

Химия твердого топлива, 2021, № 6, стр. 50-58
ВЛИЯНИЕ СОСТАВА ТВЕРДОГО ТОПЛИВА НА РАВНОВЕСНЫЕ ХАРАКТЕРИСТИКИ ПРОЦЕССА ГАЗИФИКАЦИИ В СМЕСЯХ КИСЛОРОДА И ДИОКСИДА УГЛЕРОДА
1 ФГБУН Институт систем энергетики имени Л.А. Мелентьева СО РАН
664033 Иркутск, Россия
* E-mail: donskoy.chem@mail.ru
Поступила в редакцию 05.02.2021
После доработки 17.03.2021
Принята к публикации 06.04.2021
Аннотация
С помощью равновесного термодинамического моделирования исследован процесс газификации твердых топлив разного элементного состава в смесях O2/N2 и O2/CO2. Получены расчетные зависимости характеристик процесса (температура, выход углеродного остатка) от состава дутья и стехиометрического соотношения. Добавление углекислого газа, с одной стороны, способствует конверсии углерода за счет увеличения концентрации газифицирующих агентов, с другой – снижает температуру процесса из-за увеличения теплоемкости и эндотермического эффекта реакции газификации. Эффективность применения смесей O2/CO2 для газификации топлив растет с увеличением содержания углерода. Наибольший химический к.п.д. газификации кокса и каменного угля достигается при начальной концентрации СО2 порядка 40–60 об. %.
Повышение экологической эффективности использования твердого топлива требует развития новых технологий его термохимической конверсии. Одной из основных проблем использования твердого топлива является неизбежное образование значительных количеств углекислого газа [1]. Перспективы снижения выбросов СО2 (carbon capture and storage, CCS) открываются с использованием новых угольных технологий, таких как ТЭС с внутрицикловой газификацией (IGCC) и переход на обогащенное кислородом дутье (oxyfuel) [2, 3]. При этом СО2 может быть извлечен двумя возможными способами. Первый – удаление углерода до камеры сгорания (pre-combustion), когда генераторный газ проходит конверсию водяным паром, что позволяет использовать СО для получения дополнительного Н2, а продукты конверсии разделяются (абсорбционными, мембранными и химическими методами [4, 5]. После этого обогащенный водородом газ сжигается, а СО2 концентрируется и отправляется на захоронение. Второй способ – удаление углерода из продуктов сгорания (post-combustion), аналогичен таковому для традиционных систем CCS: газовая смесь охлаждается и компримируется до условий конденсации, после чего СО2 отделяется от других газов и захоранивается. Упростить выделение СО2 в варианте post-combustion можно с помощью oxyfuel-сжигания, когда концентрация кислорода в дутье обычно выше, чем в воздухе, а роль балласта вместо азота играет смесь продуктов сгорания – СО2 и Н2О. Сжигание в смеси кислорода и продуктов сгорания имеет лучшие экологические характеристики по сравнению с традиционным сжиганием в воздухе: во-первых, очевидно, что при этом образуется меньше оксидов азота (только топливные); во-вторых, полученные дымовые газы могут быть очищены до высоких концентраций СО2, допускающих его простую утилизацию. Технологии сжигания в смесях с продуктами сгорания считаются самым технически эффективным, но при этом одним из наиболее дорогих вариантов систем CCS [6, 7]. Среди других проблем, помимо дороговизны, можно отметить требования к очистке дымовых газов от оксидов серы и азота [8]. Исследуются комбинированные технологии, например сжигание топлив с разбавлением горячими продуктами сгорания [9], использование СО2 в качестве дутьевого агента при пылеугольной газификации [10, 11].
В работах [12, 13] рассматривается сжигание генераторного газа с рециркуляцией СО2 из продуктов сгорания. В [14, 15] показано, что избыток СО2 в составе рабочего тела снижает к.п.д. газотурбинной установки. Предлагается также использование СО2 в качестве рабочего тела в комбинированных циклах [16, 17].
При кислородной газификации угля использование СО2 в качестве транспортной среды для угольных частиц повышает среднюю степень конверсии топлива [18, 19], но при этом сужается область устойчивости факела [20]. В атмосфере углекислого газа диффузия кислорода к области горения замедляется, концентрация активных частиц снижается. Интенсивность тепловыделения на фронте горения может уменьшиться на 20–25% [21]. Для описания выгорания одиночных частиц, как правило, применяются широко используемые диффузионно-кинетические модели с разными поправками на структуру приповерхностного слоя, однако при высокой концентрации диоксида углерода вклад отдельных реакций может существенно изменяться [22, 23]. Изменение состава среды приводит к изменению условий тепломассопереноса, которые также влияют на поведение топливных частиц [24, 25]. В работе [26] рассматривается задача о воспламенении угольной частицы с более подробной детализацией химической кинетики разложения органической массы в одномерной. Влияние концентрации кислорода на профиль температуры в реакторе исследовалось в [27].
Для упрощения субмоделей термохимических превращений часто применяют равновесные термодинамические модели и их сочетания с моделями гетерогенной кинетики [28, 29]. В работе [30] один из вариантов кинетико-термодинамических моделей применялся для исследования эффективности высокотемпературной газификации угольной пыли в смесях О2/СО2: расчеты показали, что высокая концентрация СО2 позволяет эффективнее использовать теплоту сгорания топлива даже при снижении температуры реакции (из-за высокой теплоемкости СО2). В публикуемой работе рассматривается процесс газификации без нагрева дутья.
МОДЕЛЬ ТЕРМОХИМИЧЕСКОГО РАВНОВЕСИЯ И ИСХОДНЫЕ ДАННЫЕ
Для исследования границ термохимической эффективности углекислотной конверсии твердых топлив было проведено термодинамическое моделирование. Подобная модель была применена в работе [31] для оценки влияния избытка водяных паров на эффективность процесса газификации угля.
При газификации твердого топлива определяющее значение имеют следующие брутто-реакции:
(1)
$\begin{gathered} {\text{C}}{{{\text{H}}}_{a}}{{{\text{O}}}_{b}}{{{\text{N}}}_{c}}{{{\text{S}}}_{d}} = {{\beta }_{1}}{\text{C}} + {{\beta }_{2}}{{{\text{H}}}_{2}} + {{\beta }_{3}}{{{\text{N}}}_{2}} + \\ \, + {{\beta }_{4}}{{{\text{H}}}_{2}}{\text{S}} + {{\beta }_{5}}{\text{N}}{{{\text{H}}}_{{\text{3}}}} + {{\beta }_{6}}{\text{CO}} + {{\beta }_{7}}{\text{C}}{{{\text{O}}}_{2}} + {{\beta }_{8}}{{{\text{H}}}_{2}}{\text{O,}} \\ \end{gathered} $(7)
${\text{CO}} + {{{\text{H}}}_{2}}{\text{O}} = {\text{C}}{{{\text{O}}}_{2}} + {{{\text{H}}}_{2}}.$Реакция (1) моделирует процесс пиролиза: состав продуктов и тепловой эффект этой реакции неоднозначно определяются исходным составом, а зависят от условий протекания (в том числе и от динамических характеристик, но при равновесном моделировании эти параметры не учитываются). Реакции (2), (3) и (5) в сумме дают полное сгорание углерода до диоксида углерода. При высокой концентрации диоксида углерода в начальном состоянии равновесие в реакции (3) смещается в сторону продуктов, а в реакциях (5) и (7), наоборот, – в сторону реагентов, поэтому в режимах газификации с разбавлением кислорода диоксидом углерода можно ожидать повышения полноты конверсии топлива и уменьшения выхода водорода.
Для газовой фазы принимается уравнение состояния идеального газа, конденсированные вещества образуют идеальные однокомпонентные фазы. Тогда задача расчета химического состояния газовой фазы выглядит следующим образом [32]:
Найти
при условиях:Решение задачи равновесия в принятых приближениях существует и единственно, что следует из выпуклости энтропии для таких систем [34, 35]. Для численного решения используется один из вариантов метода оптимального спуска [34].
Индикаторами эффективности режимов являются адиабатическая температура процесса, выход твердого остатка и химический к.п.д. газификации, равный отношению теплот сгорания генераторного газа и топлива, из которого он был получен:
Для оценки влияния сорта топлива расчеты проведены для четырех топлив с разным составом. Кокс – пример “идеального” топлива с высоким содержанием углерода, биомасса – низкокалорийного топлива. Для расчетов взяты кузнецкий каменный уголь и бурый уголь Березовского месторождения [37]. Состав топлив приведен в табл. 1.
Таблица 1.
Состав топлив
| Характеристика топлива | Обозначение | Кокс | Каменный уголь | Бурый уголь | Биомасса |
|---|---|---|---|---|---|
| Влажность на рабочую массу | W r, % | 5 | 9 | 12 | 10 |
| Зольность на сухую массу | Ad, % | 10 | 20 | 9 | 1 |
| Выход летучих веществ на сухую беззольную массу | Vdaf, % | 9 | 30 | 48 | 85 |
| Содержание углерода на сухую беззольную массу | Cdaf, % | 92 | 81 | 69.4 | 49.5 |
| Содержание водорода на сухую беззольную массу | Hdaf, % | 2.5 | 3.5 | 4.9 | 6.3 |
| Содержание кислорода на сухую беззольную массу | Odaf, % | 4.99 | 13.2 | 24.3 | 44.15 |
| Содержание азота на сухую беззольную массу | Ndaf, % | 0.01 | 2.0 | 0.9 | 0.05 |
| Содержание серы на сухую беззольную массу | Sdaf, % | 0.5 | 0.3 | 0.5 | 0 |
Помимо состава топлива переменными параметрами в расчетах являются относительный расход окислителя α (рассчитанный по отношению к стехиометрическому расходу кислорода), концентрация кислорода в дутье (от 21 до 90 об. %); разбавители – азот (традиционные режимы) и углекислый газ (перспективные режимы).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ РАСЧЕТОВ
Равновесные характеристики процесса газификации кокса (рис. 1) в смесях O2/N2 слабо зависят от концентрации кислорода: изолинии химического к.п.д. параллельны оси абсцисс до значений α порядка 0.5. При высоких температурах ход изолиний меняется, что связано с термической диссоциацией продуктов сгорания (смещение изолиний наблюдается для всех топлив). Отметим, что термохимические данные для температур выше 3000 К определялись экстраполяцией данных [33], однако эта область параметров практически нереализуема из-за требований к материалам. Максимальный химический в смесях O2/N2 к.п.д. равен 72% и почти не зависит от концентрации кислорода. В смесях O2/CO2 максимальный химический к.п.д. резко возрастает до 89% благодаря полезному использованию теплоты в эндотермических реакциях газификации за счет СО2, который в избытке присутствует в газовой фазе. Область полной конверсии также смещается в область меньших расходов окислителя.
Рис. 1.
Расчетные характеристики процесса газификации кокса в смесях O2/N2 (а)–(в) и O2/CO2 (г)–(е): адиабатическая температура, К (а), (г); химический к.п.д., % (б), (д); выход углерода, моль/кг (в), (е).
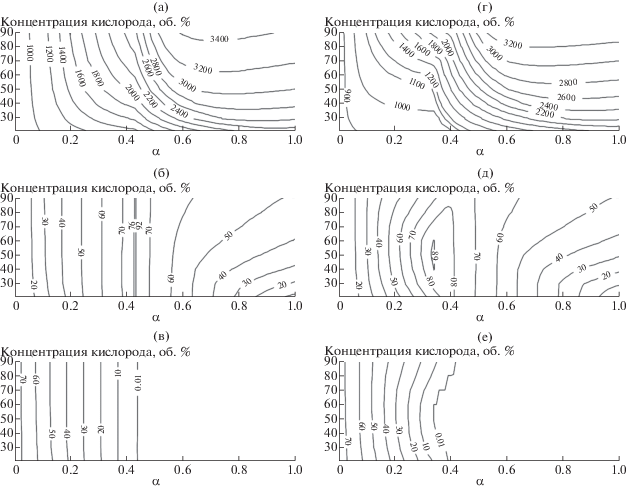
Из-за того, что удельная молярная теплоемкость СО2 выше, чем у N2, а реакция газификации имеет эндотермический эффект, адиабатическая температура газификации кокса в условиях разбавления кислорода продуктами сгорания оказывается намного ниже. С увеличением концентрации кислорода влияние разбавителя уменьшается, поэтому поведение характеристик в верхней части диаграмм практически совпадает.
Похожие закономерности наблюдаются при газификации каменного угля (рис. 2), однако из-за меньшего содержания углерода в составе максимальный химический к.п.д. в смесях O2/N2 оказывается порядка 83%. Из-за меньшей теплотворной способности по сравнению с коксом температура газификации оказывается меньше. При газификации в смесях O2/CO2 максимальный химический к.п.д. составляет 88% – почти на том же уровне, что и в случае кокса.
Рис. 2.
Расчетные характеристики процесса газификации каменного угля в смесях O2/N2 (а)–(в) и O2/CO2 (г)–(е): адиабатическая температура, К (а), (г); химический к.п.д., % (б), (д); выход углерода, моль/кг (в), (е).
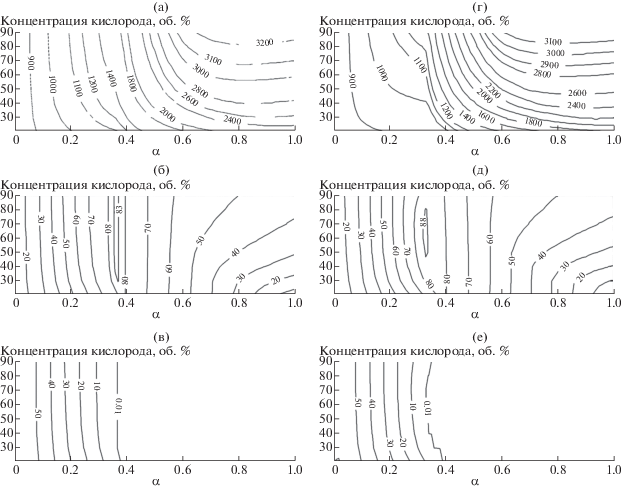
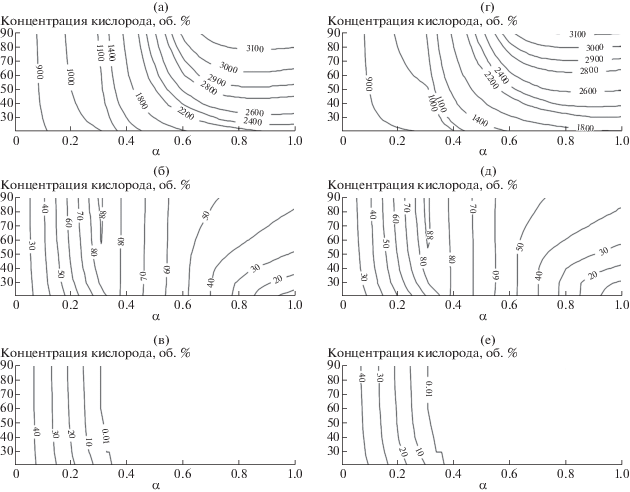
Для бурого угля (рис. 3) максимальные значения к.п.д. в смесях O2/N2 и O2/CO2 оказываются близки: примерно 88% для обоих разбавителей. Топология химического к.п.д. для обоих типов смесей довольно близка (в т.ч. максимальные значения достигаются при одних и тех же параметрах), хотя отличие температур в этой же области параметров (α = 0.4–0.6), наоборот, оказывается наибольшим. Бурый уголь как топливо с высоким выходом летучих можно газифицировать при меньших значениях α по сравнению с каменным углем в смесях O2/N2. Однако из-за его меньшей калорийности температура реакции недостаточна для эффективной утилизации теплоты в реакции углерода топлива с углекислым газом, поэтому газификация бурого угля в смесях O2/CO2 требует немного больших значений α и образование горючих компонентов за счет добавления СО2 нивелируется добавлением окислителя.
Рис. 3.
Расчетные характеристики процесса газификации бурого угля в смесях O2/N2 (а)–(в) и O2/CO2 (г)–(е): адиабатическая температура, К (а), (г); химический к.п.д., % (б), (д); выход углерода, моль/кг (в), (е).
Для газификации биомассы (рис. 4) наблюдаются те же закономерности, что и для бурого угля. Биомасса – это топливо с самым высоким выходом летучих веществ (из рассмотренных в данной работе), поэтому изменение состава смеси с O2/N2 на O2/CO2 приводит к ухудшению характеристик газа при низких концентрациях кислорода и почти не меняет их при высоких концентрациях кислорода.
Рис. 4.
Расчетные характеристики процесса газификации биомассы в смесях O2/N2 (а)–(в) и O2/CO2 (г)–(е): адиабатическая температура, К (а), (г); химический к.п.д., % (б), (д); выход углерода, моль/кг (в), (е).
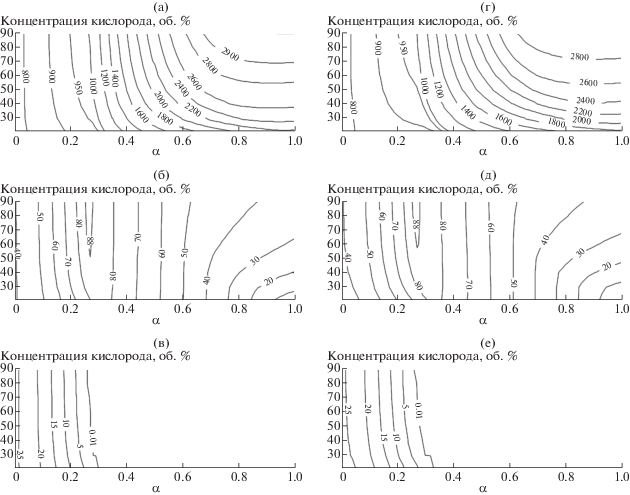
Максимальные значения равновесного химического к.п.д. для разных концентраций окислителя приведены на рис. 5. С увеличением выхода летучих и уменьшением массового содержания углерода в топливе различия между зависимостями химического к.п.д. в O2/N2 и O2/CO2 становятся все менее и менее заметными (хотя и не исчезают). Чем выше содержание углерода в топливе, тем больше положительный эффект от использования СО2 в качестве разбавителя: при окислении летучих веществ бурого угля и биомассы образуется достаточно газообразных окислителей для регенерации выделяющейся при окислении теплоты. Таким образом, высокоуглеродистые топлива, такие как каменный уголь и кокс, оказываются наиболее подходящим топливом для газификации в смесях О2/СО2.

ВЫВОДЫ
С помощью равновесного термодинамического моделирования исследована предельная эффективность процессов газификации разных сортов твердого топлива в смесях O2/CO2. В отличие от процессов газификации в смесях O2/N2, при использовании смесей O2/CO2 стехиометрические ограничения на конверсию углерода ослабляются. Присутствующий в избытке СО2 вступает в реакцию с углеродом, повышая выход горючих газов, но существенно снижая температуру (на 100–200 К). Эффект от использования СО2 зависит также от концентрации кислорода: при низких концентрациях кислорода температура процесса оказывается недостаточно высокой из-за разбавления (при этом использование СО2, наоборот, снижает химический к.п.д. газификации бурого угля и биомассы); при высоких концентрациях кислорода концентрация СО2 оказывается недостаточно высокой для эффективной конверсии углерода. В таких условиях использование смесей О2/СО2 в качестве газифицирующего агента оказывается благоприятным только для высокоуглеродистых топлив. Наибольший эффект наблюдается при концентрации кислорода 50–60 об. %. Углекислый газ может также выступать в качестве добавки для управления температурой процесса, например при использовании комбинированного дутья.
Список литературы
Зайченко В.М., Штеренберг В.Я. // Теплоэнергетика. 2019. № 6. С. 75. [Thermal Engineering. 2019. V. 66. P. 440] https://doi.org/10.1134/S0040601519060107
Thattai A.T., Oldenbroek V., Schoenmakers L., Woudstra T., Aravind P.V. // Appl. Thermal Eng. 2017. V. 114. P. 170. https://doi.org/10.1016/j.applthermaleng.2016.11.167
Chen L., Yong S.Z., Ghoniem A.F. // Progr. Energy Combust. Sci. 2012. V. 38. P. 156. https://doi.org/10.1016/j.pecs.2011.09.003
Kunze C., Spliethoff H. // Appl. Energy. 2012. V. 94. P. 109. https://doi.org/10.1016/j.apenergy.2012.01.013
Shi B., Xu W., Wu W., Kuo P.-C. // Energy Conv. Manag. 2019. V. 195. P. 290. https://doi.org/10.1016/j.enconman.2019.05.006
Cau G., Tola V., Ferrara F., Porcu A., Pettinau A. // Fuel. 2018. V. 214. P. 423. https://doi.org/10.1016/j.fuel.2017.10.023
Zhou C., Shah K., Song H., Zanganeh J., Doroodchi E., Moghtaderi B. // Energy Fuels. 2016. V. 30. № 3. P. 1741. https://doi.org/10.1021/acs.energyfuels.5b02209
Toftegaard M.B., Brix J., Jensen P.A., Glarborg P., Jensen A.D. // Progr. Energy Combust. Sci. 2010. V. 36. P. 581. https://doi.org/10.1016/j.pecs.2010.02.001
Zhang Z., Li X., Zhang L., Luo C., Lu B., Xu Y., Liu J., Chen A., Zheng C. // Fuel Proc. Tech. 2019. V. 184. P. 27. https://doi.org/10.1016/j.fuproc.2018.11.011
Kidoguchi K., Hara S., Oki Y., Kajitani S., Umemoto S., Inumaru J. // Proc. ASME 2011 Power Conference (Denver, USA, July 12–14 2011). 2011. V. 2. P. 485.
Ishi H., Hayashi T., Tada H., Yokohama K., Takashima R., Hayashi J. // Appl. Energy. 2019. V. 233–234. P. 156. https://doi.org/10.1016/j.apenergy.2018.10.021
Romano M.C., Lozza G.G. // Int. J. Greenhouse Gas Control. 2010. V. 4. P. 469. https://doi.org/10.1016/j.ijggc.2009.11.010
Oki Y., Hara S., Umemoto S., Kidoguchi K., Hamada H., Kobayashi M., Nakao Y. // Energy Proc. 2014. V. 63. P. 471. https://doi.org/10.1016/j.egypro.2014.11.050
Prawobo B., Aziz M., Umeki K., Susanto H., Yan M., Yoshikawa K. // Appl. Energy. 2015. V. 158. P. 97. https://doi.org/10.1016/j.apenergy.2015.08.060
Filippov P.S., Lazebniy I.P., Ryzhkov A.F. // J. Phys. Conf. Ser. 2020. V. 1565. P. 012096. https://doi.org/10.1088/1742-6596/1565/1/012096
Allam R., Martin S., Forrest B., Fetvedt J., Lu X., Freed D., Brown G.W.Jr., Sasaki T., Itoh M., Manning J. // Energy Proc. 2017. V. 114. P. 5948. https://doi.org/10.1016/j.egypro.2017.03.1731
Weiland N.T., White C.W. // Fuel. 2018. V. 212. P. 613. https://doi.org/10.1016/j.fuel.2017.10.022
Botero C., Field R.P., Herzog H.J., Ghoniem A.F. // Appl. Energy. 2013. V. 104. P. 408–417. https://doi.org/10.1016/j.apenergy.2012.11.028
Tupsakhare S., Dooher J., Modroukas D., Castaldi M. // Fuel. 2019. V. 256. P. 115848. https://doi.org/10.1016/j.fuel.2019.115848
Watanabe H., Ahn S., Tanno K. // Energy. 2017. V. 118. P. 181. https://doi.org/10.1016/j.energy.2016.12.031
Jimenez S., Gonzalo-Tirado C. // Combust. Flame. 2017. V. 176. P. 94. https://doi.org/10.1016/j.combustflame.2016.09.024
Yin C., Yan J. // Appl. Energy. 2016. V. 162. P. 742. https://doi.org/10.1016/j.apenergy.2015.10.149
Yu J., Ou W., Zhou K. // Fuel. 2014. V. 124. P. 173. https://doi.org/10.1016/j.fuel.2014.01.101
Senneca O., Scala F., Chirone R., Salatino P. // Fuel. 2017. V. 201. P. 65. https://doi.org/10.1016/j.fuel.2016.11.034
Kriebitzsch S., Richter A. // Combust. Flame. 2020. V. 211. P. 185. https://doi.org/10.1016/j.combustflame.2019.08.028
Maffei T., Khatami R., Perucci S., Faravelli T., Ranzi E., Levendis Y.A. // Combust. Flame. 2013. V. 160. P. 2559. https://doi.org/10.1016/j.combustflame.2013.06.002
Ralnikov P.A., Abaimov N.A., Ryzhkov A.F. // J. Phys. Conf. Ser. 2018. V. 1128. P. 012007. https://doi.org/10.1088/1742-6596/1128/1/012007
Valero A., Uson S. // Energy. 2006. V. 31. P. 1643. https://doi.org/10.1016/j.energy.2006.01.005
Донской И.Г. // ХТТ. 2016. № 3. С. 54. [Solid Fuel Chem. 2016. V. 50. P. 191] https://doi.org/10.3103/S0361521916030034
Donskoy I. // E3S Web Conf. 2020. V. 209. P. 03011. https://doi.org/10.1051/e3sconf/202020903011
Свищев Д.А., Кейко А.В. // Теплоэнергетика. 2010. № 6. С. 33.
Каганович Б.М., Филиппов С.П., Кейко А.В., Шаманский В.А. // Теплоэнергетика. 2011. № 2. С. 51. [Thermal Engineering. 2011. V. 58. P. 143] https://doi.org/10.1134/S0040601511020054
Глушко В.П. Термодинамические и теплофизические свойства продуктов сгорания. Т. 1. М.: ВИНИТИ АН СССР, 1971.
Каганович Б.М., Кейко А.В., Шаманский В.А., Ширкалин И.А., Зароднюк М.С. Технология термодинамического моделирования. Редукция моделей движения к моделям покоя. Новосибирск: Наука, 2010. 236 с.
Rossi C.C.R.S., Berezuk M.E., Cardozo-Filho L., Guirardello R. // Comp. Chem. Eng. 2011. V. 35. P. 1226. https://doi.org/10.1016/j.compchemeng.2010.07.019
Асланян Г.С., Гиневская И.Ю., Шпильрайн Э.Э. // ХТТ. 1984. № 1. С. 90.
Вдовенко В.С., Мартынова М.И., Новицкий Н.В., Юшина Г.Д. Энергетическое топливо СССР (ископаемые угля, горючие сланцы, торф, мазут и горючий природный газ). М.: Энергоатомиздат, 1991. 184 с.
Дополнительные материалы отсутствуют.
Инструменты
Химия твердого топлива