

Кинетика и катализ, 2019, T. 60, № 2, стр. 193-205
Влияние способа введения бора на формирование и активность катализаторов гидроочистки
А. Н. Логинова 1, И. М. Круковский 1, *, Я. В. Морозова 1, А. Х. Купцов 1, В. В. Фадеев 1
1 ООО “Объединенный центр исследований и разработок”
119333 Москва, Ленинский просп., 55/1, стр. 2, Россия
* E-mail: KrukovskyIM@rn-rdc.ru
Поступила в редакцию 11.09.2018
После доработки 24.10.2018
Принята к публикации 04.12.2018
Аннотация
Представлены экспериментальные данные о влиянии способа введения бора и соединений Ni(Co) и Mo на физико-химические свойства и гидродесульфирующую активность катализаторов в гидроочистке прямогонной дизельной фракции. Установлена зависимость прочности гранул, их удельной поверхности, спектральных и микроструктурных характеристик от способа приготовления образцов катализатора, содержащих 0.7–0.8 мас. % B2O3.
ВВЕДЕНИЕ
Катализаторы гидрообессеривания дизельных топлив (ГО ДТ), содержащие сульфиды Ni, Co, Mo и W, применяются в промышленности свыше 80 лет [1, 2]. Однако до настоящего времени предпринимаются успешные попытки их совершенствования и предел в этом направлении еще не достигнут. Анализ литературы показывает, что одним из основных путей создания новых высокоэффективных катализаторов является оптимизация состава и структурных характеристик гранулированного носителя [3–6]. В качестве компонентов носителей наиболее часто используют оксиды алюминия, кремния, титана и циркония как в чистом виде, так и в составе смешанных оксидов [7–9], а также модифицирующие добавки галогенов, фосфора и бора [10–13].
Одними из первых процессов, где начали применять алюмоборатные катализаторы, были гидрокрекинг и изомеризация углеводородных фракций [14, 15]. Для процессов гидрогенизационного удаления серо- и азотсодержащих соединений из углеводородных фракций стали использовать гранулы Al2O3–B2O3 с нанесенными сульфидными компонентами [16, 17]. Введение бора в катализаторы гидродесульфуризации производится на стадии приготовления гранулированного носителя (добавление борной кислоты в массу гидроксидa алюминия [18, 19]), а также пропиткой прокаленного носителя борной кислотой или ее аммонийными солями [10, 11, 20–22]. Растворимость борной кислоты невелика при комнатной температуре и возрастает с ее повышением [23]. Поэтому для введения пропиткой более чем 1–2 мас. % B2O3 требуется нагрев раствора H3BO3 вплоть до температуры ~80°C в зависимости от количества растворяемой борной кислоты [15, 24]. После нанесения активных компонентов на гранулированный алюмоборатный носитель гранулы просушивают и прокаливают. Полученный катализатор переводят из оксидной формы в сульфидную под действием сероводорода или диметилдисульфида.
Борсодержащие катализаторы нашли использование в процессах гидроочистки широкого спектра сырья – от модельных смесей, содержащих тиофен [19, 20], до тяжелых масел [12, 18] и газойля каталитического крекинга [13]. При приготовлении борсодержащих катализаторов гидродесульфуризации, как правило, применяется введение борной кислоты на стадии пептизации гидроксида алюминия [25–27].
Анализ литературных данных о физико-химических свойствах борсодержащих катализаторов гидроочистки показывает, что соединения бора выступают в качестве модификаторов кислотности носителей [12, 18, 29, 30], влияют на взаимодействие носителя с нанесенными активными компонентами и, как следствие, на каталитическую активность.
В случае малой концентрации оксида бора (менее 1%), введенного пропиткой, достигается улучшенное распределение оксидов переходных металлов на поверхности носителя [29]. Добавка оксида бора уменьшает образование каталитически неактивного Co3O4 [16, 20] и снижает содержание Mo, Ni и Co в тетраэдрической координации с Al3+ [21]. Молибдат-ионы связаны с ионами B3+ более прочно, чем с Al3+ [18]. Это обеспечивает повышенную гидрирующую и десульфирующую активность катализаторов [15, 18, 29].
В большинстве случаев в литературе представлены данные о формировании и активности Co–Mo-содержащих катализаторов. Сравнение с Ni–Mo-катализаторами проводилось лишь в работе [21], где была отмечена бóльшая активность никельсодержащих образцов в удалении 2,6-диметилдибензотиофена и меньшая степень их закоксовывания. Кроме того, практически отсутствуют данные о влиянии способа введения бора в катализатор на его гидродесульфирующую активность. В настоящей работе мы постарались восполнить этот пробел.
Целью настоящего исследования является изучение влияния метода введения бора и соединений активных компонентов на физико-химические свойства и активность катализаторов гидродесульфуризации (ГДС) нефтяных фракций. Для этого были синтезированы образцы, содержащиe одинаковое количество B2O3, NiO и MoO3, но различающиeся способом приготовления.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтезированные и изученные в настоящей работе образцы катализатора ГО ДТ различались последовательностью внесения оксида бора и активных компонентов (табл. 1), но имели одинаковый состав (мас. %): 18–20 MoO3, 5–6 NiO(CoO), 0.7–0.8 B2O3, остальное – Al2O3. Синтез образцов предусматривал следующие стадии: получение гранулированного носителя и введение активных компонентов и модификатора. Каждая стадия завершалась сушкой по ступенчатой программе (выдержка по 2 ч при температуре 60, 80 и 110°C) и прокаливанием при 550°C.
Таблица 1.
Схема приготовления борсодержащих катализаторов и образцов сравнения
| Первая пропитка | |||
|---|---|---|---|
| прокаленный носитель | компоненты, вводимые пропиткой | высушенный образец | прокаленный образец |
| B2O3 + MoO3 + Al2O3 | (NH4)6Mo7O24 | MoO3/B2O3 + MoO3 + Al2O3 (60°C) | MoO3/B2O3 + MoO3 + Al2O3 |
| (NH4)6Mo7O24 + Ni(NO3)2 + H2O2 | NiO + MoO3/B2O3 + MoO3 + Al2O3 (60°C) | NiO + MoO3/B2O3 + MoO3 + Al2O3 | |
| MoO3 + Al2O3 | (NH4)6Mo7O24 + H3BO3 + Ni(NO3)2 + H2O2 | NiO + MoO3 + B2O3/MoO3 + Al2O3 (60°C) | NiO + MoO3 + B2O3/MoO3 + Al2O3 |
| (NH4)6Mo7O24 + H3BO3 | MoO3 + B2O3/MoO3 + Al2O3 (60°C) | MoO3 + B2O3/MoO3 + Al2O3 | |
| H3BO3 | B2O3/MoO3 + Al2O3 (60°C) | B2O3/MoO3 + Al2O3 | |
| (NH4)6Mo7O24 + Ni(NO3)2 + H2O2 | NiO + MoO3/MoO3 + Al2O3 (60°C) | NiO + MoO3/MoO3 + Al2O3 | |
| Al2O3 | H3BO3 | B2O3/Al2O3 (60°C) | B2O3/Al2O3 |
| Вторая пропитка | |||
| прокаленный образец | компоненты, вводимые пропиткой по влагоемкости | высушенный образец | прокаленный образец |
| MoO3/B2O3 + MoO3 + Al2O3 | Ni(NO3)2 | NiO/MoO3/B2O3 + MoO3 + Al2O3 (60°C) | NiO/MoO3/B2O3 + MoO3 + Al2O3 |
| MoO3 + B2O3/MoO3 + Al2O3 | Ni(NO3)2 | NiO/MoO3 + B2O3/MoO3 + Al2O3 (60°C) | NiO/MoO3 + B2O3/MoO3 + Al2O3 |
| B2O3/MoO3 + Al2O3 | (NH4)6Mo7O24 + Ni(NO3)2 + H2O2 | NiO + MoO3/B2O3/MoO3 + Al2O3 (60°C) | NiO + MoO3/B2O3/MoO3 + Al2O3 |
| (NH4)6Mo7O24 + Co(NO3)2 + H2O2 | CoO + MoO3/B2O3/MoO3 + Al2O3 (60°C) | CoO + MoO3/B2O3/MoO3 + Al2O3 | |
| B2O3/Al2O3 | (NH4)6Mo7O24 + Ni(NO3)2 + H2O2 | NiO + MoO3/B2O3/Al2O3 (60°C) | NiO + MoO3/B2O3/Al2O3 |
| Сульфидирование | |||
| прокаленный образец | сульфидированный образец | ||
| NiO/MoO3/B2O3 + MoO3 + Al2O3 | NiO/MoO3/B2O3 + MoO3 + Al2O3 (Sulf) | ||
| NiO + MoO3/B2O3 + MoO3 + Al2O3 | NiO + MoO3/B2O3 + MoO3 + Al2O3 (Sulf) | ||
| NiO + MoO3 + B2O3/MoO3 + Al2O3 | NiO + MoO3 + B2O3/MoO3 + Al2O3 (Sulf) | ||
| NiO/MoO3 + B2O3/MoO3 + Al2O3 | NiO/MoO3 + B2O3/MoO3 + Al2O3 (Sulf) | ||
| NiO + MoO3/B2O3/MoO3 + Al2O3 | NiO + MoO3/B2O3/MoO3 + Al2O3 (Sulf) | ||
| CoO + MoO3/B2O3/MoO3 + Al2O3 | CoO + MoO3/B2O3/MoO3 + Al2O3 (Sulf) | ||
| NiO + MoO3/MoO3 + Al2O3 | NiO + MoO3/MoO3 + Al2O3 (Sulf) | ||
Для получения гранулированного носителя MoO3 + Al2O3 порошок гидроксида алюминия марки Pural SB (“Sasol AG”) пептизировали раствором азотной кислоты, вводили в пластичную массу раствор парамолибдата аммония (ПМА) и пластификатор (триэтиленгликоль), после чего формовали продавливанием через круглую фильеру, сушили и прокаливали. Для получения носителя B2O3 + MoO3 + Al2O3 выполняли те же операции, только пептизацию производили раствором азотной и борной кислот. Содержание MoO3 в носителях MoO3 + Al2O3 либо B2O3 + MoO3 + + Al2O3 составляло 13 мас. %.
Бор вводили из раствора борной кислоты двумя способами: 1) на стадии приготовления носителя B2O3 + MoO3 + Al2O3 добавлением в пластичную массу перед ее формовкой и 2) пропиткой гранулированного прокаленного носителя MoO3 + + Al2O3. Оксид молибдена MoO3 вносили из раствора ПМА соэкструзией и пропиткой. Соотношение количества молибдена, вносимого на стадии приготовления носителя B2O3 + MoO3 + + Al2O3 и вводимого путем пропитки, составляло 11 : 9 и было подобрано таким образом, чтобы время устойчивости перекисного раствора (стабилизатора, позволяющего наносить пропиткой активные компоненты в высокой концентрации без существенного увеличения вязкости раствора) соответствовало минимальным требованиям по его хранению в условиях промышленного производства (не менее 12 ч при 20°C и 2 ч при 40°C). Повышенная температура в случае пропитки требуется для поддержания борной кислоты в растворенном состоянии. По истечении указанного времени на дне емкости с раствором начинал образовываться осадок. Добавление стабилизатора в раствор требовалось и при одновременном введении пропиткой соединений Ni (Со) и Mo.
Пропитку носителя растворами борной кислоты, нитрата никеля (кобальта) и ПМА выполняли в одну или две стадии. Было приготовлено две группы образцов. В образцы I группы бор, молибден и никель (кобальт) вводили путем двух пропиток по влагоемкости с промежуточным прокаливанием. Первой пропиткой вносили MoO3 и/или B2O3 (MoO3/B2O3 + MoO3 + Al2O3, MoO3 + + B2O3/MoO3 + Al2O3, B2O3/MoO3 + Al2O3,) второй пропиткой – NiO (СоО) и/или MoO3 в зависимости от состава обрацов, полученных на первой стадии (NiO/MoO3/B2O3 + MoO3 + Al2O3, NiO/MoO3 + + B2O3/MoO3 + Al2O3, NiO + MoO3/B2O3/MoO3 + + Al2O3 и CoO + MoO3/B2O3/MoO3 + Al2O3) (табл. 1).
Образцы II группы приготовлены одной пропиткой перекисным Ni–Mo- или В–Ni–Mo-раствором в зависимости от состава носителя. К ним относятся катализаторы NiO + MoO3/B2O3 + + MoO3 + Al2O3 и NiO + MoO3 + B2O3/MoO3 + + Al2O3. (табл. 1).
После стадий просушивания и прокаливания все введенные активные компоненты и модификатор находились в оксидной форме.
Часть образцов (в их обозначении указана температура обработки 60°C) проанализирована сразу после пропитки после высушивания гранул. Образцы, прошедшие последующую прокалку при 550°C, не содержат указания на температуру обработки.
Для образцов в оксидной форме определяли прочность гранул на раздавливание по ОСТ 153-39.2-020-2002 на тензиметре Hounsfield H5KS (“Tinius Olsen”, Англия). Структурные характеристики (удельную площадь поверхности по БЭT (SБЭТ), объем пор (Vпор) и их распределение по диаметру по методу BJH) находили методом низкотемпературной азотной порометрии на приборе ASAP 2020 (“Micromeritics”, США).
Кислотность поверхности порошкообразных образцов изучали методом термопрограммированной десорбции аммиака. Использовали следующее отнесение силы кислотных центров по температуре десорбции (T, °C): T < 200 – слабые; 200 ≤ T < 300 – средние; T ≥ 300 – сильные кислотные центры [31].
Содержание молибдена, никеля и бора определяли на атомно-эмиссионном анализаторе iCAP-6500 Duo (“Thermo Fisher Scientific”, США) с плазменным источником излучения (ИСП-АЭС). Анализируемый образец переводили в растворимое состояние сплавлением с тетраборатом лития с последующим растворением в разбавленной азотной кислоте.
Спектры комбинационного рассеяния (КР) порошкообразных образцов регистрировали на спектрометре DXR (“Thermo-Nicolet”, США). Возбуждение на линии 532 нм с мощностью 10 мВт, голографическая решетка высокого разрешения, входная щель 25 или 50 мкм (спектральные разрешения 1.6–2.0 и 3.2–3.9 см–1 соответственно). Размер анализируемого участка ~2.1 мкм при сфокусированном луче лазера. Длительность накопления сигнала – 1000 с в восьми повторах с усреднением полученных спектров.
Перед экспериментами для определения активности синтезированных катализаторов образцы переводили из оксидной формы в сульфидную. Гранулы образца объемом 20 см3 смачивали 10 мл смеси диметилдисульфида и петролейного эфира, взятых в объемном отношении 1 : 1. Далее переносили в трубчатый кварцевый реактор и нагревали в токе водорода (6 л/ч) по следующей программе:
− нагрев до 300°C со скоростью 150°C/ч;
− выдержка при 300°C в течение 15 мин.
После сульфидирования испытываемый образец (гранулы диаметром 1 мм, плотная засыпка) помещали между двумя слоями инертных керамических шариков в вертикально расположенный стальной трубчатый реактор. Каталитические испытания проводили на проточной установке в токе водорода при температуре 380°C, давлении 5.0 МПа, объемной скорости подачи сырья 1.0 ч–1, объемном отношении водород : сырье при нормальных условиях 350 л/л. Сырье, подвергаемое гидроочистке на борсодержащих катализаторах, представляет собой смесь прямогонных дизельных фракций с установки АВТ-11 АО “Новокуйбышевский НПЗ”. Характеристики сырья представлены в табл. 2.
Таблица 2.
Физико-химические свойства сырья, подвергаемого гидроочистке на лабораторной проточной установке
| Наименование показателя | Метод определения | Единица измерения | Значение показателя |
|---|---|---|---|
| Фракционный состав: | EN ISO 3405 | °С | |
| − начало кипения | 186.0 | ||
| − конец кипения | 357.0 | ||
| Содержание серы | ASTM D4294 | мас. % | 1.03 |
| Содержание ароматических углеводородов: − моноароматические − диароматические − три + ароматические − полициклические (сумма ди- и три + ароматических) |
IP 391/01 | мас. % |
18.5 7.4 0.8 8.2 |
| Плотность при 20°C | ASTM D4052 | кг/м3 | 837 |
| Температура вспышки в закрытом тигле | ГОСТ 6356 | °С | 66 |
Программа испытания представлена в табл. 3. По достижении заданной температуры начинали подачу сырья и отсчет времени накопления проб. Длительность накопления каждой пробы составляла 8 ч. По окончании накопления пробы проводили ее отбор из сепаратора.
Таблица 3.
Условия испытания образцов катализаторов в гидроочистке дизельной фракции
| Номер пробы | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| Температура, °C | 320 | 320 | 340 | 360 | 380 | 380 | 380 | 380 | 380 | 380 | 380 | 380 |
Длительность испытания каждого образца составила 80–96 ч. Пробы очищали от образовавшегося в ходе реакции сероводорода путем обработки 10%-ным раствором NaOH, после чего сушили над гранулами CaCl2. В каждой отобранной пробе находили количество остаточной общей серы. В случае высокой концентрации серы определение производили методом рентгеновской флуоресценции по ASTM D 4294 на приборе Спектроскан SL (НПО “Спектрон”, Россия); при ее содержании менее 50 ppm применяли метод окислительной микрокулонометрии, используя прибор Thermo Euroglas ECS 3000 (“Thermo Fisher Scientific”, США). Жидкую пробу инжектировали в поток газа, состоящий из 80% кислорода и 20% инертного газа (аргона или гелия). При сгорании сера окислялась до диоксида, который поступал в титровальную ячейку, заполненную электролитом (водный раствор уксусной кислоты с добавлением иодида калия и азида натрия). Диоксид серы титровали трииодид-ионами, генерируемыми в ячейке под действием тока [32].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Носители
Механическая прочность гранул борсодержащих образцов. Результаты тестирования гранул катализаторов на механическую прочность в зависимости от способа внесения оксида бора представлены в табл. 4. Прочность изученных образцов определялась составом носителя (MoO3 + + Al2O3 либо B2O3 + MoO3 + Al2O3). При нанесении B2O3 на гранулированный носитель MoO3 + + Al2O3 пропиткой получались высокопрочные образцы. Напротив, введение оксида бора в пластичную массу на стадии приготовления B2O3 + + MoO3 + Al2O3 почти в три раза снижало прочность сформованных из нее и прокаленных гранул, и для ее доведения до значения на уровне 4.0 кг/мм (как в случае гранул B2O3/MoO3 + + Al2O3) требовалось добавление дополнительного количества азотной кислоты. По всей видимости, необходимость повышенного содержания HNO3 при приготовлении носителя является следствием протекания реакции между слабоамфотерным H3BO3 и азотной кислотой: H3BO3 + + HNO3 ⇌ B(OH)2NO3 + H2O [33]. При дальнейшем увеличении содержания пептизирующего агента прочность гранул B2O3 + MoO3 + Al2O3 проходила через максимум, после чего наблюдалось ее снижение (табл. 4).
Таблица 4.
Прочность гранул образцов борсодержащего катализатора
| Способ внесения B2O3 | Концентрация HNO3 в пептизирующем растворе, % | Прочность, кг/мм |
|---|---|---|
| Пропитка | 8.2 | 4.1 |
| Введение в массу носителя |
8.2 | 1.5 |
| 9.1 | 2.1 | |
| 9.7 | 4.3 | |
| 10.2 | 6.3 | |
| 11.3 | 4.0 |
Кислотность. Данные о концентрации кислотных центров различной силы, полученные методом ТПД аммиака, представлены в табл. 5. Гранулированный оксид алюминия, использованный для приготовления образцов сравнения, содержит слабые, средние и сильные кислотные центры в соотношении 1 : 2 : 1. Пропитка его борной кислотой с последующим прокаливанием (B2O3/Al2O3) приводит к некоторому снижению кислотности, главным образом, за счет сокращения концентрации сильных кислотных центров.
Таблица 5.
Данные ТПД аммиака по концентрации (мкмоль/г) кислотных центров различной силы в образцах
| Образец | Слабые центры |
Средние центры |
Сильные центры |
Всего |
|---|---|---|---|---|
| Al2O3 | 139 | 301 | 152 | 592 |
| B2O3/Al2O3 | 129 | 286 | 113 | 528 |
| MoO3 + Al2O3 | 191 | 341 | 198 | 730 |
| B2O3/MoO3 + + Al2O3 | 182 | 343 | 170 | 695 |
| B2O3 + MoO3 + + Al2O3 | 416 | 195 | 100 | 711 |
В результате введения оксида молибдена в состав носителя (MoO3 + Al2O3) увеличивается общее число кислотных центров примерно на треть, что согласуется с литературными данными [34]. Пропитка его борной кислотой с последующим прокаливанием (B2O3/MoO3 + Al2O3) понижает кислотность. Самые заметные изменения кислотности происходят при внесении бора на стадии приготовления носителя B2O3 + MoO3 + + Al2O3 добавлением в пластичную массу в ходе пептизации гидроксида алюминия. Сформованный и прокаленный носитель B2O3 + MoO3 + + Al2O3 отличается наибольшей концентрацией слабых центров (свыше 400 мкмоль/г) и наименьшей – сильных центров (100 мкмоль/г).
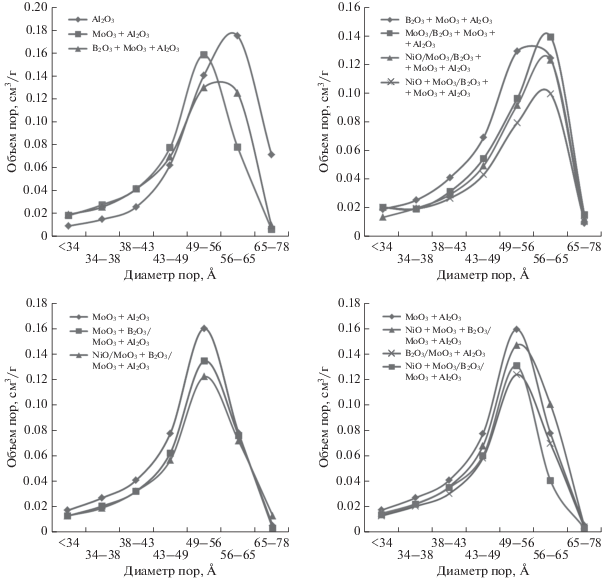
Структурные характеристики. Данные азотной порометрии представлены в табл. 6 и на рис. 1. Исходные гранулированные носители (Al2O3, MoO3 + Al2O3 и B2O3 + MoO3 + Al2O3) характеризуются объемом пор 0.4–0.5 см3/г. Удельная площадь поверхности чистого оксида алюминия, полученного из порошка Pural SB, составляет 259 м2/г. При введении соэкструзией соединений молибдена и бора (MoO3 + Al2O3 и B2O3 + MoO3 + + Al2O3) SБЭТ увеличивается. Противоположная зависимость наблюдается для распределения пор по диаметру.
Таблица 6.
Данные азотной порометрии носителей и катализаторов
| Образец | SБЭТ, м2/г | Vпор, см3/г |
|---|---|---|
| Al2O3 | 259 | 0.49 |
| MoO3 + Al2O3 | 308 | 0.43 |
| B2O3 + MoO3 + Al2O3 | 320 | 0.45 |
| MoO3/B2O3 + MoO3 + Al2O3 | 267 | 0.40 |
| NiO/MoO3/B2O3 + MoO3 + Al2O3 | 244 | 0.36 |
| NiO + MoO3/B2O3 + MoO3 + Al2O3 | 207 | 0.31 |
| NiO + MoO3 + B2O3/MoO3 + Al2O3 | 230 | 0.32 |
| MoO3 + B2O3/MoO3 + Al2O3 | 250 | 0.36 |
| NiO/MoO3 + B2O3/MoO3 + Al2O3 | 235 | 0.34 |
| B2O3/MoO3 + Al2O3 | 278 | 0.41 |
| NiO + MoO3/B2O3/MoO3 + Al2O3 | 228 | 0.33 |
Данные спектроскопии комбинационного рассеяния света. Спектр КР Mo-содержащих носителей характеризуются интенсивной широкой полосой с максимумом при 950 см–1, соответствующей валентным колебаниям терминальных Mo=O-групп, и плечом около 858 см–1 , относящимся к симметричным валентным колебаниям Мо–О–Мо [35, 36]. Фаза оксида молибдена (частоты 667, 820 и 996 см–1) отсутствует.
Образцы после пропитки
Спектры КР непрокаленных образцов I группы после первой пропитки представлены на рис. 2; спектры КР непрокаленных образцов II группы и непрокаленных образцов I группы после второй пропитки представлены на рис. 3.
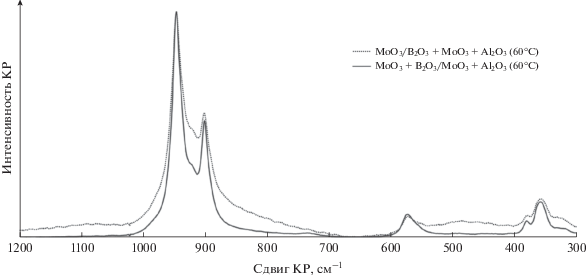
Рис. 3.
Спектры КР непрокаленных образцов II группы и непрокаленных образцов I группы после второй пропитки.
По сравнению с носителями MoO3 + Al2O3 и B2O3 + MoO3 + Al2O3 в спектре образцов I группы после пропитки раствором ПМА появляются интенсивные полосы в области 945, 918 (плечо), 901 (валентные колебания Mo=O) и 570 см–1 (колебания Al–O–Mo), которые соответствуют парамолибдату аммония и продуктам его хемосорбции – анионам ${\text{MoO}}_{4}^{ - }$ и [Al(OH)6Mo6O18]3– [37]. Введение борной кислоты в пропиточный раствор изменяет его кислотность, что способствует увеличению доли анионов ${\text{MoO}}_{4}^{ - }$ (901 см–1) в образце MoO3 + B2O3/MoO3 + Al2O3 (60°C) по сравнению с аналогичным по составу MoO3/B2O3 + MoO3 + + Al2O3 (60°C).
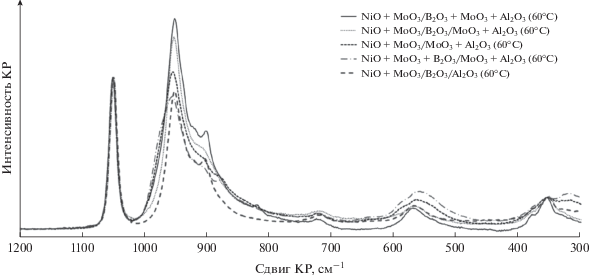
Высушенные после первой пропитки образцы I группы прокаливали, после чего пропитывали раствором нитрата никеля. После второй пропитки гранулы имели снаружи светло-зеленую окраску, а на поперечном срезе – темно-зеленую. Разница в окраске обусловлена образованием слоя MoO3 при разложении малорастворимой соли (NH4)3[Al(OH)6Mo6O18] на поверхности гранул. Спектры образцов I группы после второй пропитки приведены на рис. 3.
Использование пероксида водорода позволяет существенно повысить растворимость парамолибдата аммония и увеличить время стабильности совместного никель-молибденового раствора, использованного для приготовления образцов II группы. После пропитки гранулы высушенных образцов имели зеленоватую окраску различных оттенков. В отличие от образцов I группы их окраска была более яркая и однородная как на внешней, так и на внутренней поверхности гранул при том же содержании Ni2+. Более равномерное распределение активных компонентов при их одновременном введении было отмечено также для Co–Mo-содержащих катализаторов [38]. Гранулы борсодержащих образцов NiO + + MoO3 + B2O3/MoO3 + Al2O3 (60°C) и NiO + + MoO3/B2O3/MoO3 + Al2O3 (60°C) помимо основного зеленого цвета имели желтоватый оттенок, близкий к цвету пропиточного перекисного раствора.
В спектрах КР образцов II группы появляется интенсивная полоса валентных колебаний нитрогруппы в области 1050 см–1 (рис. 3). Так как содержание нитрата никеля было одинаково для всех образцов, представленных на рис. 3, эту полосу использовали для нормировки интенсивности. Спектр NiO + MoO3/B2O3/Al2O3 (60°C) наиболее простой в этой серии, поскольку образец не содержит соединений молибдена в носителе. Другие спектры представляют собой наложение полос колебаний Mo=O и Mo–O–Al носителя и адсорбированных форм парамолибдата аммония и нитрата никеля.
Наблюдается явная зависимость относительной интенсивности полосы при 950 см-1 от природы носителя. Наибольшая интенсивность, и, соответственно, более сильное взаимодействие раствора с носителем отмечено для образца NiO + + MoO3/B2O3 + MoO3 + Al2O3 (60°C), в который бор был введен в носитель соэкструзией, что привело к преобладанию в нем слабых кислотных центров. Минимальная интенсивность пика при 950 см–1 характерна для NiO + MoO3/B2O3/Al2O3 (60°C) и NiO + MoO3 + B2O3/MoO3 + Al2O3 (60°C). Интересно отметить, что последние два образца существенно различаются содержанием молибдена (соответственно, 9 и 20% в прокаленных образцах в пересчете на MoO3). В образце с меньшим содержанием молибдена перекисные соединения отсутствуют, что согласуется с литературными данными [39, 40]. При использовании молибденсодержащих носителей и, особенно, при введении H3BO3 в пропиточный раствор происходит асимметричное высокочастотное уширение пиков с максимумом при 950 и 550 см–1, что может указывать на присутствие в NiO + MoO3 + B2O3/MoO3 + Al2O3 (60°C) перекисных соединений молибдена. Эти особенности спектров согласуются со сделанными ранее наблюдениями об изменении цвета гранул изучаемых образцов. Плечо в области 880 см–1 в спектре этого образца, по всей видимости, отвечает валентным колебаниям В–О свободной борной кислоты [41].
Полоса при 910 см–1 наиболее выражена для образцов II группы, приготовленных с применением наименее кислотных носителей – NiO + + MoO3/B2O3/Al2O3 (60°C) и NiO + MoO3/B2O3 + + MoO3 + Al2O3 (60°C), однако ее относительная интенсивность заметно меньше, чем в случае пропитки молибденовым раствором (образцы MoO3 + B2O3/MoO3 + Al2O3(60°C) и MoO3/B2O3 + + MoO3 + Al2O3 (60°C)) (рис. 2).
Прокаленные образцы
Данные спектроскопии комбинационного рассеяния света. На рис. 4 представлены спектры КР прокаленных при 550°C образцов. Основной особенностью прокаленных образцов является наличие фазы оксида молибдена, имеющей характеристические пики с максимумами при 667, 820 и 995 см–1. Наблюдается корреляция количества MoO3 с интенсивностью полос ${\text{MoO}}_{4}^{ - }$ и [Al(OH)6Mo6O18]3– в непрокаленных образцах (рис. 1 и 2). Помимо широких полос с максимумами при 960 и 860 см–1 (колебания Mo=O и Mo–O–Mo соответственно) присутствует линия с частотой 1026 см–1, характерной для молибдата алюминия [35]. Дополнительная дублетная структура около 962 и 945 см–1 в спектре КР образца NiO + MoO3/B2O3 + MoO3 + + Al2O3 отвечает биметаллическим оксидам α,β-NiMoO4 [38, 40]. При совместной пропитке Ni–Mo-раствором получаются образцы, спектральные характеристики которых (кроме интенсивности полос) во многом совпадают с исходным носителем состава 13% MoO3 + Al2O3, что указывает на близость состояний Mo, введенного пропиткой и соэкструзией. Согласно литературным данным [42] катализаторы гидроочистки, полученные соэкструзией гидроксида алюминия с парамолибдатом аммония и пропиткой гранул оксида алюминия раствором ПМА имеют близкую активность.
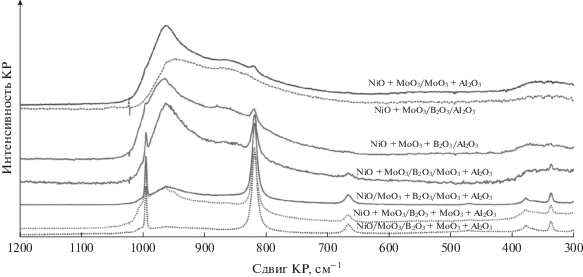
Отметим, что в настоящей работе количество MoO3, вводимого пропиткой, составляет 9%, что позволило избежать интенсивного образования фазы Al2(MoO4)3. По данным [43] образование этого каталитически малоактивного соединения молибдена преимущественно происходит при нанесении пропиткой свыше 13 мас. % MoO3.
Структурные характеристики. В зависимости от вводимых на стадии замеса модификаторов SБЭТ и Vпор носителя могут существенно меняться (табл. 6). В ходе последующего нанесения NiO и MoO3 площадь поверхности и объем пор постепенно снижались, достигая значений 207–244 м2/г и 0.31–0.36 см3/г соответственно. Средний диаметр пор, определенный по кривой десорбции, составлял 50–60 Å для всех изученных образцов. Характер распределения пор по диаметру в прокаленных образцах определяется составом носителя (рис. 1). Наименьшей SБЭТ и объемом пор обладал образец NiO + MoO3/B2O3–MoO3–Al2O3, приготовленный совместной пропиткой Mo–Ni-раствором борсодержащего носителя, который был синтезирован введением борной кислоты на стадии пептизации. При раздельном нанесении Mo и Ni на тот же носитель (образец NiO/MoO3/B2O3–MoO3–Al2O3) площадь поверхности оказалась больше на ~17%.
При одновременном введении пропиткой соединений бора, никеля и молибдена (образец NiO + + MoO3 + B2O3/MoO3–Al2O3) площадь поверхности оказывалась ниже, чем при их последовательном нанесении (NiO/MoO3 + B2O3/MoO3–Al2O3 и NiO + + MoO3/B2O3/MoO3–Al2O3), что может указывать на большее заполнение пор в первом случае.
Образцы в сульфидной форме
Данные спектроскопии комбинационного рассеяния света. Выдержка прокаленных образцов в токе водорода, содержащего пары диметилдисульфида, существенно меняет вид их спектров комбинационного рассеяния (рис. 5). Пики MoO3 (820 и 995 см–1) имеют меньшую интенсивность, чем до сульфидирования, а для ряда сульфидных образцов вообще отсутствуют. Широкая асимметричная полоса, отвечающая валентным колебаниям Mo=O и Mo–O–Мо (максимумы при 960 и 860 см–1 на рис. 4) изменяет форму и приобретает максимум в области 830 см–1, соответствующий различным формам валентных колебаний О–Мо=S в полиоксисульфидах [44]. В спектрах образцов NiO + MoO3 + B2O3/MoO3 + + Al2O3(sulf) и NiO + MoO3/B2O3/MoO3 + Al2O3(sulf) появляется небольшой пик при 410 см–1 (следы слоистого дисульфида молибдена) [44].
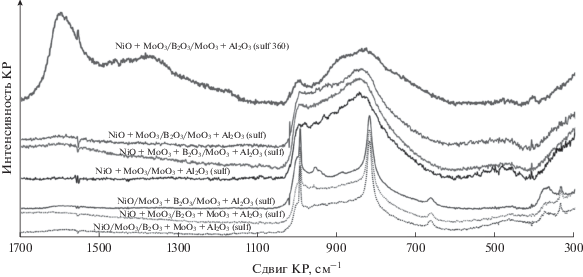
Увеличение продолжительности и температуры обработки в парах диметилдисульфида (2 ч при 360°C) повышает степень сульфидирования и приводит к частичному зауглероживанию, что видно на примере образца NiO + MoO3/B2O3/MoO3 + + Al2O3(sulf 360). Полосы с максимумами при 1600 и 1370 см–1 характерны для графитизированной (sp2-гибридизация) и неупорядоченной (sp3-гибридизация) форм углерода соответственно. Их относительно высокая интенсивность обусловлена бóльшим сечением рассеяния по сравнению с оксисульфидными структурами на поверхности катализатора, но действительное содержание углерода по данным термогравиметрического анализа составляет ~1 мас. %.
Таким образом, исследование синтезированных образцов сульфидированных и несульфидированных борсодержащих алюмоникельмолибденовых катализаторов методом КР-спектроскопии показало, что в ходе сульфидирования происходит значительное изменение состава исследуемых образцов с частичным переходом оксидов внесенных молибдена и никеля в сульфиды.
Каталитическая активность. После сульфидирования образцы были испытаны в процессе гидроочистки прямогонной дизельной фракции, содержащей 1.0 мас. % серы. Физико-химические свойства сырья и программа испытания указаны в табл. 2 и табл. 3 соответственно. На рис. 6 представлены данные по содержанию остаточной серы в стабильном гидрогенизате, полученном при температуре 380°C, давлении 5.0 МПа и объемной скорости подачи сырья 1.0 ч–1.
Рис. 6.
Влияние способа введения бора в состав катализатора на содержание остаточной серы в пробах стабильного гидрогенизата. Условия реакции: 380°C, 5.0 МПа, объемная скорость подачи сырья – 1.0 ч–1, содержание серы в сырье – 1.0%.
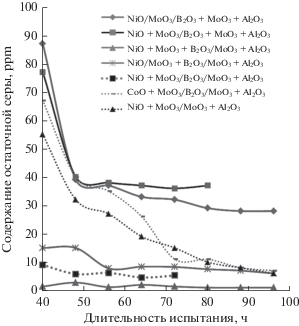
Активность изученных образцов постепенно растет со временем, достигая определенного предела, что может указывать на неполное сульфидирование катализатора на момент начала проведения процесса гидроочистки. Наблюдается явная зависимость десульфирующей способности катализатора от способа введения активных компонентов. Наибольшей активностью обладал образец NiO + MoO3 + B2O3/MoO3–Al2O3, полученный одновременным внесением соединений бора, никеля и молибдена в носитель MoO3–Al2O3. Он характеризуется малым временем достижением максимальной активности (менее 40 ч) и высокой стабильностью работы с получением дизельной фракции с ультранизким содержанием серы (1–2 ppm). Несколько меньшую активность проявляли образцы NiO/MoO3 + + B2O3/MoO3–Al2O3 и NiO + MoO3/B2O3/MoO3–Al2O3, приготовленные двухстадийной пропиткой, причем катализатор, в который бор вводили первой пропиткой, был активнее. Также для этих образцов требовалось большее время для выхода в режим максимальной активности (на ~20 ч) по сравнению с образцом NiO + MoO3 + B2O3/MoO3–Al2O3, синтезированным методом одностадийной совместной пропитки. Аналогичный по составу и способу приготовления кобальтсодержащий катализатор CoO + MoO3/B2O3/MoO3–Al2O3 оказался менее активен, чем Ni-содержащий образец. Кроме того, для Co–Mo-катализатора время приработки для получения гидрогенизата с содержанием остаточной серы менее 10 ppm составляло ~80 ч. Причины различий в активности Со–Мо- и Ni–Mo-катализаторов сходного состава можно объяснить тем, что содержание B2O3 (0.8 мас. %) является необходимым и достаточным для Ni–Mo-катализатора и избыточным для Со–Мо-образца [45–47].
Введение бора в состав носителя на стадии пептизации приводит к образованию малоактивных в реакциях гидрогенолиза серосодержащих соединений образцов NiO/MoO3/B2O3–MoO3–Al2O3 и NiO + MoO3/B2O3–MoO3–Al2O3. В течение всего периода испытания эти катализаторы показывали невысокую активность в реакциях десульфуризации. Несмотря на близкие спектральные характеристики (наличие интенсивных пиков валентных колебаний MoO3, рис. 5), образец NiO/MoO3 + B2O3/MoO3–Al2O3 оказался значительно более активным. После 50 ч испытания он вышел на уровень постоянной активности – 7–8 ppm остаточной серы в продукте.
Интересно отметить, что катализатор сравнения NiO + MoO3/MoO3–Al2O3, не содержащий добавки B2O3, занял промежуточное положение по активности между образцами, полученными добавлением бора на стадии пептизации в носитель, и образцами, в которых B2O3 был внесен пропиткой. Он характеризуется длительным выходом на уровень максимальной активности (порядка 90 ч) и по этому показателю близок к кобальтсодержащему образцу CoO + MoO3/B2O3/MoO3–Al2O3. Сопоставление перечисленных фактов указывает на то, что введение бора пропиткой (как совместным, так и последовательным нанесением компонентов) ускоряет выход катализаторов гидроочистки в режим наибольшей активности. Причиной этого, по-видимому, является подвижность ионов бора, что ускоряет перестройку внесенных в состав катализатора компонентов, расположенных на поверхности катализатора, из оксидной формы в сульфидную под действием серосодержащих соединений за счет ослабленного взаимодействия активной фазы с носителем [48, 49], а также изменяет электронные свойства самих сульфидных центров [7, 14, 50].
ЗАКЛЮЧЕНИЕ
В настоящей работе было исследовано влияние способа введения 0.7–0.8 мас. % оксида бора на физико-химические свойства и активность Ni(Co)–Mo-катализаторов в гидроочистке прямогонной дизельной фракции. На основании полученных результатов можно сделать следующие выводы:
1. Влияние на кислотность носителя. При введении H3BO3 на стадии пептизации в молибденсодержащий носитель происходит резкое увеличение числа слабых кислотных центров. Пропитка молибденсодержащего носителя борной кислотой заметного воздействия на кислотность не оказывает.
2. Влияние на фазовый состав непрокаленных образцов. Введение H3BO3 соэкструзией усиливает взаимодействие носителя с пропиточным раствором, а введение пропиткой – ослабляет. Взаимодействие носителя с раствором приводит к образованию малорастворимых соединений молибдена, расположенных преимущественно на внешней поверхности гранул. Совместное введение Ni2+ и Mo6+ обеспечивает более однородную пропитку по сечению гранулы.
3. Влияние на образование сульфидной фазы и каталитическую активность. Сульфидирование образцов, полученных пропиткой носителя раствором борной кислоты, а также полученных при введении ее в пропиточный раствор, протекает более глубоко. Такие образцы характеризуются самым быстрым выходом на уровень максимальной активности в десульфуризации. По всей видимости, наличие оксида бора, введенного пропиткой носителя, способствует ускоренному превращению оксидной фазы в сульфидную. Внесение такого же количества B2O3 (0.8–1.0 мас. %) путем соэкструзии в носитель, напротив, замедляет сульфидирование и снижает активность катализатора. Образец сравнения, не содержащий B2O3, занимает промежуточное положение по скорости выхода на максимальную активность. В целом, введение бора пропиткой (как совместным, так и последовательным нанесением компонентов) ускоряет выход катализаторов гидроочистки в режим наибольшей активности.
На основе совокупности представленных данных о превращениях, происходящих при формировании катализатора, сделано предположение о том, что повышенная активность борсодержащих алюмоникельмолибденовых катализаторов определяется способом внесения бора и связана с подвижностью анионов борной кислоты при ее введении пропиткой. Борная кислота ускоряет перестройку поверхности катализатора под действием серосодержащих соединений из оксидной формы в сульфидную, а также изменяет электронные свойства самих сульфидных центров.
Список литературы
Ландау М.В., Нефедов Б.К., Алексеенко Л.Н. Переработка нефти. М.: ЦНИИТЭнефтехим, 1985. 81 с.
Topsøe H., Clausen B.S., Massoth F.E. // Catalysis: Science and Technology. Berlin, Springer-Verlag. 1996. V. 11. 312 p.
Breysse M., Portefaix J.L., Vrinat M. // Catal. Today. 1991. V. 10. P. 489.
Dhar G.M., Massoth F.E., Shabtai J. // J. Catal. 1984. V. 85. P. 44.
Breysse M., Afanasiev P., Geantet C., Vrinat M. // Catal. Today. 2003. V. 86. P. 5.
Michaud P., Lemberton J.L., Perot G. // Appl. Catal. A: General. 1998. V. 169. P. 343.
Dhar G.M., Srinivas B.N., Rana M.S., Kumar M., Maity S.K. // Catal. Today. 2003. V. 86. P. 45.
Hanafi S.A., El-Syed H.A., Soltan E.-S.A. // Energy Sources. Part A. 2008. V. 30. P. 698.
Rayoa P., Ancheyta J., Ramirez J., Gutierrez-Alejandre A. // Catal. Today. 2004. V. 98. P. 171.
Пaт. 2569682 PФ, 2015.
Никульшин П.А., Томина Н.Н., Ишутенко Д.И., Пимерзин А.А. // Изв. ВУЗов. Химия и хим. технология. 2008. Т. 51. Вып. 9. С. 51.
Ferdous D., Dalai A.K., Adjaye J. // Appl. Catal. A: General. 2004. V. 260. P. 137.
Lecrenay E., Sakanishi K., Mochida I., Suzuka T. // Appl. Catal. A: General. 1998. V. 175. P. 237.
Izumi Y., Shiba T. // Bull. Chem. Soc. Japan. 1964. V. 37. P. 1797.
Giraldo S.A., Centeno A. // Catal. Today. 2008. V. 133–135. P. 255.
Houalla M., Delmon B. // Appl. Catal. 1981. V. 1. P. 285.
Stranick M.A., Houalla M., Hercules D.M. // J. Catal. 1987. V. 104. P. 396.
Chen Y.-W., Tsai M.-C. // Catal. Today. 1999. V. 50. P. 57.
Dumeignil F., Sato K., Imamura M., Matsubayashi N., Payen E., Shimada H. // Appl. Catal. A: General. 2006. V. 315. P. 18.
Saih Y., Segawa K. // Appl. Catal. A: General. 2009. V. 353. P. 258.
Torres-Mancera P., Ramirez J., Cuevas R., Gutierrez-Alejandre A., Murrieta F., Luna R. // Catal. Today. 2005. V. 107–108. P. 551.
Пaт. 2620089 PФ, 2017.
Blasdale W., Slansky C.M. // J. Am. Chem. Soc. 1939. V. 61. P. 917.
Карпова Т.Р., Булучевский Е.А., Лавренов А.В., Леонтьева Н.Н., Тренихин М.В., Гуляева Т.И., Талзи В.П. // Химия в интересах устойчивого развития. 2013. Т. 21. С. 61.
Пaт. 2478428 PФ, 2013.
Пaт. 2286846 PФ, 2006.
Пaт. 2197323 PФ, 2003.
Ramirez J., Castillo P., Cedeno L., Cuevas R., Castillo M., Palacios J.M., Agudo A.L. // Appl. Catal. A: General. 1995. V. 132. P. 317.
Rashidi F., Sasaki T., Rashidi A.M., Kharat A.N., Jozani K.J. // J. Catal. 2013. V. 299. P. 321.
Lewandowski M., Sarbak Z. // Fuel. 2000. V. 79. № 5. P. 487.
Ющенко В.В. // Журн. физ. химии. 1997. Т. 71. № 4. С. 628.
Новиков Е.А. // Мир нефтепродуктов. 2008. № 5. С. 1.
Карапетьянц М.Х., Дракин С.И. // Общая и неорганическая химия. М.: Химия, 1981, С. 330.
Петров И.Я., Трясунов Б.Г. // Вестник КузГТУ. 2007. № 2. С. 69.
Payen E., Kasztelan S., Grimblot J., Bonnelle J.P. // J. Raman Spectroscopy. V. 17 (1986). P. 233.
Batista A.T. / Innovative preparations of heterogeneous catalysts for the production of (bio)fuels. Thesis Master of Science Degree. Universidade Tecnica de Lisboa, 2016. P. 37.
Carrier X., Lambert J.-F., Kuba S., Knözinger H., Che M. // J. Mol. Struct. 2003. V. 656. P. 231.
Bergwerff J.A., Visser T., Weckhuysen B.M. // Catal. Today. 2008. V. 130. P. 117.
Campbell N.J., Dengel A.C., Edwards C.J., Griffith W.P. // J. Chem. Soc. Dalton Trans. 1989. V. 1203. P. 1203.
Rochet A., Baubet B., Moizan V., Devers E., Hugon A., Pichon C., Payen E., Briois V. // J. Phys. Chem. C. 2015. V. 119. P. 23928.
Demetriou A., Pashalidis I., Nicolaides A.V., Kumke M.U. // Desalination and Water Treatment. 2013. V. 51. P. 6130.
Luck F., Viez F. // Stud. Surf. Sci. Catal. 1989. V. 48. P. 611.
Петров И.Я., Трясунов Б.Г. // Вестник КузГТУ. 2012. № 4. С. 93.
Schrader G.L., Cheng C.P. // J. Catal. 1983. V. 80. P. 369.
Usman T., Kubota I., Hiromitsu Y., Okamoto Y. // J. Catal. 2007. V. 247. P. 78.
Kubota T., Rinaldi N., Okumura K., Honma T., Hirayama S., Okamoto Y. // Appl. Catal. A: General. 2010. V. 373. P. 214.
Li D., Sato T., Imamura M., Shimada H., Nishijima A. // Appl. Catal. B: Environ. 1998. V. 16. № 3. P. 255.
Weber T., Muijsers J.C., Wolput J.H.M.C., Verhagen C.P.J., Niemantsverdriet J.W. // J. Phys. Chem. 1996. V. 100. P. 14144.
Vatutina Yu.V., Klimov O.V., Nadeina K.A., Danilova I.G., Gerasimov E.Yu., Prosvirin I.P., Noskov A.S. // Appl. Catal. B Environ. 2016. V. 199. P. 23.
Chen W., Mauge F., van Gestel J., Nie H., Li D., Long X. // J. Catal. 2013. V. 304. P. 47.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ