

Химическая физика, 2023, T. 42, № 7, стр. 59-69
Механизм однореакторной стереоселективной сборки спирокеталевых производных из циклогексанона и фенилацетилена в среде KOH/ДМСО: квантовохимическое исследование
В. Б. Орел 1, *, А. А. Манжуева 1
1 Иркутский государственный университет
Иркутск, Россия
* E-mail: orelv@isu.ru
Поступила в редакцию 16.01.2023
После доработки 16.02.2023
Принята к публикации 20.02.2023
- EDN: YFNJBE
- DOI: 10.31857/S0207401X23070154
Аннотация
Механизмы реакции образования 15-[(Z)-фенилметилиден]-7,14-диоксадиспиро[5.1.5.2]пентадекана и конкурирующей с ней реакции образования ненасыщенных кетонов из циклогексанона и фенилацетилена исследованы с использованием квантовохимического подхода B2PLYP-D2/6-311+G**//B3LYP/6-31+G* с учетом сольватационных эффектов в рамках модели IEFPCM. В рамках анионной модели (ANIONGAS) рассмотрены все стадии сборки диспирокеталя и устойчивость различных конформеров и изомеров интермедиатов и продукта. В рамках более детальной моносольватной модели (MONOPCM) оценены активационные барьеры сборки диспирокеталя и конкурирующей реакции С-винилирования. Полученные результаты квантовохимического расчета хорошо согласуются с данными эксперимента.
ВВЕДЕНИЕ
Большинство встречающихся в природе спирокеталей представляют собой биологически активные соединения [1, 2]. Например, реверомицины [3, 4], которые содержат спирокетальные скелеты, являются ингибиторами митогенной активности эпидермального фактора роста; рутиенноцин представляет собой ионофорный антибиотик [5, 6]; окадаиковая кислота и таутомицин – ингибиторы протеинфосфатазы [7]; интеграмицин представляет собой ингибитор интегразы ВИЧ-1 [8]. Мильбемицины и авермектины являются глистогонными и инсектицидными агентами [2], а спонгистатины представляют собой морские антимикотические макролиды [9]. Более того, ингибирующая теломеразу активность гризеородина и рубромицина также объясняется наличием спирокетального фрагмента в этих природных соединениях [10, 11]. Многие простые спирокетали насекомых являются неустойчивыми и действуют как феромоны [2, 12]. Многогранность биологически активных свойств, проявляемых спирокеталями, связывают с их структурной гибкостью, которая открывает доступ к различным конформациям спирокетального фрагмента [13]. Это было доказано на примере девяти разных авермектинов [14], которые похожи структурно, но имеют очень разное биологическое действие.
Высокая частота, с которой спирокеталевые фрагменты встречаются среди природных продуктов, обладающих биологической активностью, делает синтез содержащих такие фрагменты соединений актуальной задачей при разработке новых, перспективных лекарственных препаратов. Действительно, много усилий было приложено к синтезу соединений, содержащих спирокетали. Некоторые наиболее универсальные стратегии синтеза рассмотрены в работах [15–17]. Большинство из этих подходов включают использование жестких кислотных условий, в которых осуществляется конденсация дигидроксикетонов или их синтетических аналогов [18]. Это связано с тем, что осуществление синтеза в данных условиях приводит непосредственно к наиболее термодинамически стабильной конфигурации, присутствующей в природе и, следовательно, обладающей биологической активностью. Так, в случае с объемными заместителями преобладающей конфигурацией обычно является конфигурация с экваториальным расположением заместителей в циклах (Схема 1 ).
Схема 1 . Конденсация дигидроксикетонов в присутствии кислоты
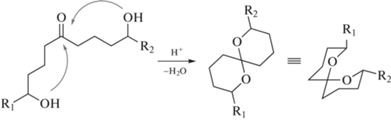
Но присутствие кислотолабильных функциональных групп в молекуле существенно ограничивает применение этого метода. Чтобы преодолеть проблему жестких кислотных условий и сохранить стереоселективность реакции, применяют методики синтеза, основанные на катализе переходными металлами [19]. Однако в этом случае часто нарушается региоселективность реакции и образуется смесь спирокеталей (Схема 2 ).
Схема 2 . Циклизация нонин-4-диола-1,9 под действием палладиевого катализатора PdCl2(PdCN)2
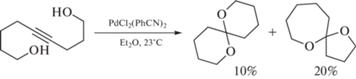
Отметим также, что синтез спирокеталей, в особенности биологически активных и родственных природным аналогам, часто оказывается многостадийным процессом [20, 21]. Жесткие кислотные условия и/или использование катализаторов на основе дорогих переходных металлов (Pd, Pt, Ir, Rh), а также многостадийность делают актуальной задачей поиск новых подходов к синтезу спирокеталей. В этой связи могут быть интересны осуществляемые в суперосновных средах взаимопревращения кетонов с ацетиленами, которые послужили трамплином для запуска свободных от переходных металлов однореакторных сборок целого ряда особо значимых в синтетическом отношении карбо- и гетероциклов [22]. Одна из “ветвей” этих реакций относится к взаимопревращениям ацетиленов с циклоалифатическими кетонами, приводящим к образованию тетрациклических производных фронталина [23], диспирокеталей [24] и гексагидроазуленонов [25]. Так, взаимодействие циклоалифатических кетонов с фенилацетиленом при 80 °С в течение 1 ч приводит, помимо ожидаемых по реакции С-винилирования β,γ- и α,β-ненасыщенных кетонов, (с выходами 26–57%) к образованию диспирокеталей с выходами до 22% [24]. Несмотря на их скромный выход эта реакция может быть использована в качестве кратчайшего пути к спирокеталевым структурам.
Помимо самостоятельного интереса к диспирокеталевым структурам важно изучение механизма их сборки. Это представляет практический интерес для улучшения их выхода, а кроме того, вносит вклад в развитие фундаментальных представлений о химии ацетиленов и кетонов в суперосновных средах. По причине многостадийности сборки диспирокеталей и высоких скоростей отдельных стадий исследование механизма этой реакции экспериментальными методами затруднено. Полезным и оправданным является привлечение для этих целей методов квантовой химии, которые сегодня используются в качестве одного из базовых инструментов исследования не только различных электронных и структурных свойств сложных молекулярных систем [26–29], но и механизмов реакций [30, 31].
Цель данной работы – сравнительное квантовохимическое исследование механизма сборки 15-[(Z)-фенилметилиден]-7,14-диоксадиспиро[5.1.5.2] пентадекана из циклогексанона и фенилацетилена и конкурирующей реакции С-винилирования циклогексанона (Схема 3 ).
Схема 3 . Реакции образования 15-[(Z)-фенилметилиден]-7,14-диоксадиспиро[5.1.5.2]пентадекана (I) и ненасыщенных кетонов (II)
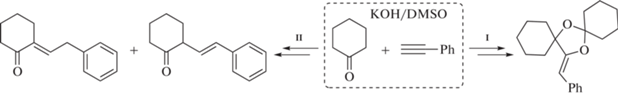
МЕТОДЫ И ПОДХОДЫ
Для проведения расчетов использовался пакет квантовохимических программ GAUSSIAN-16 [32]. Оптимизация структурных параметров молекулярных частиц, участвующих в реакции, выполнялась в рамках метода функционала плотности B3LYP [33, 34] с применением базисного набора 6-31+G*. В том же приближении, B3LYP/6-31+G*, при стандартной температуре 298.15 K рассчитывали колебательные поправки и термодинамические функции. Для всех стационарных точек исследовали число отрицательных собственных значений матрицы Гессе. Связь полученных переходных состояний с соответствующими минимумами на поверхности потенциальной энергии доказывалась спуском по координате реакции (IRC) с использованием алгоритма квадратичной аппроксимации (LQA) [35].
Энергии стационарных форм уточнялись в рамках дважды гибридного функционала B2PLYP [36] с расширенным базисом 6-311+G** и с применением дисперсионной поправки Гримме D2 [37]. Значение энергии сольватации в диметилсульфоксиде (ДМСО) рассчитывались в рамках модели поляризуемого диэлектрического континуума в интегральной формулировке (IEFPCM) [38]. Моделирование полости проводилось в рамках подхода GEPOL [39] с радиусами универсального силового поля (Universal Force Field (UFF)) и универсальным масштабирующим множителем растворителя α = 1.1 [40]. При расчете энергии сольватации использовалось следующее значение диэлектрической постоянной ДМСО: ε = 46.8.
Для оценки свободной энергии активации в растворе мы использовали подход, основанный на результатах работы [41]. Применительно к растворам ДМСО [42] в этот подходе предполагается, что энтропия в растворе диметилсульфоксида, Ssol, может быть получена из значения энтропии Sharm, найденного в гармоническом приближении для идеального газа:
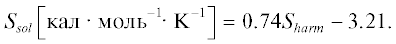
Все стадии сборки 15-[(Z)-фенилметилиден]-7,14-диоксадиспиро[5.1.5.2]пентадекана, а также термодинамическая устойчивость различных конформеров и изомеров интермедиатов и продуктов этой сборки предварительно исследовались в рамках анионной модели (ANIONGAS). В этой модели пренебрегается присутствием катиона, а растворитель учитывается только в виде поправки на энергию сольватации в рамках IEFPCM. Отметим, что на реакционном профиле рассматривались структуры только с наиболее устойчивыми конформациями типа “кресло” циклогексановых фрагментов [43, 44]. Далее, для изучения механизма сборки диспирокеталя с учетом влияния конкурирующей реакции С-винилирования использовалась более детальная моносольватная модель (MONOPCM). В этой модели в явном виде учитываются недиссоциированная молекула щелочи KOH и одна молекула ДМСО, обеспечивающие описание специфической сольватации, а оптимизация геометрических параметров происходит уже с учетом влияния растворителя в рамках модели поляризуемого континуума и описывает неспецифические взаимодействия. В нашей недавней работе [45] приведено более подробное описание обеих моделей, ANIONGAS и MONOPCM, а также продемонстрированы их согласие с наиболее полной моделью суперосновного комплекса (модель PENTAGAS) и применимость для исследования реакций ацетиленов в суперосновных средах.
Все кинетические кривые моделировались при T = 353 K уравнением Аррениуса k(T) = = (kBT/h)exp(–ΔG‡/RT) с использованием программы KINET [46].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Образование диспирокеталя из циклогексанона (1) и фенилацетилена (2) в среде KOH/ДМСО рассмотрено согласно схеме, которую предложили авторы статьи [24] (Схема 4 ).
Схема 4 . Механизм образования диспирокеталя из циклогексанона и фенилацетилена

Сборка диспирокеталя начинается с нуклеофильного присоединения фенилэтинид-иона (3) по карбонильной группе соединения 1 с образованием 1-(2-фенилэтинил)циклогексан-1-олята (5) – реакция этинилирования Фаворского. Присоединение фенилэтинид-иона может осуществляться в аксиальное (a) и в экваториальное (e) положения циклогексанона (рис. 1).
Рис. 1.
Строение переходных состояний реакции этинилирования циклогексанона. На рисунке в Å и см–1 указаны длины связей и мнимые частоты соответственно.
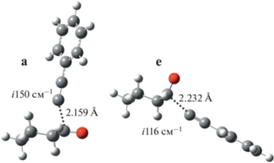
В рамках анионной модели алкоголят 5a образуется с понижением энергии ΔG = –3.1 ккал/моль относительно суммы энергий ΔG изолированных соединений 3 и 1, тогда как образование 5e связано с меньшим понижением свободной энергии: ΔG = –1.7 ккал/моль. Кроме того, аксиальная атака 1 соединением 3 осуществляется с энергией активации Δ$G_{{{\mathbf{1}}{\text{ + }}{\mathbf{3}} \to {\mathbf{5a}}}}^{\ddag }$ = 12.7 ккал/моль, что на ΔΔG‡ = 1.3 ккал/моль меньше энергии активации необходимой для экваториальной атаки (рис. 2).
Рис. 2.
Схема реакционного профиля образования диспирокеталя из двух молекул циклогексанона и одной молекулы фенилацетилена в рамках модели ANIONGAS; ΔG – свободная энергия Гиббса (в ккал/моль).
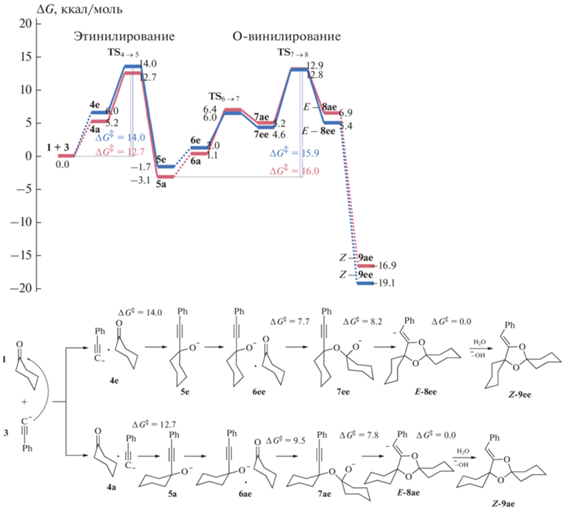
Необходимо отметить, что 5e может изомеризоваться в термодинамически более устойчивый алкоголят-ион 5a с небольшими энергиями активации, Δ$G_{{{\mathbf{5e}} \to {\mathbf{5a}}}}^{\ddag }$ = 11.2 ккал/моль, через алкоголят с твист-конформацией циклогексильного заместителя.
Алкоголят-ионы 5a и 5е способны присоединяться по двойной связи C=O к следующей молекуле кетона. Согласно результатам расчета вначале происходит образование комплексов 6a и 6e (ΔG = 2.0 и 1.1 ккал/моль соответственно). Далее комплексы 6 могут преобразоваться в анионы полукеталя (7) через соответствующие переходные состояния TS6→7. В зависимости от стороны атаки молекулы циклогексанона могут образоваться четыре конформера аниона полукеталя – 7аа, 7ае, 7еа и 7ее (Схема 5 ).
Схема 5 . Нуклеофильное присоединение алкоголят-иона 5 по карбонильной связи циклогексанона; ΔG‡ – энергия активации Гиббса (в ккал/моль)
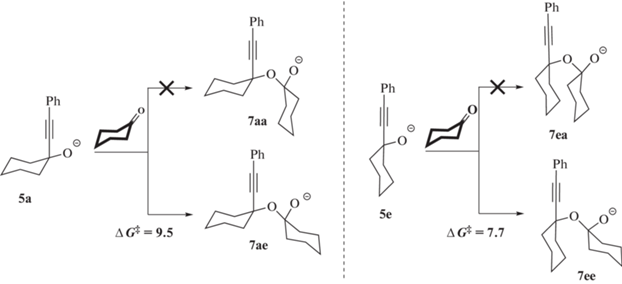
Однако удается локализовать только два переходных состояния TS6→7, связанные с присоединением алкоголят-ионов 5a и 5e только в экваториальное положение соединения 1. В результате образуются два экваториальных конформера анионов полукеталя: 7ае и 7ее, с повышением энергии на ΔG = 4.6 и 5.2 ккал/моль относительно 5 и 1 соответственно. Несмотря на термодинамическую невыгодность их образования, анионы 7 являются промежуточными на пути циклизации в диспирокеталь (рис. 2). Для образования анионов полукеталя 7 необходимо преодолеть активационный барьер, не превышающий Δ$G_{{{\mathbf{5a}}{\text{ + }}1 \to {\mathbf{7ae}}}}^{\ddag }$ = = 9.5 ккал/моль. Конформеры 7, которые образуются в результате присоединения соединения 5 в аксиальное положение кетона, неустойчивы и при оптимизации геометрических параметров распадаются на исходные алкоголят-ионы и циклогексанон.
Заключительная стадия внутримолекулярного присоединения O-аниона по тройной связи С≡С в анионе 7 приводит к образованию аниона диспирокеталя (8), который далее протонируется молекулой воды с образованием конечного продукта 9 – диспирокеталя (реакция O-винилирования, рис. 2). Как было продемонстрировано нами ранее [47, 48], в переходном состоянии нуклеофильного присоединения по тройной связи фенилацетиленовый фрагмент может иметь только транс-искаженное строение, что наблюдается и в TS7→8 (рис. 3). Такое переходное состояние приводит к E-изомерам 8, при протонировании которых образуются Z-изомеры 9, что объясняет наблюдаемую Z-стереоселективность сборки диспирокеталей.
Энергии активации внутримолекулярного O-винилирования с образованием диспирокеталей E-8ее и E-8aе составляют Δ$G_{{{\mathbf{7ee}} \to E{\mathbf{ - 8ee}}}}^{\ddag }$ = 8.2 ккал/моль и Δ$G_{{{\mathbf{7ae}} \to E{\mathbf{ - 8ae}}}}^{\ddag }$ = 7.8 ккал/моль, соответственно. При этом суммарная энергия активации на пути к E-8ее из наиболее устойчивого конформера 5а (напомним, что 5e легко может переходить в 5a) и соединения 1 составляет Δ$G_{{{\mathbf{5a}} \to E{\mathbf{ - 8ee}}}}^{\ddag }$ = 15.9 ккал/моль. Энергия активации, связанная с превращением 5а → E-8ае, оказывается незначительно выше (ΔΔG‡ = 0.1 ккал/моль). Таким образом, на реакционном профиле лимитирующий процесс сборки диспирокеталя включает две последовательные стадии: 5 → 7 → E-8 (рис. 2). Близость энергий активации превращения 5 → E-8 предсказывает образование двух конформеров аниона диспирокеталя – E-8ае и E-8eе. Протонирование анионов 8 происходит без активационного барьера и приводит к образованию конечных диспирокеталей Z-9ае и Z-9ее с понижением свободной энергии относительно исходных соединений 1 и 3 на ΔG = –16.9 и –19.1 ккал/моль соответственно (рис. 2).
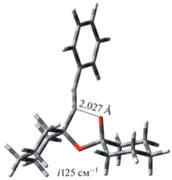
По реакционному профилю (рис. 2) получены диспирокетали Z-9ee и Z-9ае, которые могут изомеризоваться друг в друга и еще в две другие конформации – Z-9ea и Z-9aa. Конформер Z-9ea оказывается термодинамически наиболее устойчивым из всех четырех и существует в равновесной смеси с конформером Z-9ее; количественное соотношение этих конформеров составляет ~1 : 1 (рис. 4).
Рис. 4.
Конформации Z-изомера диспирокеталя 9 и их относительная термодинамическая устойчивость (ΔG, ккал/моль).
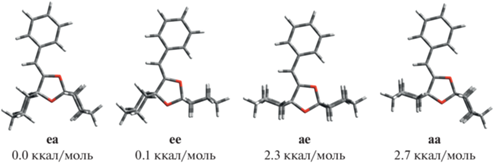
В случае перехода Z-9ае → Z-9ее меняется конформация циклогексильного заместителя бывшего алкоголят-иона, изомеризация которого уже была рассмотрена выше и происходит относительно легко (Δ$G_{{{\mathbf{5e}} \to {\mathbf{5a}}}}^{\ddag }$ = 11.2 ккал/моль). Далее при изомеризации Z-9ee в Z-9ea осуществляется переход второго циклогексильного заместителя также через твист-конформацию с суммарной энергией активации Δ$G_{{Z{\mathbf{ - 9ee}} \to Z{\mathbf{ - 9ea}}}}^{\ddag }$ = 11.8 ккал/моль. Механизм изомеризации Z-9aa мы не рассматривали, так как этот изомер самый неустойчивый.
Конформационные переходы между анионами полукеталей 7 и анионами диспирокеталей 8 не рассматривались, так как 7 легко распадаются обратно на 1 и 5, а 8 легко протонируется с образованием 9 (рис. 2). Отметим, что наиболее выгодная конформация диспирокеталя Z-9ea оказывается на ΔΔG = 5.4 ккал/моль термодинамически более устойчивой, чем соответствующая наиболее выгодная конформация E-изомера диспирокеталя. Таким образом, экспериментально наблюдаемая Z-стереоселективность реакции объясняется кинетической и термодинамической предпочтительностью Z-изомера.
Наряду с реакцией сборки диспирокеталей в суперосновной среде осуществляется нуклеофильное присоединение аниона циклогексанона к фенилацетилену (реакция С-винилирования). Эта реакция приводит к образованию смеси изомеров β,γ- и α,β-ненасыщенных кетонов в соотношении 9 : 1 с суммарным выходом в 57% [24].
Модель ANIONGAS адекватно передает качественные различия в барьерах реакций этинилирования и винилирования [45], между которыми возникает конкуренция. Однако влияние катионного центра и его ближайшего окружения может быть существенным, а кроме того, в различной степени выраженным для этих реакций. При сравнительном исследовании механизмов этих двух направлений принципиально важно, какой состав исходных реагентов предпочтительнее образуется под действием супероснования: анион циклогексанона/фенилацетилен или циклогексанон/фенилэтинид-ион. Поэтому дальнейшее исследование было выполнено с включением в расчет комплекса KOH·ДМСО (модель MONOPCM) [45]; при этом на пути сборки диспирокеталей рассматривались только наиболее устойчивые конформации интермедиатов и продуктов.
Образование диспирокеталей, как подробно было показано выше в рамках анионной модели, запускается реакцией этинилирования циклогексанона фенилэтинид-ионом. Формирование исходного комплекса фенилэтинида калия (11) происходит в результате депротонирования фенилацетилена в предреакционном комплексе KOH·ДМСО·PhCCH (10). Это превращение осуществляется с незначительным понижением энергии ΔG = –0.2 ккал/моль и связано с небольшой энергией активации Δ$G_{{{\mathbf{10}} \to {\mathbf{11}}}}^{\ddag }$ = 2.4 ккал/моль (рис. 5).
Рис. 5.
Схема реакционного профиля конкурирующих направлений: реакции образования диспирокеталя 9 и реакции C-винилирования в рамках модели MONOPCM. Теоретические кинетические кривые накопления продуктов реакции взаимодействия циклогексанона с фенилацетиленом в присутствии KOH/ДМСО при различных температурах.
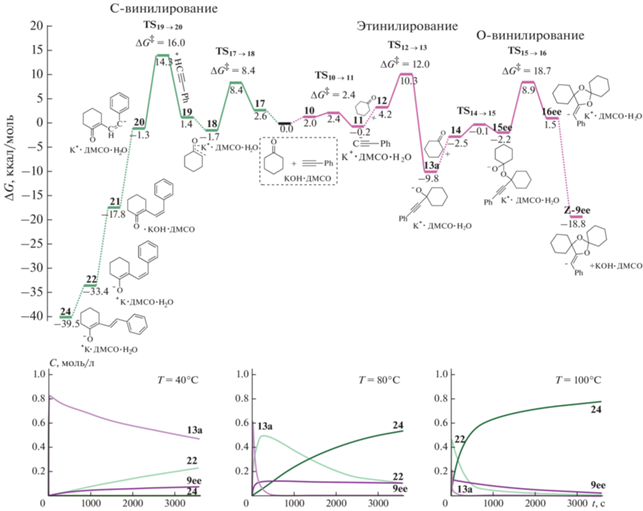
Координация циклогексанона к комплексу 11 приводит к образованию предреакционного комплекса 12. Нуклеофильная атака фенилэтинид-ионом карбонильной группы молекулы циклогексанона в комплексе 12 осуществляется далее с энергией активации Δ$G_{{{\mathbf{12}} \to {\mathbf{13a}}}}^{\ddag }$ = 12.0 ккал/моль и приводит к комплексу алкоголята 13а. Достигаемое в результате образования комплекса 13а понижение энергии системы составляет ΔG = –9.8 ккал/моль относительно исходных молекул 1 и 2 (рис. 5). Близкое значение энергии активации реакции этинилирования циклогексанона (ΔG‡ = 12.9 ккал/моль) предсказывалось в рамках анионной модели, при этом образование алкоголят-иона 5а сопровождалось понижением энергии лишь на 2.9 ккал/моль. Бóльшая устойчивость комплекса 13а в модели MONOPCM объясняется его стабилизацией катионом калия.
Завершают сборку диспирокеталя стадии присоединения еще одной молекулы циклогексанона в предреакционном комплексе 14 с образованием комплекса полукеталя 15ee (Δ$G_{{{\mathbf{13a}} \to {\mathbf{15ee}}}}^{\ddag }$ = = 9.7 ккал/моль, ΔG = 7.6 ккал/моль) и последующее внутримолекулярное O-винилирование, приводящее к карбаниону диспирокеталя 16ee (Δ$G_{{{\mathbf{15ee}} \to {\mathbf{16ee}}}}^{\ddag }$ = 11.1 ккал/моль, ΔG = 3.7 ккал/моль). В моносольватной модели MONOPCM энергия активации этого превращения составляет Δ$G_{{{\mathbf{13a}} \to {\mathbf{16ee}}}}^{\ddag }$ = = 18.7 ккал/моль, что несколько больше, чем в анионной модели, и также является лимитирующим. Винильный карбанион в комплексе 16ее протонируется молекулой воды без активационного барьера с образованием молекулы Z-9ее и регенерацией комплекса основания KOH·ДМСО. При этом общее понижение энергии при образовании Z-9ее относительно исходных соединений 1 и 2 составляет ΔG = –18.8 ккал/моль (рис. 5). Схожее понижение энергии относительно соединений 1 и 3 предсказывала анионная модель (ΔG = –19.1 ккал/моль).
В присутствии супероснования KOH/ДМСО может также осуществляться депротонирование молекулы циклогексанона с образованием карбаниона циклогексанона. Так, взаимодействие молекулы 1 с KOH·ДМСО с повышением энергии ΔG = 2.6 ккал/моль приводит к комплексу 17. В комплексе 17 через переходное состояние TS17→18 с активационным барьером ΔG‡ = 8.4 ккал/моль осуществляется перенос протона α-СH2-группы молекулы циклогексанона к гидроксид-иону с образованием комплекса енолята калия (18). Депротонирование циклогексанона кинетически менее выгодно на ΔΔG‡ = 6.0 ккал/моль, чем депротонирование молекулы фенилацетилена. С другой стороны, образование комплекса 18 термодинамически более выгодно на ΔΔG = 1.5 ккал/моль (рис. 5). Относительная термодинамическая выгодность комплекса енолята хорошо согласуется с большей кислотностью циклогексанона (pKa = = 26.4 [49]) по сравнению с фенилацетиленом (pKa = 28.8 [50]).
Присоединение комплекса 18 по тройной связи молекулы 2 приводит к неустойчивому предреакционному комплексу 19 (ΔG = 1.4 ккал/моль). В комплексе 19 осуществляется атака енолят-ионом тройной ацетиленовой связи с образованием комплекса E-аниона β,γ-ненасыщенного кетона 20 (ΔG = 0.4 ккал/моль). Это превращение связано с энергией активации Δ$G_{{{\mathbf{19}} \to {\mathbf{20}}}}^{\ddag }$ = 16.0 ккал/моль, что на ΔΔG‡ = 4.0 ккал/моль больше энергии активации конкурирующей реакции этинилирования циклогексанона комплексом 11. Карбанионный центр в комплексе 20 без активационного барьера протонируется присутствующей молекулой воды с образованием комплекса 21 – Z-изомера β,γ-ненасыщенного кетона. Образование комплекса 21 приводит к понижению энергии системы на ΔG = = 16.1 ккал/моль (относительно 2 и 18). Далее в комплексе 21 относительно легко (Δ$G_{{{\mathbf{21}} \to {\mathbf{22}}}}^{\ddag }$ = = 1.1 ккал/моль) происходит перенос протона CH-группы α-положения ненасыщенного кетона на гидроксильную группу основания KOH с образованием комплекса 22 – 1Z,3Z-диенолята калия (рис. 6). В результате такого превращения свободная энергия системы понижается еще на 15.6 ккал/моль.
Рис. 6.
Схема реакционного профиля образования комплекса 22 и его Z-E-изомеризации в комплекс 24 в рамках модели MONOPCM.
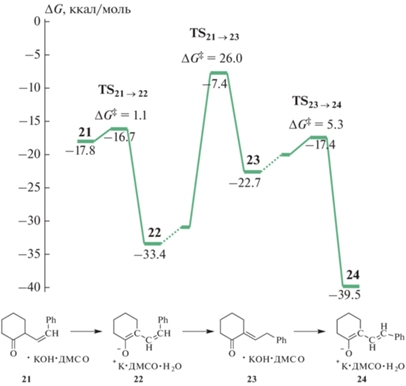
Отметим, что ненасыщенные кетоны выделяются при обработке реакционной смеси водой [24]. Протонирование Z,Z-диенолята будет приводить к Z-изомеру β,γ-ненасыщенного кетона, а не к наблюдаемому Е-изомеру. Предшественником Е-изомера β,γ-ненасыщенного кетона является Z,E-диенолят. Кроме того, Z,Е-конфигурация диенолята имеет более эффективное сопряжение, а в Z,Z-диеноляте стерическое отталкивание атомов водорода дестабилизирует этот изомер [51].
Z-E-Изомеризация может происходить через стадию конформационного SP–AC-вращения вокруг связи Cβ–Cγ в промежуточном α,β-ненасыщенном кетоне. Образование α,β-ненасыщенного кетона в составе комплекса 23 связано с высокой энергией активации Δ$G_{{{\mathbf{22}} \to {\mathbf{23}}}}^{\ddag }$ = 26.0 ккал/моль и повышением энергии системы на 10.7 ккал/моль относительно 22. По спуску из переходного состояния TS22→23 образуется SP-конформер, однако оптимизация этой стационарной точки приводит в необходимую AC-конформацию α,β-ненасыщенного кетона в комплексе 23. Завершает Z-E-изомеризацию стадия отрыва протона γ-СH2-группы α,β-ненасыщенного кетона в комплексе 23. Это превращение связано с энергией активации Δ$G_{{{\mathbf{23}} \to {\mathbf{24}}}}^{\ddag }$ = 5.3 ккал/моль и приводит к образованию комплекса Z,Е-диенолята (24), на 6.1 ккал/моль более устойчивому, чем комплекс 22 (рис. 6).
Таким образом, в присутствии супероснования взаимодействие комплекса 18 с фенилацетиленом приводит к образованию комплекса 24. При дальнейшей обработке комплекса 24 водой будет протонироваться как γ-, так и α-положение с образованием α,β-ненасыщенного кетона и E-изомера β,γ-ненасыщенного кетона соответственно. Оценки относительной термодинамической устойчивости этих двух форм ненасыщенных кетонов с использованием параметров континуальной модели IEFPCM для воды (α = 1.1 и ε = 78.4) показывают, что термодинамически наиболее выгодной на 1.0 ккал/моль является β,γ-форма. Это хорошо согласуется с экспериментально наблюдаемым соотношением β,γ- к α,β-ненасыщенному кетону, составляющим 9 : 1 [24].
Термодинамические оценки предсказывают, что образование комплекса 24 (ΔG = –39.5 ккал/моль) является более предпочтительным, чем сборка диспирокеталя Z-9ee (ΔG = –18.8 ккал/моль). С другой стороны, наименьшим активационным барьером Δ$G_{{{\mathbf{12}} \to {\mathbf{13a}}}}^{\ddag }$= 12.0 ккал/моль характеризуется образование комплекса 13a. Именно его дальнейшие превращения либо со следующей молекулой циклогексанона в сторону диспирокеталя (Δ$G_{{{\mathbf{13a + 1}} \to {\mathbf{9ee}}}}^{\ddag }$ = 18.7 ккал/моль), либо распад на анион циклогексанона и фенилацетилен (Δ$G_{{{\mathbf{13a}} \to {\mathbf{18 + 2}}}}^{\ddag }$ = 20.1 ккал/моль) с последующим С-винилированием (Δ$G_{{{\mathbf{18 + 2}} \to {\mathbf{20}}}}^{\ddag }$ = 16.0 ккал/моль) определяют конечный состав продуктов (рис. 5).
При этом энергии активации описанных стадий преодолимы в условиях эксперимента (80 °С и 1 ч). Смоделированные кинетические кривые (рис. 5) действительно показывают, что изначально образуется алкоголят-ион 13а, который расходуется на образование диспирокеталя Z-9ее и продуктов C-винилирования 22 и 24. При более высоких температурах (100 °С) равновесие полностью смещается в сторону образования продуктов С-винилирования 24, а при низких (40 °С) температурах преобладают кинетически наиболее выгодные алкоголят-ионы 13а (рис. 5).
ЗАКЛЮЧЕНИЕ
В рамках метода B2PLYP-D2/6-311+G**// B3LYP/6-31+G* построен реакционный профиль сборки 15-[(Z)-фенилметилиден]-7,14-диоксадиспиро[5.1.5.2]пентадекана из циклогексанона и фенилацетилена и для каждого интермедиата определены кинетически и термодинамически наиболее выгодные конформации расположения заместителей в циклогексановом фрагменте.
Показано, что сборку диспирокеталя запускает стадия этинилирования циклогексанона фенилацетиленом с образованием алкоголята. Тогда как скорость-определяющим является завершающий сборку двухстадийный процесс: 1) образование O-аниона полукеталя в результате взаимодействия алкоголята с циклогексаноном и 2) внутримолекулярное O-винилирование в полукетале.
Конкурирующая реакция С-винилирования с образованием ненасыщенных кетонов является термодинамически более предпочтительной, чем реакция этинилирования с образованием алкоголятов и последующее их превращение в диспирокетали. Однако С-винилирование кинетически менее выгодно, чем образование алкоголята. Это определяет ненасыщенные кетоны как термодинамические продукты при более высоких температурах (от 100 °С), а ацетиленовые спирты – как кинетические продукты при более низких температурах (до 40 °С). При этом наибольший выход диспирокеталей наблюдается лишь в узком интервале температур (60–80 °С) из-за близости энергий активации превращения алкоголята в диспирокеталь (Δ$G_{{{\mathbf{13a}} \to {\mathbf{9ee}}}}^{\ddag }$ = 18.7 ккал/моль) и его распада на анион циклогексанона и фенилацетилен (Δ$G_{{{\mathbf{13a}} \to {\mathbf{18 + 2}}}}^{\ddag }$ = 20.1 ккал/моль) с последующим С-винилированием (Δ$G_{{{\mathbf{18 + 2}} \to {\mathbf{20}}}}^{\ddag }$ = 16.0 ккал/моль). Эти выводы подтверждены видом кинетических кривых, смоделированных с учетом всех рассматриваемых стадий на пути образования ненасыщенных кетонов и диспирокеталей.
Авторы выражают благодарность д.х.н., проф. Н.М. Витковской за полезные обсуждения и рекомендации.
Работа выполнена в рамках госзадания Министерства образования и науки РФ (тема FZZE-2020-0025).
Список литературы
Brimble M., Furkert D. // Curr. Org. Chem. 2003. V. 7. № 14. P. 1461; https://doi.org/10.2174/1385272033486404
Perron F., Albizati K.F. // Chem. Rev. 1989. V. 89. № 7. P. 1617; https://doi.org/10.1021/cr00097a015
Koshino H., Takahashi H., Osada H. et al. // J. Antibiot. (Tokyo). 1992. V. 45. № 9. P. 1420; https://doi.org/10.7164/antibiotics.45.1420
Shimizu T., Usui T., Machida K. et al. // Bioorg. Med. Chem. Lett. 2002. V. 12. № 23. P. 3363; https://doi.org/10.1016/S0960-894X(02)00782-5
Cullen W.P., Celmer W.D., Chappel L.R. et al. // J. Ind. Microbiol. 1988. V. 2. № 6. P. 349; https://doi.org/10.1007/BF01569573
Kotecha N.R., Ley S.V., Mantegani S. // Synlett. 1992. № 05. P. 395; https://doi.org/10.1055/s-1992-21357
Tachibana K., Scheuer P.J., Tsukitani Y. et al. // J. Amer. Chem. Soc. 1981. V. 103. № 9. P. 2469; https://doi.org/10.1021/ja00399a082
Singh S.B., Zink D.L., Heimbach B. et al. // Org. Lett. 2002. V. 4. № 7. P. 1123; https://doi.org/10.1021/ol025539b
Pettit G.R., Chicacz Z.A., Gao F. et al. // J. Org. Chem. 1993. V. 58. № 6. P. 1302; https://doi.org/10.1021/jo00058a004
Ueno T., Takahashi H., Oda M. et al. // Biochemistry 2000. V. 39. № 20. P. 5995; https://doi.org/10.1021/bi992661i
Li A., Piel J. // Chem. Biol. 2002. V. 9. № 9. P. 1017; https://doi.org/10.1016/S1074-5521(02)00223-5
Francke W., Kitching W. // Curr. Org. Chem. 2001. V. 5. № 2. P. 233; https://doi.org/10.2174/1385272013375652
Lenci E. // Small Molecule Drug Discovery. Elsevier, 2020. P. 225–245; https://doi.org/10.1016/B978-0-12-818349-6.00008-X
Sun P., Zhao Q., Zhang H. et al. // ChemBioChem 2014. V. 15. № 5. P. 660; https://doi.org/10.1002/cbic.201300616
Zarganes-Tzitzikas T., Dömling A. // Org. Chem. Front. 2014. V. 1. № 7. P. 834; https://doi.org/10.1039/C4QO00088A
Ramachary D.B., Mondal R., Venkaiah C. // Org. Biomol. Chem. 2010. V. 8. № 2. P. 321; https://doi.org/10.1039/B920152A
Sydnes M.O. // Curr. Green Chem. 2014. V. 1. P. 216; https://doi.org/10.2174/2213346101666140221225404
Mead K.T., Brewer B.N. // Curr. Org. Chem. 2003. V. 7. № 3. P. 227; https://doi.org/10.2174/1385272033372969
Palmes J.A., Aponick A. // Synthesis (Stuttg). 2012. V. 44. № 24. P. 3699; https://doi.org/10.1055/s-0032-1317489
Raju B.R., Saikia A.K. // Molecules. 2008. V. 13. № 8. P. 1942; https://doi.org/10.3390/molecules13081942
Yadav J.S., Raghavendra Rao K.V., Ravindar K. et al. // Synlett. 2010. № 1. P. 51; https://doi.org/10.1055/s-0029-1218546
Trofimov B.A., Schmidt E.Y. // Acc. Chem. Res. 2018. V. 51. № 5. P. 1117; https://doi.org/10.1021/acs.accounts.7b00618
Schmidt E.Y., Zorina N.V., Skitaltseva E.V. et al. // Tetrahedron Lett. 2011. V. 52. № 29. P. 3772; https://doi.org/10.1016/j.tetlet.2011.05.056
Schmidt E.Y., Zorina N.V., Skitaltseva E.V. et al. // Tetrahedron Lett. 2011. V. 52. № 29. P. 3772; https://doi.org/10.1016/j.tetlet.2011.05.056
Trofimov B.A., Schmidt E.Y., Skitaltseva E.V. et al. // Ibid. 2011. V. 52. № 33. P. 4285; https://doi.org/10.1016/j.tetlet.2011.06.019
Siyamak Shahab, Masoome Sheikhi // Russ. J. Phys. Chem. B. 2020. V. 14. № 1. P. 15–18. https://doi.org/10.1134/S1990793120010145
Zhiyan Wu, Zhang L., Liao Y. // Russ. J. Phys. Chem. B. 2021. V. 15. № S1. P. S81–S91. https://doi.org/10.1134/S1990793121090153
Breslavskaya N.N., Wasserman L.A., Barashkova I.I., Buchachenko A.L. // Russ. J. Phys. Chem. B. 2019. V. 13. № 4. P. 569; https://doi.org/10.1134/S199079311904002X
Ramakrishnan R., Dral P.O., Rupp M. et al. // Sci. Data. 2014. V. 1. P. 1; https://doi.org/10.1038/sdata.2014.22
Забалов М.В., Левина М.А., Тигер Р.П. // Хим. физика 2019. Т. 38. № 9. С. 3; https://doi.org/10.1134/S0207401X19090127
Grambow C.A., Pattanaik L., Green W.H. // Sci. Data. 2020. V. 7. № 1. P. 1–8. https://doi.org/10.1038/s41597-020-0460-4
Frisch M., Trucks G., Schlegel H., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Petersson G.A., Nakatsuji H., Li X., Caricato M., Marenich A.V., Bloino J., Janesko B.G., Gomperts R., Mennucci B., Hratchian H.P., Ortiz J.V., Izmaylov A.F., Sonnenberg J.L., Williams-Young D., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V.G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J.A., Jr., Peralta J.E., Ogliaro F., Bearpark M.J., Heyd J.J., Brothers E.N., Kudin K.N., Staroverov V.N., Keith T.A., Kobayashi R., Normand J., Raghavachari K., Rendell A.P., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Millam J.M., Klene M., Adamo C., Cammi R., Ochterski J.W., Martin R.L., Morokuma K., Farkas O., Foresman J.B., Fox D.J. Gaussian 16. Wallingford CT: Gaussian Inc., 2019.
Becke A.D. // Phys. Rev. A. 1988. V. 38. № 6. P. 3098; https://doi.org/10.1103/PhysRevA.38.3098
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1988. V. 37. № 2. P. 785; https://doi.org/10.1103/PhysRevB.37.785
Page M., Doubleday C., McIver J.W. // J. Chem. Phys. 1990. V. 93. № 8. P. 5634; https://doi.org/10.1063/1.459634
Grimme S. // Ibid. 2006. V. 124. № 3. P. 034108; https://doi.org/10.1063/1.2148954
Grimme S., Ehrlich S., Goerigk L. // J. Comput. Chem. 2011. V. 32. № 7. P. 1456; https://doi.org/10.1002/jcc.21759
Tomasi J., Mennucci B., Cancès E. // J. Mol. Struct: THEOCHEM. 1999. V. 464. № 1–3. P. 211; https://doi.org/10.1016/S0166-1280(98)00553-3
Pascual-ahuir J.L., Silla E., Tuñon I. // J. Comput. Chem. 1994. V. 15. № 10. P. 1127; https://doi.org/10.1002/jcc.540151009
Bondi A. // J. Phys. Chem. 1964. V. 68. № 3. P. 441; https://doi.org/10.1021/j100785a001
Wertz D.H. // J. Amer. Chem. Soc. 1980. V. 102. № 16. P. 5316; https://doi.org/10.1021/ja00536a033
Vitkovskaya N.M., Kobychev V.B., Bobkov A.S. et al. // J. Org. Chem. 2017. V. 82. № 23. P. 12467; https://doi.org/10.1021/acs.joc.7b02263
Allinger N.L. // J. Amer. Chem. Soc. 1959. V. 81. № 21. P. 5727; https://doi.org/10.1021/ja01530a049
Stortz C.A. // J. Phys. Org. Chem. 2010. V. 23. № 12. P. 1173; https://doi.org/10.1002/poc.1689
Vitkovskaya N.M., Orel V.B., Kobychev V.B. et al. // Intern. J. Quantum Chem. 2020. V. 120. № 9. P. 1; https://doi.org/10.1002/qua.26158
Abramenkov A.V. Kinet for Windows. 2012. Ver. 0.8.
Ларионова Е.Ю., Витковская Н.М., Кобычев В.Б. и др. // Журн. структур. химии 2007. Т. 48. № S7. С. 101.
Ларионова Е.Ю., Витковская Н.М., Кобычев В.Б. и др. // Докл. РАН 2011. Т. 438. № 6. С. 765.
Bordwell F.G., Fried H.E. // J. Org. Chem. 1991. V. 56. № 13. P. 4218; https://doi.org/10.1021/jo00013a027
Matthews W.S., Bares J.E., Bartmess J.E. et al. // J. Amer. Chem. Soc. 1975. V. 97. № 24. P. 7006; https://doi.org/10.1021/ja00857a010
Trofimov B.A., Schmidt E.Y., Zorina N.V. et al. // Adv. Synth. Catal. 2012. V. 354. № 9. P. 1813; https://doi.org/10.1002/adsc.201200210
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика