

Кинетика и катализ, 2019, T. 60, № 1, стр. 45-53
Роль кислоты в распаде смешанного бензоилзамещенного фосфониево-иодониевого илида
Т. Д. Некипелова 1, *, Т. А. Подругина 2, Д. С. Виноградов 2, П. И. Демьянов 2, В. А. Кузьмин 1
1 ФГБУН Институт биохимической физики им. Н.М. Эмануэля РАН
119334 Москва, ул. Косыгина, 4, Россия
2 ФГБОУ ВО Московский государственный университет им. М.В. Ломоносова,
Химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
* E-mail: nekip@sky.chph.ras.ru
Поступила в редакцию 18.05.2018
После доработки 14.09.2018
Принята к публикации 17.08.2018
Аннотация
Смешанные фосфониево-иодониевые илиды позволяют синтезировать труднодоступные и новые гетероциклические соединения. Фотоинициируемые реакции фосфониево-иодониевых илидов с ацетиленами происходят с периодом индукции и катализируются кислотами, в том числе образующимися в реакции. Методами УФ-видимой спектрофотометрии исследованы кинетические особенности взаимодействия бензоилзамещенного фосфониево-иодониевого илида с трифторуксусной кислотой. Определены кинетические параметры реакции. Предложен механизм автокатализа кислотами, заключающийся в образовании протонированной формы исходного илида, активной в распаде на катион-радикалы. Более активный распад протонированной формы илида подтвержден теоретическими термохимическими расчетами.
ВВЕДЕНИЕ
В настоящее время смешанные фосфониево-иодониевых илиды, соединения с несколькими реакционными центрами, общая формула которых представлена ниже, исследуются с целью синтеза на их основе труднодоступных и новых гетероциклических соединений [1–3]:
где R1 = Ph или пятичленный ароматический гетероцикл, а R2 = CO(Ph), CO(OMe), CO(OEt), CN, PO(OEt)2, S(O)2(.п-MeC6H4)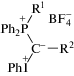
Фенилиодониевая группа в составе смешанных илидов определяет их способность к разложению под действием света с образованием активных ионных и ион-радикальных интермедиатов, как это наблюдали для диарилиодониевых солей [4, 5]. В последнее время открыты две новые фотохимические реакции смешанных фосфониево-иодониевых илидов с соединениями, содержащими тройную связь (С≡N и С≡С), в результате которых синтезируются замещенные оксазолы (с нитрилами) и фурановые производные и фосфорсодержащие гетероциклические соединения с ацетиленами. Реакция с ацетиленами является общей для фосфониево-иодониевых илидов различного строения [6–11] и приведена на схеме 1 для бензоилзамещенного илида 1 (R1 = = Ph, R2 = CO(Ph)), используемого в настоящем исследовании.
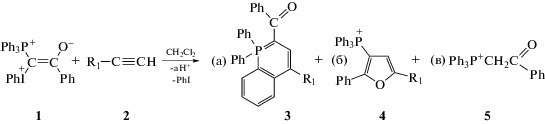
Схема 1.
Основные продукты реакции – λ5-фосфинолин (3), производное фурана (4) и фосфониевая соль (5) (схема 1, в этой и последующих схемах противоион ${\text{BF}}_{4}^{ - }$ опущен). Фотохимические реакции ацетиленов с фосфониево-иодониевым илидом 1 происходят только в хлористом метилене при концентрации илида, превышающей 0.01 моль/л, с периодом индукции [12, 13]. В процессе фотолиза илидов и их смесей с ацетиленами в CH2Cl2 образуется кислота, поэтому было предположено, что она может играть роль катализатора [13, 14]. В [13] было показано, что при добавлении сильных органических кислот (трифторуксусной кислоты и толуолсульфокислоты) период индукции в реакции илида 1 с фенилацетиленом сокращается, реакция переходит в термический режим, т.е. происходит без облучения. Важно заметить, что для получения в этих условиях целевого продукта 3 концентрация добавленной кислоты не должна превышать концентрацию илида.
Известно, что хлористый метилен подвергается фотоинициированным радикальным реакциям с образованием HCl и фосгена [15–17]. Поэтому была предложена схема распада исходного илида, включающая вовлечение растворителя (CH2Cl2) в радикальные реакции [14, 18]. Было сделано предположение, что образовавшийся хлористый водород может катализировать фотолиз и ускорять реакцию [14].
В связи с этим в настоящей работе исследовано взаимодействие илида 1 с трифторуксусной кислотой (ТФК) и рассмотрен механизм автокатализа с участием кислот, включающий промежуточное образование протонированной формы илида, т. е. фосфониево-иодониевого дикатиона, активного в распаде на радикалы и катион-радикалы. Более легкий распад протонированной формы илида 1, приводящий к образованию катион-радикалов, подтвержден теоретическими расчетами в рамках теории функционала плотности.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Илид 1 получали согласно методике, предложенной в [1, 7, 8]. Илид тщательно отмывали эфиром от кислоты (HBF4), которая используется в его синтезе. Хлористый метилен (Эталонный, “Компонент реактив” Россия), не содержащий стабилизатора и HCl, и ацетонитрил (о.ч., “Компонент реактив”, Россия) применяли без дополнительной очистки.
Спектры поглощения измеряли на спектрофотометре UV3101 PC (“Shimadzu”, Япония) в кварцевых кюветах с длиной оптического пути 1.0 или 0.4 см. Растворы илида 1 (1 × 10–4 моль/л) готовили в CH2Cl2 или ацетонитриле. Рассчитанное количество раствора ТФК ([ТФК] = 1.35 моль/л) в соответствующем растворителе (1 : 10), прикапывали микропипеткой в раствор илида, при этом добавка кислоты уменьшала концентрацию илида не более, чем на 1%. Далее по всему тексту указана окончательная концентрация ТФК в реакционной смеси. Все измерения проводили при комнатной температуре на воздухе.
Кинетику реакции илида 1 с ТФК в CH2Cl2 регистрировали на спектрофотометре UV3101 PC (“Shimadzu”, Япония) в кинетическом режиме на длине волны 270 нм в кварцевых кюветах с длиной оптического пути 1.0 см.
Масс-спектры были получены на масс-спектрометре 7T LTQ FT Ultra (“Thermo Finnigan”, Германия) с использованием прямого ввода проб и универсального источника ионизации Finnigan Ion MaxSource в режиме электроспрея (ESI) (см. детали в [14]). Спектрофотометрические исследования в УФ-видимой области и масс-спектрометрические исследования выполнены в Центре коллективного пользования ИБХФ РАН “Новые материалы и технологии”.
Для квантово-химических расчетов в рамках теории функционала плотности использовали программы GAMESS US [19] с применением функционала PBE0 [20], базисных наборов 6-311++G(d,p) (встроенных в программу GAMESS US) для атомов H, C, O и P, а также базисного набора aug-cc-pVDZ-pp с эффективным остовным потенциалом для атома I [21]. Оптимизацию геометрии илида 1, его протонированной формы, всех других частиц, образующихся при распаде обоих форм исследуемого илида, проводили с учетом сольватации хлористым метиленом с использованием модели поляризованного континуума [22]. Все изученные системы оптимизировали без наложения каких-либо ограничений за исключением того, что рассчитывали только дублетные состояния частиц с неспаренным электроном.
Колебательный анализ, выполненный также с учетом сольватации хлористым метиленом, подтвердил, что всем оптимизированным системам соответствуют реальные минимумы на поверхности потенциальной энергии. Термохимический анализ для всех теоретически изученных систем проводили при 298.15 К (25°С). Изменения свободных энергий Гиббса (ΔG298) при 25°С, соответствующих распаду илида 1 и его протонированной формы, оценивали как разность между суммой свободных энергий продуктов распада и свободной энергией исходного илида. Для моделирования вертикальных возбуждений 1 и его протонированной формы в их соответствующие синглетные состояния применяли метод нестационарной теории функционала плотности (TD-DFT-метод [23]) в сочетании с PCM-моделированием сольватации хлористым метиленом.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Взаимодействие илида 1 с трифторуксусной кислотой
При добавлении избытка ТФК (5 × 10‒3 моль/л) в раствор илида 1 (1 × 10‒4 моль/л) в CH2Cl2 или ацетонитриле без облучения образуется продукт со спектром поглощения, близким к наблюдаемому спектру промежуточного продукта при фотолизе раствора илида 1 такой же концентрации в CH2Cl2 (ср. рис. 1 с рис. 1A в [14]) При этом в отсутствие облучения скорость реакции с ТФК в CH2Cl2 более чем на порядок выше, чем в ацетонитриле (рис. 1, вставка). Следует отметить, что при фотолизе илида в ацетонитриле в присутствии кислоты не обнаружено производного оксазола, который является основным продуктом в реакции фотогетероциклизации 1 с ацетонитрилом в отсутствие кислоты [10]. Продукт, образующийся при разложении 1 в кислых растворах, устойчив без облучения, но при облучении реакционной смеси светом с длиной волны λirr < 365 нм фотолизуется с образованием соли 5. Спектральные изменения при фотолизе 1 и в реакциях 1 с ТФК в отсутствие облучения аналогичны: наблюдается увеличение поглощения в области длин волн 260–290 нм и уменьшение поглощения в области длин волн >290 нм. Однако масс-спектрометрический анализ фотолизата 1 в CH2Cl2 после окончания фотолиза [14] и растворов, полученных в реакциях 1 с ТФК в CH2Cl2 и ацетонитриле без облучения, показал, что в этих реакциях продукты различаются. В обоих случаях основными являются продукты взаимодействия илида с кислотами: HCl при фотолизе и ТФК при добавлении кислоты.
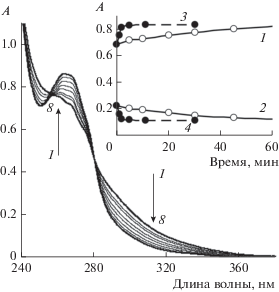
Рис. 1.
Изменение спектров поглощения илида 1 ([1] = 1 × 10–4 моль/л) в присутствии ТФК ([ТФК] = = 5 × 10–3 моль/л) в ацетонитриле; время, мин: 0 (1), 5 (2), 10 (3), 20 (4), 30 (5), 45 (6), 63 (7), 90 (8). Вставка: кинетика изменения поглощения в ацетонитриле (1, 2) и CH2Cl2 (3, 4); длина волны регистрации, нм: 270 (1, 3) и 300 нм (2, 4).
Основной продукт реакции 1 с ТФК, которому в масс-спектре соответствует ион с m/z = 493.12 Да (1 + ТФК–PhI)+, – это, по-видимому, соединение 8b, образующееся в результате превращений, показанных на схеме 2. При масс-спектрометрическом анализе продуктов взаимодействии 1 с ТФК в CH2Cl2 был обнаружен также молекулярный ион с m/z = 721.15 Да, который не был идентифицирован. В следовых количествах в обоих растворителях присутствует молекулярный ион с m/z = 697.04 Да (1 + ТФК)+, содержащий фрагмент PhI в своем составе. Этот ион относится к продукту присоединения ТФК к илиду 1 (7b). Соединение 7b при анализе методом ESI-MS может путем выброса PhI также давать молекулярный ион с m/z = 493.12 Да. Следует сказать, что при проведении реакции илида 1 с ТФК в CH2Cl2 не образуются хлорсодержащие соединения 7a и 8a, хотя эти соединения были идентифицированы при фотолизе разбавленных растворов илида 1 в CH2Cl2 [14].
На основании экспериментальных данных настоящей работы по темновому взаимодействию 1 с ТФК и результатов, полученных ранее при фотолизе 1 в CH2Cl2 [14], предложена следующая схема (схема 2) взаимодействия кислот (HA) с илидом 1:
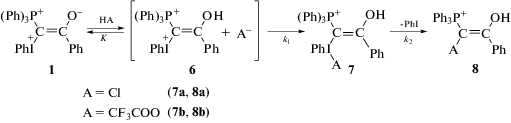
Схема 2.
Фосфониево-иодониевый дикатион 6 является продуктом обратимого присоединения протона к илиду 1. Дикатион в ионной паре [6 + A–] вступает в реакцию с анионом кислоты с образованием соединения 7. Об этом свидетельствует наличие в масс-спектрах реакционных смесей следовых количеств молекулярного иона с m/z = = 619.10 Да (1 + HCl)+ при фотолизе 1 в CH2Cl2 [14] (соединение 7а), и следовых количеств молекулярного иона с m/z = 697.04 Да (1 + ТФК)+ (соединение 7b) в реакции 1 с трифторуксусной кислотой. Из наших исследований следует, что при анализе фенилиодониевых соединений методом ESI-MS в основном регистрируются ионы, образующиеся в результате отщепления PhI. Соединения 8 представляют собой продукты дальнейшего превращения 7. Образование соединений, подобных 7 и 8, было предложено в [24] при изучении SN2 замещения в алкилвинил(фенил)иодониевых солях. В настоящей работе мы показали, что продукт присоединения аниона (A–) к иодониевой группе 7, неустойчив, выбрасывает PhI и быстро превращается в 8. На основании этого мы полагаем, что лимитирующей стадией процесса превращения дикатиона 6 в конечный продукт 8 является реакция дикатиона 6 с анионом кислоты с образованием 7.
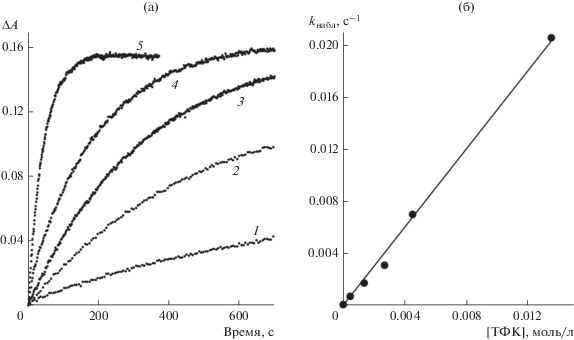
Спектрофотометрическое исследование кинетики взаимодействия илида 1 с избытком ТФК (регистрация при 270 нм) (рис. 2а) показало, что кривые изменения поглощения хорошо аппроксимируются экспоненциальными зависимостями, при этом константа скорости образования продуктов реакции (kнабл) линейно зависит от концентрации ТФК (рис. 2б) и бимолекулярная эффективная константа скорости образования продуктов реакции илида 1 с ТФК (kэфф) равна 1.5 ± 0.1 л моль–1 с–1. В рамках предложенного механизма взаимодействия смешанного фосфониево-иодониевого илида 1 с кислотами (схема 2) линейная зависимость константы скорости от концентрации кислоты означает, что K[HA] $ \ll $ 1, а определенная экспериментально kэфф = k1K. Максимальная использованная концентрация кислоты в наших экспериментах [ТФК] = 0.014 моль/л, т.е. K $ \ll $ 70 л моль‒1. Низкое значение константы равновесия означает, что равновесные концентрации дикатиона 6 в смеси незначительны. Действительно, если принять, что K = 5 л моль‒1, то равновесная концентрация 6 будет ~5 × 10–6 моль/л при [1] = 1 × 10–4 моль/л и [ТФК] = 1 × 10–2 моль/л, т.е. только 5% от исходной концентрации илида 1. Это связано, по-видимому, с тем, что вместо катиона 1 образуется дикатион 6 с двойным положительным зарядом. Можно ожидать, что устойчивость такого дикатиона не должна быть очень высокой. Возможно, поэтому 6 либо вновь превращается в 1, либо распадается на 10 и PhI+· (см. далее, реакция (II)) или стабилизируется путем превращения в 7, несущем меньший (+1) положительный заряд.
Рис. 2.
а – Кинетика изменения поглощения ΔA = At – A0 при λрег = 270 нм в смеси илид 1 ([1] =1 × 10–4 моль/л) и ТФК в CH2Cl2 в зависимости от концентрации ТФК (моль/л): 0.00045 (1), 0.00135 (2), 0.0027 (3), 0.0045 (4), 0.0135 (5); б – зависимость константы скорости изменения поглощения (kнабл) от концентрации ТФК.
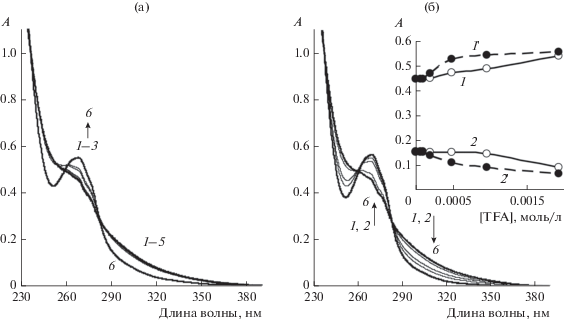
Сложный характер реакции илида 1 с кислотой и последующих превращений подтверждается зависимостью спектров поглощения смеси илида с ТФК от концентрации ТФК (рис. 3). Если в нулевой момент времени в исследуемой смеси концентрация кислоты не превышает концентрацию илида, спектр поглощения практически не изменяется в течение 15 мин, что подтверждает низкую концентрацию образовавшегося дикатиона 6. Дальнейшее увеличение концентрации кислоты приводит к росту поглощения в области 260–286 нм. При этом в области длин волн λ > 286 нм повышение концентрации кислоты до десятикратного избытка по сравнению с концентрацией илида практически не изменяет поглощение раствора в течение нескольких минут (рис. 3а). В течение 15 мин спектр поглощения меняется в зависимости от концентрации ТФК: при возрастании концентрации ТФК поглощение при λ > 286 нм уменьшается аналогично тому, как это происходит во времени после добавления большего избытка кислоты (ср. рис. 3б и рис. 1). При увеличении времени наблюдения происходит смещение изобестических точек (рис. 3а и 3б), что также указывает на сложный характер взаимодействия илида 1 с кислотой.
Рис. 3.
Спектры поглощения илида 1 ([1] =1.0 × 10–4 моль/л) через 1 (а) и 15 (б) мин после добавления ТФК в концентрации (104, моль/л): 0 (1), 0.6 (2), 2.0 (3), 5.0 (4), 10 (5), 20 (6). Вставка: зависимость поглощения от концентрации ТФК на длинах волн (нм): 270 (1, 1'), 300 (2, 2 ') через 1 (1, 2) и 15 мин (1', 2'); растворитель – CH2Cl2.
Теоретический термохимический анализ распада илида 1 и его протонированной формы 6
Ранее было сделано предположение, что фосфониево-иодониевый дикатион 6, представляющий собой протонированный илид 1, активнее в распаде на радикальные интермедиаты, чем исходный илид [14] (здесь и далее мы пренебрегали влиянием противоанионов ${\text{BF}}_{4}^{ - }$ на свойства 1 и дикатиона 6), и его образование in situ при взаимодействии 1 с HCl (генерируемым из хлористого метилена как растворителя) или со специально добавленной кислотой ускоряет синтез конечных продуктов за счет возрастания скорости инициирования. Для подтверждения предположения о более легком по сравнению с илидом 1 распаде дикатиона 6 на радикальные и ион-радикальные частицы был проведен теоретический термохимический анализ реакций (I) и (II) с учетом сольватации хлористым метиленом с использованием модели поляризованного континуума.
Проведенные PBE0-расчеты показывают, что изменение свободной энергии Гиббса (ΔG298) в реакциях (I) и (II) при 298 К (25°С) согласно термохимическому анализу составляет 42.6 ккал моль–1 (178 кДж) и 21.2 ккал моль–1 (89 кДж) соответственно. Энтальпия для реакций (I) и (II) при 25°С увеличивается на 58.1 ккал моль–1 (244 кДж) и 38.5 ккал моль–1 (161 кДж) соответственно. Таким образом, данные расчетов качественно подтверждают заметно меньшую устойчивость дикатиона 6 (реакция (II)) к распаду по сравнению с распадом исходного илида 1 (реакция (I)). Дополнительными свидетельствами в пользу бóльшей склонности 6 к распаду (то есть к выбросу катион-радикала (PhI)+·) является удлинение связи между атомом иода и атомом углерода (С2–I), связанным с PPh3-группой в 6, от 2.063 Å в 1 до 2.094 Å в 6, тогда как длина связи между атомом иода и атомом углерода фенильной группы в 6 короче (2.128 Å), чем в 1 (2.137 Å). Заряд (по Малликену) на атоме иода в 6 и 1 равен 0.594 и 0.606 соответственно. В (PhI)+· длина связи С–I составляет 2.038 Å, и заряд на атоме иода равен 0.578. Таким образом, расчеты показывают, что 6 более подготовлен к выбросу (PhI)+·, чем 1, с чем и связан, по-видимому, легкий распад протонированной формы 1 на два катион-радикала.
Механизм кислотного автокатализа при фотолизе илида 1
Поскольку теоретический анализ распада илида 1 и образующегося из него дикатиона 6 проводили для термической реакции, то возникает вопрос, насколько эти данные могут быть применены к фотолитическому распаду илида 1. Хорошо очищенный кристаллический илид 1 стабилен и сохраняется практически без изменений в течение нескольких месяцев. Однако, будучи растворенным в CH2Cl2, илид 1 за одни или несколько суток в темноте превращается в продукты, из которых основным является соль 5. Темновой процесс такого превращения ускоряется следами кислоты, что соответствует расчетным данным для распада дикатиона 6. При облучении раствора илид 1 (0.05 моль/л) полностью расходуется за 1 час, при этом основным продуктом, как и в темновом процессе, является соль 5. Таким образом, разложение илида 1 при облучении и в темновых условиях происходит с промежуточным образованием радикальных интермедиатов (под радикальными интермедиатами имеются ввиду как нейтральные радикалы, так и катион-радикалы) и одинаковых конечных продуктов, но скорость их образования существенно выше при фотолизе.
Распад 1 в присутствии следов кислоты ускоряется и при фотолизе. Фотолиз 1 и дикатиона 6 должен приводить к их возбуждению в синглетные состоянию. TD DFT-моделирование оптического вертикального возбуждения илида 1 и дикатиона 6 в соответствующие синглетные состояния 1* и 6* в хлористом метилене показывает, что при довольно близких энергиях возбуждения 6 и 1 вероятность вертикального возбуждения 1 в первое синглетное состояние почти в два раза ниже по сравнению с таковой для 6 (табл. 1), и потому ожидаемо, что при облучении смеси 1 и 6 образование первого синглетно-возбужденного состояния 6 и его дальнейший распад будет более вероятным. Судя по величинам Eex и f (табл. 1), возбуждение 6 в синглетные состояния 2–4 по сравнению с аналогичным возбуждением 1 еще более вероятно.
Таблица 1.
TD-DFT-моделирование оптического вертикального возбуждения илида 1 и его протонированной формы 6 в хлористом метилене в соответствующие синглетные состояния*
| Синг-летное состояние | 1 | 6 | ||||
|---|---|---|---|---|---|---|
| λex, нм | Eex, эВ | f | λex, нм | Eex, эВ | f | |
| 1 | 355.25 | 3.4900 | 0.0038 | 363.71 | 3.4088 | 0.0067 |
| 2 | 316.45 | 3.9180 | 0.0159 | 354.65 | 3.4960 | 0.0094 |
| 3 | 293.94 | 4.2180 | 0.0861 | 338.58 | 3.6619 | 0.0166 |
| 4 | 292.09 | 4.2448 | 0.0029 | 330.69 | 3.7493 | 0.0944 |
| 5 | 276.50 | 4.4841 | 0.0136 | 290.32 | 4.2706 | 0.0012 |
Можно предположить, что, как и в случае темновых реакций (I) и (II), энергии Гиббса для превращения синглетов 6* в (PhI)+· и катион-радикал 10 будут заметно меньше энергий Гиббса для превращения синглетов 1* в (PhI)+· и 9. Это означает, что и фотолитическое разложение 1 в хлористом метилене должно ускоряться кислотами, что подтверждается экспериментальными данными. Вполне разумно ожидать, что значения ΔG298 для синглетно-возбужденных состояний 6* и 1* будут значительно меньше ΔG298 для распада 6 и 1 в основном состоянии, то есть распад 1 при его фотолизе должен происходить заметно быстрее. Для подтверждения этих предположений необходимо провести дополнительное квантово-химическое изучение 6* и 1* и термохимических параметров их распада, что представляет собой отдельную задачу.
Дальнейшие превращения радикальных интермедиатов (PhI)∙+, 9 и 10 происходят по реакциям (III)–(VI):
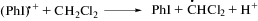
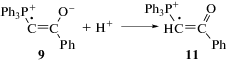

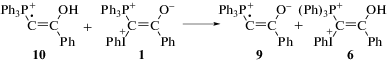
Результатом реакций (III)–(VI) является превращение промежуточных радикальных интермедиатов 9 и 10 в катион-радикал 11. Как показывают PBE0-расчеты с учетом сольватации хлористым метиленом, свободная энергия катион-радикала 11 при 25°С на 22.4 ккал моль–1 (94 кДж) ниже, чем таковая для 10. Другими словами, 11 на 22.4 ккал моль–1 по сравнению с 10 термодинамически более предпочтителен.
Катион-радикал 11 действительно относительно стабилен, его строение было предложено на основании квантово-химического расчета возможной структуры катион-радикала, регистрируемого методом ЭПР при фотолизе илида 1 [18]. В реакции (VII) этого радикала с растворителем образуется фосфониевая соль 5, основной продукт превращений илида [18], и радикал С•HCl2:
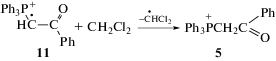
Автокатализ фотолиза илида 1 хлористым водородом может осуществляться, по-видимому, за счет фотолитических и темновых превращений радикала растворителя •СHCl2 (образовавшегося в реакциях (III) и (VII)) по реакциям (VIII)‒(X), в которых генерируется хлористый водород [15–17]:
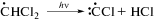
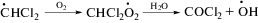
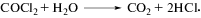
Избыток добавленной кислоты выводит илид из реакции, поскольку способствует сдвигу равновесия (схема 2) в сторону дальнейшего превращения реакционно-способного дикатиона 6 в соединения 7 и 8, которые не являются инициаторами радикальных реакций. Это связано с тем, что превращение 6 в 7 происходит в бимолекулярной реакции с анионом кислоты. Увеличение концентрации кислоты повышает концентрацию последнего и, соответственно, ускоряет эту реакцию. Это объясняет экспериментальное наблюдение, что при избытке кислоты не происходит образования целевых гетероциклов 3 и 4 в присутствии ацетиленов [13]. Подробно закономерности реакции смешанных фосфониево-иодониевых илидов с ацетиленами будут рассмотрены в следующих публикациях.
При использовании идеально чистого илида 1 триггером реакции теоретически должен быть распад самого илида, который запускает последовательность реакций (I)–(X). Однако нельзя исключить, что инициирование реакции при облучении и без него в основном обусловлено именно радикальным распадом дикатиона 6, а не исходного илида 1. Об этом свидетельствует зависимость начальной скорости фотолиза от степени очистки различных фосфониево-иодониевых илидов от следов HBF4, которая применяется для их получения. В синтезированном илиде 1 даже после очень тщательной отмывки эфиром от HBF4 могут содержаться следы протонированного илида, дикатиона 6, на котором происходит первичное инициирование за счет образования катион-радикалов (PhI)+• и 10. В реакции (III) катион-радикал (PhI)+• и в реакции (V) катион-радикал 10 генерируют новые протоны, которые способствуют появлению дикатиона 6, а далее процесс развивается в автокаталитическом режиме за счет образующегося из растворителя хлористого водорода в реакциях (III), (VIII), (X). Добавление основания (Et)3N в раствор илида 1 ингибирует его фотолитические и темновые реакции, что также подтверждает роль кислот в инициировании реакции.
ЗАКЛЮЧЕНИЕ
Экспериментальное спектрофотометрическое исследование взаимодействия бензоилзамещенного смешанного фосфониево-иодониевого илида 1 с трифторуксусной кислотой и теоретические термохимические расчеты показали, что фосфониево-иодониевый дикатион существенно более активен в распаде на катион-радикалы, чем исходный илид. Автокаталитический характер фотолиза фосфониево-иодониевого илида обусловлен образованием хлористого водорода в сопряженной радикальной реакции хлористого метилена. При взаимодействии хлористого водорода с исходным илидом образуется более реакционно-способный фосфониево-иодониевый дикатион 6, что приводит к увеличению скорости образования катион-радикалов, активных в дальнейших превращениях илида и одновременно индуцирующих радикальные реакции растворителя, приводящие к появлению HCl. Закономерности, выявленные для фотолитического разложения 1, по-видимому, приложимы и к разложению других смешанных фосфониево-иодониевых илидов.
Показано, что протонированная форма илида 1, образующаяся при его взаимодействии с кислотой, в присутствии избытка последней превращается в соединение 8, не участвующее в дальнейших реакциях илида. Предложен механизм взаимодействия кислот со смешанными илидами, охарактеризованы продукты взаимодействия и оценены кинетические параметры реакции превращения илида 1 в соединение 8.
Список литературы
Матвеева Е.Д., Подругина Т.А., Гришин Ю.К., Ткачев В.В., Жданкин В.В., Алдошин С.М., Зефиров Н.С. // Журн. Oрг. Химии. 2003. Т. 39. № 4. С. 572.
Матвеева Е.Д., Подругина Т.А., Гришин Ю.К., Павлова А.С., Зефиров Н.С. // Журн. Oрг. Химии. 2007. Т. 43. № 2. С. 209.
Матвеева Е.Д., Подругина Т.А., Павлова А.С., Миронов А В., Зефиров Н.С. // Изв. АН. Сер. хим. 2008. № 2. С. 391.
Dektar J.L., Hacker N.P. // J. Org. Chem. 1990. V. 55. P. 639.
Narewska J., Strzelczyk R., Podsiadły R. // J. Photochem. Photobiol. A. 2010. V. 212. P. 68.
Матвеева Е.Д., Подругина Т.А., Павлова А.С., Миронов А.В., Зефиров Н.С. // Изв. АН. Сер. хим. 2008. № 10. С. 2195.
Matveeva E.D., Podrugina T.A., Pavlova A.S., Mironov A.V., Gleiter R., Zefirov N.S. // Eur. J. Org. Chem. 2009. № 14. P. 2323.
Matveeva E.D., Podrugina T.A., Pavlova A.S., Mironov A.V., Borisenko A.A., Gleiter R., Zefirov N.S. // J. Org. Chem. 2009.V. 74. P. 9428.
Matveeva E.D., Podrugina T.A., Taranova M.A., Borisenko A.A., Mironov A.V., Gleiter R., Zefirov N.S. // J. Org. Chem. 2011. V. 76. P. 566.
Nekipelova T.D., Kuzmin V.A., Matveeva E.D., Gleiter R., Zefirov N.S. // J. Phys. Org. Chem. 2013. V. 26. P. 137.
Matveeva E.D., Vinogradov D.S., Podrugina T.A., Nekipelova T.D., Mironov A.V., Gleiter R., Zefirov N.S. // Eur. J. Org. Chem. 2015. P. 7324.
Некипелова Т.Д., Таранова М.А., Матвеева Е.Д., Подругина Т.А., Кузьмин В.А., Зефиров Н.С. // Докл. АН. 2012. Т. 447. № 3. С. 296.
Некипелова Т.Д., Таранова М.А., Матвеева Е.Д., Кузьмин В.А., Зефиров Н.С. // Кинетика и катализ. 2015. Т. 56. С. 411.
Levina I.I., Klimovich O.N., Bormotov D.S., Vinogradov D.S., Kononikhin A.S., Dement’eva O.V., Senchikhin I.N., Podrugina T.A., Nikolaev E.N., Kuzmin V.A., Nekipelova T.D. // J. Phys. Org. Chem. 2018. V. 31. P. e3844. doi 10.1002/poc.3844
Symons J.P., Yarwood A.J. // Trans. Faraday Soc. 1963. V. 59. P. 90.
Михеев Ю.А., Пустошный В.П., Топтыгин Д.Я. // Изв. АН СССР. Сер. хим. 1976. № 9. С. 2094.
Park H.-R., Jeong Y.-T., Kim M.-S., Woo H.-G., Ham H.-S. // Bull. Korean Chem. Soc. 1997. V. 18. P. 287.
Некипелова Т.Д., Каспаров В.В., Коварский А.Л., Воробьев А.Х., Подругина Т.А., Виноградов Д.С., Кузьмин В.А., Зефиров Н.С. // Докл. АН. 2017. Т. 474. № 6. С. 707.
Schmidt M.W., Baldridge K.K., Boatz J.A., Elbert S.T., Gordon M.S., Jensen J.H., Koseki S., Matsunaga N., Nguyen K.A., Su S., Windus T.L., Dupuis M., Montgomery J.A., Jr. // J. Comput. Chem. 1993. V. 14. P. 1347.
Adamo C., Barone V. // J. Chem. Phys. 1999. V. 110. P. 6158.
Schuchardt K.L., Didier B.T., Elsethagen T., Sun L., Gurumoorthi V., Chase J., Li J., Windus T.L. // J. Chem. Inf. Model. 2007. V. 47. P. 1045.
Mennucci B. // WIREs Comput. Mol. Sci. 2012. V. 2. P. 386.
Casida M.E., Huix-Rotllant M. // Annu. Rev. Phys. Chem. 2012. V. 63. P. 287.
Okuyama T., Takino T., Sato K., Ochiai M. // J. Am. Chem. Soc. 1998. V. 120. P. 2275.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ