной работе. А именно, комбинированный базис гауссовых функций и плоских волн, использованный в работе [10], хуже по уровню точности, чем набор атомно-центрированных функций. Отметим также, что применение гибридных функционалов, как в данной работе, имеет преимущество перед обобщенно-градиентными (GGA) функционалами для моделирования реакций нуклеофильного присоединения [13]. А также важно отметить, что применение менее надежного метода сказывается на качестве описания нуклеофильного и электрофильного сайтов взаимодействующих частиц, что вызвано отсутствием нелокального точного хартри-фоковского обмена. Такие эффекты уже наблюдались в реакциях нуклеофильного присоединения к электрофильному атому углерода [13]. Это приводит к значительному искажению других геометрических параметров, а не только распределения расстояний PG…Ow. Для нативной формы фермента среднее значение длины связи с GGA-функционалом составляет 3.4 Å, что соответствует нереакционной конформации, найденной с использованием гибридного функционала. Для G12VRas использование GGA-функционала приводит к среднему значению этого расстояния, равному около 3.7 Å, что на 0.5 Å превышает значение, полученное в расчетах с гибридным функционалом.
Применение метода молекулярной динамики с потенциалами КM/MM позволяет получить существенную информацию о каталитической активности, проводя анализ только фермент-субстратного комплекса и не прибегая к затратным расчетам всего энергетического профиля реакции. На сегодняшний день уже накоплен обширный расчетный материал, в частности по структурам ES-комплексов, который может быть использован для динамического анализа. Такие структуры хранятся в базе данных интермедиатов ферментативных реакций [22] или в дополнительных материалах к оригинальным работам [23, 24].
ЗАКЛЮЧЕНИЕ
По результатам суперкомпьютерного молекулярного моделирования динамических свойств фермент-субстратных комплексов Ras–GAP и G12VRas-GAP методами КM/MM MД показано, что расположение молекулярных групп в активном центре ферментативной системы существенно меняется при замене аминокислотного остатка глицина в позиции 12 на валин G12V, что приводит к снижению доли реакционных конформаций в реакции гидролиза гуанозинтрифосфата по механизму с участием глутамина. Основным методическим результатом работы является заключение о важности использования атомно-центрированных базисов и гибридных функционалов при расчетах потенциалов в методе КM(ТФП)/MM MД с использованием программ TeraChem–NAMD. Сравнение с результатами предшествующих расчетов более низкого уровня точности по программе CP2K с использованием смешанного базиса плоских волн и гауссовых функций и обобщенно-градиентного функционала BLYP показывает, что применение методики более высокого уровня приводит к заметным изменениям в рассчитанных распределениях ансамблей фермент-субстратных комплексов.
Работа выполнена при финансовой поддержке Российским фондом фундаментальных исследований (проект 19-73-20032) с использованием оборудования Центра коллективного пользования сверхвысокопроизводительными вычислительными ресурсами МГУ им. М.В. Ломоносова и Межведомственного суперкомпьютерного центра РАН.
Список литературы
Senn H., Thiel W. // Angew. Chem. Intern. Ed. 2009. V. 48. P. 1198; https://doi.org/10.1002/anie.200802019
Adamo C., Barone V. // J. Chem. Phys. 1999. V. 110. P. 6158; https://doi.org/10.1063/1.478522
Grimme S., Antony J., Ehrlich S. et al. // J. Chem. Phys. 2010. V. 132. 154104; https://doi.org/10.1063/1.3382344
Melo M.C.R., Bernardi R.C., Rudack T. et al. // Nat. Methods. 2018. V. 15. P. 351; https://doi.org/10.1038/nmeth.4638
Phillips J.C., Braun R., Wang W. et al. // J. Comput. Chem. 2005. V. 26. P. 1781; https://doi.org/10.1002/jcc.20289
Seritan S., Bannwarth C., Fales B.S. et al. // WIREs Comput. Mol. Sci. 2021. V. 11. e1494; https://doi.org/10.1002/wcms.1494
Cox A.D., Der C.J. // Small GTPases. 2010. V. 1. P. 2; https://doi.org/10.4161/sgtp.1.1.12178
Wey M., Lee J., Jeong S.S. et al. // Biochemistry. 2013. V. 52. P. 8465; https://doi.org/10.1021/bi400679q
Scheffzek K., Ahmadian M.R., Kabsch W. et al. // Science. 1997. V. 277. P. 333; doi: 1997https://doi.org/10.1126/science.277.5324.33
Khrenova M.G., Mironov M.V., Grigorenko B.L. et al. // Biochemistry. 2014. V. 53. P. 7093; https://doi.org/10.1021/bi5011333
Mironov M.V., Khrenova M.G., Lychko L.A. et al. // Proteins. 2015. V. 83. P.1046; https://doi.org/10.1002/prot.24802
VandeVondele J., Krack M., Mohamed F. et al. // Comput. Phys. Commun. 2005. V. 167. P. 103; https://doi.org/10.1016/j.cpc.2004.12.014
Khrenova M.G., Tsirelson V.G., Nemukhin A.V. // Phys. Chem. Chem. Phys. 2020. V. 22. P. 19069; https://doi.org/10.1039/d0cp03560b
Best R.B., Zhu X., Shim J. et al. // J. Chem. Theory Comput. 2012. V. 8. P. 3257; https://doi.org/10.1021/ct300400x
Glennon T.M., Villà J., Warshel A. // Biochemistry. 2000. V. 39. P. 9641; https://doi.org/10.1021/bi000640e
Grigorenko B.L., Nemukhin A.V., Topol I.A. et al. // Proteins. 2005. V. 60. P. 495; https://doi.org/10.1002/prot.20472
Mishra A.K., Lambright D.G. // Biopolymers. 2016. V. 105. P. 431; https://doi.org/10.1002/bip.22833
Khrenova M.G., Grigorenko B.L., Kolomeisky A.B. et al. // J. Phys. Chem. B. 2015. V. 119. P. 12838; https://doi.org/10.1021/acs.jpcb.5b07238
Grigorenko B.L., Kots E.D., Nemukhin A.V. // Org. Biomol. Chem. 2019. V. 17. P. 4879; https://doi.org/10.1039/c9ob00463g
Khrenova M.G., Kulakova A.M., Nemukhin A.V. // J. Chem. Inf. Model. 2021. V. 61. P. 1215; https://doi.org/10.1021/acs.jcim.0c01308
Khrenova M.G., Grigorenko B.L., Nemukhin A.V. // ACS Catal. 2021. V. 11. P. 8985; https://doi.org/10.1021/acscatal.1c00582
Григоренко Б.Л., Хренова М.Г., Кулакова А.М., Немухин А.В. // Хим. физика. 2020. V. 39. P. 13; https://doi.org/10.31857/s0207401x20060023
Лущекина С.В., Григоренко Б.Л. Поляков И.В. и др. // Хим. физика. 2022. Т. 41. № 2. С. 34.
Поляков И.В., Григоренко Б.Л., Немухин А.В. // Хим. физика. 2021. Т. 40. С. 44; https://doi.org/10.31857/s0207401x21020138
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика