

Кинетика и катализ, 2019, T. 60, № 5, стр. 624-632
Исследование структуры кобальтсодержащих катализаторов, синтезированных в субкритических условиях
М. Е. Маркова a, *, А. В. Гавриленко b, А. А. Степачёва b, В. П. Молчанов b, В. Г. Матвеева b, М. Г. Сульман b, c, Э. М. Сульман b
a ФГБОУ ВО Тверской государственный университет
170100 Тверь, ул. Желябова, 33, Россия
b ФГБОУ ВО Тверской государственный технический университет
170026 Тверь, наб. А. Никитина, 22, Россия
c ФГБУН Институт элементоорганических соединений им. А.Н. Несмеянова РАН
119991 Москва, ул. Вавилова, 28, Россия
* E-mail: mashulikmarkova@gmail.com
Поступила в редакцию 20.09.2018
После доработки 29.03.2019
Принята к публикации 22.04.2019
Аннотация
Проведено физико-химическое исследование кобальтсодержащих (10 мас. %) катализаторов синтеза Фишера–Тропша, нанесенных на диоксид кремния, которые были получены в субкритических условиях (Т = 200°С, Р = 8 МПа) с использованием воды (Тс = 374.1°С, Рс = 22.1 МПа) и пропанола-2 (Тс = 235.6°С, Рс = 5.8 МПа). Полученные образцы сравнивали с катализатором 10 мас. % Co/SiO2, приготовленным методом пропитки по влагоемкости. Сравнение свойств катализаторов в жидкофазном синтезе Фишера–Тропша показало, что наиболее активным и селективным к образованию алифатических углеводородов С6–С7 является образец, полученный в субкритической воде и характеризующийся высокой площадью поверхности (131.7 м2/г), равномерным распределением частиц активной фазы со средним размером 5 нм и большей доступностью катионов кобальта для реагентов. По данным РФЭС состав активной фазы этого катализатора представлен в основном двумя соединениями: Co(OH)2и Co3O4.
ВВЕДЕНИЕ
Нанесенные металлические катализаторы приобретают все большее значение в различных областях химической технологии. Такие гетерогенные системы отличаются высокой механической прочностью, развитой поверхностью и позволяют снизить содержание используемого металла [1]. В настоящее время для нанесения металлических частиц на различные подложки используется множество техник: пропитка по влагоемкости [2], коллоидный синтез [3], микроэмульсионный метод [4], сонохимический синтез [5], осаждение и со-осаждение [6], полиольный метод [7] и т.д. В зависимости от используемого метода, физические и физико-химические свойства полученных катализаторов изменяются в значительной степени.
Наиболее часто синтез нанесенных металлических катализаторов осуществляют методом пропитки носителей раствором прекурсора металла с последующей сушкой, термической обработкой или восстановлением металлических наночастиц [2]. Однако высокие вязкость и поверхностное натяжение большинства жидкостей, используемых в качестве растворителей в процессе пропитки, приводят к медленной диффузии металла в порах носителя, и, как следствие, к низкой дисперсности металлических частиц и негомогенности получаемого образца [8]. Кроме того, последующие процессы сушки и термической активации могут вызывать значительные изменения в структуре катализатора, резко снижая площадь поверхности и агрегацию активной фазы.
В последние годы большое внимание привлекает синтез нанесенных катализаторов с использованием суб- и сверхкритических жидкостей [9]. Благодаря уникальным свойствам сверхкритические жидкости позволяют контролировать размер получаемых наночастиц, их дисперсность, структуру и морфологию. Низкое поверхностное натяжение сверхкритических жидкостей предотвращает закупорку пор носителя во время синтеза, а малая вязкость ускоряет диффузию раствора. Помимо этого, в сверхкритических условиях повышается растворимость многих соединений, что дает возможность расширить спектр используемых прекурсоров металла [10]. Сверхкритическое нанесение металлов включает несколько этапов: 1) растворение прекурсора в сверхкритической жидкости; 2) внесение пористого носителя в полученный раствор; 3) адсорбция раствора в порах и на поверхности носителя; 4) химическое или термическое восстановление прекурсора с образованием частиц активной фазы [11]. В качестве растворителей в сверхкритическом синтезе катализаторов наиболее часто используют диоксид углерода [8, 9, 12–15] и воду [16–18]. Кроме того, существуют данные о применении для этих целей метанола [19], аммиака [20], изопропилового спирта [21, 22], циклогексана [23] и пропана [24].
Так, в работах [16, 25] были изучены структура и каталитические свойства Pd/ZrO2 и Pd(Rd)/TiO2, синтезированных в сверхкритической и субкритической воде соответственно. Otsu и Oshima [26] исследовали структуру оксидов марганца, свинца и серебра, нанесенных на γ-Al2O3 в среде сверхкритической воды. Sun и соавт. [27] синтезировали никель-, кобальт- и железосодержащие катализаторы в среде субкритического этанола. Yahya и др. [28] изучали структуру и фотокаталитические свойства гексатитаната калия, полученного в сверх- и субкритической воде. Использование сверх- и субкритических условий для синтеза нанесенных катализаторов показало, что независимо от того, была ли достигнута критическая область, приготовленные каталитические системы характеризуются высокой кристалличностью, наноразмерностью и равномерным распределением активной фазы. Поскольку для полярных жидкостей в сверхкритическом состоянии наблюдается резкое изменение свойств (в частности, диэлектрической постоянной, полярности и т.д.), для синтеза металлосодержащих композитов наиболее оптимальными являются суб- и околокритические условия [17].
В настоящей работе проведено исследование структуры и физико-химических свойств кобальтовых наночастиц, нанесенных на оксид кремния, которые были получены в субкритических условиях с использованием воды и пропанола-2 в качестве растворителей, а также сравнение этих наносистем с катализатором, приготовленным методом пропитки.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходные материалы для синтеза кобальтсодержащих катализаторов
Для синтеза катализаторов использовали следующие материалы: хлорид кобальта(II) шестиводный, гидрокарбонат натрия, оксид кремния безводный, пропанол-2, этиленгликоль (все вышеперечисленные реагенты марки х. ч., производство “Реахим”, Россия) и дистиллированную воду.
Синтез катализаторов в субкритических условиях
Синтез кобальтсодержащих катализаторов в субкритических условиях проводили в реакторе высокого давления PARR-4307 (“Parr Instrument”, США). В реактор вносили 1 г оксида кремния, хлорид кобальта в расчете 10 мас. % кобальта и 0.1 г гидрокарбоната натрия в качестве осаждающего агента (для ускорения гидролиза) в 30 см3 растворителя (дистиллированной воды или пропанола-2). Реактор герметизировали и трижды продували азотом для удаления кислорода воздуха. Затем устанавливали необходимое рабочее давление азота (6.0 МПа), контролируемое по манометру, и температуру (Т = 200°С). Общее давление в реакторе после нагрева составляло: 7.5 МПа и 8 МПа для воды и пропанола-2 соответственно. Процесс проводили при непрерывном перемешивании со скоростью 750 об./мин. Время проведения синтеза составляло 15 мин. После окончания синтеза реакционную смесь охлаждали до комнатной температуры, фильтровали, промывали 15–20 см3 используемого растворителя до отсутствия реакции на хлорид-ионы и сушили на воздухе. Полученные образцы обозначены как Co/SiO2(H2O) и Co/SiO2(ИПС).
Синтез катализаторов методом пропитки
Синтез кобальтсодержащего катализатора методом пропитки проводился следующим образом: 1 г оксида кремния (SiO2), высушенного до постоянной массы, пропитывали смесью, состоящей из хлорида кобальта в расчете 10 мас. % кобальта, 8 см3 этиленгликоля и 2 см3 дистиллированной воды в течение 15 мин. Затем полученный образец отфильтровывали и обрабатывали водным раствором гидрокарбоната натрия с концентрацией 2.76 г/дм3 в течение 15 мин, промывали дистиллированной водой до отсутствия реакции на хлорид- и карбонат- ионы и вновь сушили на воздухе до полного высыхания. Полученный образец обозначен как Co/SiO2(ЭГ).
Все приготовленные образцы выдерживали в течение 3 ч в токе водорода при температуре 300°С для перевода гидроксидов кобальта в оксиды с последующим их восстановлением.
Физико-химические исследования катализаторов
Низкотемпературная адсорбция азота. Определение удельной площади поверхности и пористости проводили c использованием анализатора площади поверхности и распределения пор по размерам Beckman CoulterТМ SА 3100ТМ (“Coulter Corporation”, США). Подготовку образцов осуществляли с помощью прибора Beckman CoulterTM SA-PREPTM (“Coulter Corporation”, США) при температуре –120°С в течение 60 мин. В ходе анализа применяли модели Ленгмюра, Брунауэра–Эммета–Теллера БЭТ) и t-график. Для расчета распределения пор использовали уравнение Харкинса–Юра.
Просвечивающая электронная микроскопия. Просвечивающая электронная микроскопия (ПЭМ) образцов катализаторов была проведена с целью определения размера кобальтсодержащих частиц и их распределения. Образцы готовили методом микросреза толщиной 50 нм и анализировали при ускоряющем напряжении электронной пушки 60 кВ с использованием микроскопа JEM 1010 (“JEOL”, Япония). Микрофотографии были получены с помощью цифровой камеры Gatan (“Gatan Inc.”, США). Анализ полученных микрофотографий проводили, используя пакеты программ Adobe Photoshop и Scion Image Processing Toolkit.
Рентгеновская фотоэлектронная спектроскопия. Качественный состав поверхности и валентное состояние металла в образцах определяли методом рентгенофотоэлектронной спектроскопии (РФЭС) на фотоэлектронном спектрометре ЭС-2403, оснащенном анализатором энергий PHOIBOS-100-MCD (“Specs GmbH”, Германия) и рентгеновским источником XR-50 с твинанодом Mg/Al. Для регистрации спектров использовали линию AlKα1.2. Мощность излучения составляла 250 Вт. Обзорные спектры образцов записывали в диапазоне 0–1200 эВ с шагом 0.5 эВ и временем накопления в точке 0.3 с.
Спектры анализировали с применением программного пакета Casa XPS. Модельное разложение спектров высокого разрешения с целью выделения индивидуальных состояний осуществляли с учетом таких характеристик фотоэлектронных подуровней, как энергия связи компонентов, соотношение площадей компонентов, внутридублетное расщепление. В качестве модельного фона был выбран фон по Ширли. Минимизацию проводили по методу Левенерга–Марквардта.
Инфракрасная спектроскопия диффузного отражения. Инфракрасные спектры диффузного отражения записывали с помощью ИК-спектрометра Protege 460 (“Nicolet”, США) с использованием самодельной приставки диффузного отражения. Перед регистрацией спектров образцы в виде фракции 0.25‑0.35 мм подвергали вакуумной обработке в течение 2 ч при температуре 400°С. В качестве молекулы-теста применяли монооксид углерода. СО адсорбировали при комнатной температуре при равновесном давлении 1599.9 Па. Десорбцию СО проводили также при комнатной температуре в вакууме. Спектры записывали в диапазоне от 6000 до 400 см–1 с шагом 4 см–1.
Тестирование синтезированных катализаторов в жидкофазном синтезе Фишера‑Тропша
Синтез Фишера–Тропша в присутствии полученных каталитических систем осуществляли в стальном реакторе PARR-4307 (“Parr Instrument”, США) в присутствии додекана (растворитель). В качестве синтез-газа использовали смесь СО и Н2 в объемном соотношении 1 : 6. Высокое содержание водорода в газовой смеси обусловлено необходимостью дополнительного гидрирования олефинов и кислородсодержащих соединений, образующихся в присутствии кобальтсодержащего катализатора [29–32]. Температура процесса составляла 200°С, общее давление в реакторе – 2 МПа, масса катализатора – 0.1 г, объем растворителя – 30 cм3.
Жидкую фазу анализировали методом газовой хроматографии масс-спектрометрии с помощью газового хроматографа GC-2010 и масс-спектрометра GCMS-QP2010S (“SHIMADZU”, Япония). Пробы жидкой фазы отбирали через 3 ч реакции после охлаждения и конденсации жидких продуктов. Анализ жидкой фазы проводили при следующих условиях: объем пробы – 3 мкл; начальную температуру колонки 150°C выдерживали в течение 6 мин, затем плавно повышали до 250°C со скоростью 15°C/мин; температура инжектора – 280°C; газ-носитель – гелий; давление He – 253.5 кПа; общий поток He – 81.5 cм3/мин; линейная скорость потока He – 20.8 cм3/с; колонка HP-1MS: L = 30 м, d = 0.25 мм, толщина пленки – 0.25 мкм; температура источников ионов – 260°C; температура интерфейса – 280°C; режим сканирования – от 10 до 800 m/z; скорость сканирования – 625; ионизация электронным ударом.
Пробы газовой фазы объемом 1 cм3 отбирали через 3 ч реакции после охлаждения и конденсации жидких продуктов. Анализ газовой фазы проводили хроматографическим методом с помощью газового хроматографа Кристаллюкс 4000М (“МетаХром”, Россия), оснащенного пламенно-ионизационным детектором и катарометром, подключенными последовательно. Для разделения компонентов газовой смеси использовали насадочную колонку длиной 2.5 м и диаметром 3.0 мм, заполненную гранулами полимерного адсорбента MN270 (“Purolight Inc.”, Великобритания) с фракцией 125–250 мкм. Газовую фазу анализировали при следующих условиях: начальную температуру колонки 40°C выдерживали в течение 4 мин, затем плавно поднимали до 250°C со скоростью 15°C/мин; температура испарителя и детектора – 260°C; газ-носитель – гелий; общий поток He – 30.0 cм3/мин.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Результаты анализа удельной площади поверхности полученных образцов представлены в табл. 1. Наибольшее снижение удельной площади поверхности по сравнению с исходным оксидом кремния наблюдается для катализатора, синтезированного в среде субкритической воды. Для этого же образца отмечено значительное сокращение площади поверхности микропор. Подобное изменение пористой структуры характерно для гидротермальной обработки оксида кремния, приводящей к его рекристаллизации и исчезновению микропор [33]. Для катализатора, полученного в субкритическом пропаноле-2, также наблюдается двукратное уменьшение общей площади поверхности и шестикратное снижение площади поверхности микропор. В виду полярного характера используемого растворителя при обработке оксида кремния субкритическим пропанолом-2 также вероятна рекристаллизация носителя, что сказывается на его пористости. Наименьшее сокращение площади поверхности отмечено для образца, приготовленного методом пропитки.
Таблица 1.
Удельная площадь поверхности и общий объем пор кобальтсодержащих катализаторов
| Образец | Удельная площадь поверхности, м2/г | Общий объем пор, дм3/г | ||
|---|---|---|---|---|
| Модель Ленгмюра | Модель БЭТ | t-график | ||
| SiO2 | 391 | 389 | 269* 121** |
0.25 |
| Сo/SiO2(Н2О) | 128 | 132 | 127* 5** |
0.22 |
| Сo/SiO2(ИПС) | 205 | 207 | 179* 25** |
0.23 |
| Сo/SiO2(ЭГ) | 315 | 327 | 274* 53** |
0.21 |
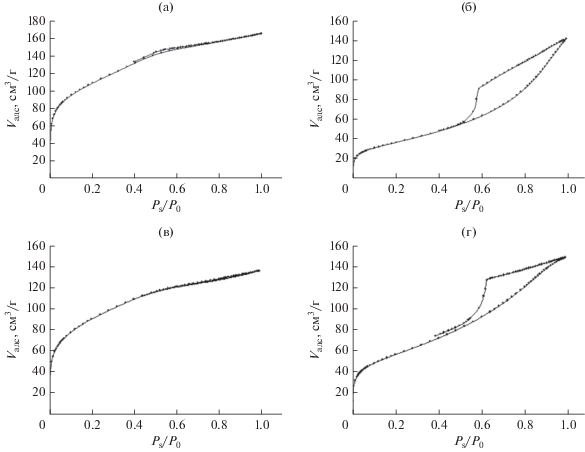
Анализ изотерм адсорбции–десорбции азота (рис. 1) показал, что изотермы образцов исходного оксида кремния (рис. 1а) и катализатора, полученного в субкритическом пропаноле-2 (рис. 1в), относятся к типу I, характерному для микропористых веществ со слабым взаимодействием адсорбат–адсорбент. Изотермы образцов, синтезированных в среде субкритической воды (рис. 1б) и методом пропитки (рис. 1г), принадлежат к типу IV с ярко выраженными петлями гистерезиса формы Н2. Такие изотермы характерны для микро-мезопористых веществ с высокой степенью взаимодействия адсорбат–адсорбент, обладающих цилиндрическими порами с широким распределением по размеру [34, 35].
Рис. 1.
Изотермы адсорбции–десорбции азота для образцов: SiO2 (а), Co/SiO2(H2O) (б), Co/SiO2(ИПС) (в), Co/SiO2(ЭГ) (г).
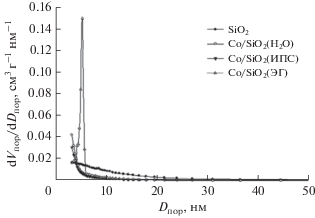
Анализ распределения пор по размерам для исследуемых образцов (рис. 2) свидетельствует, что по сравнению с носителем для катализаторов, приготовленных в субкритических условиях, снижается количество пор с размером менее 6 нм. Это указывает на формирование частиц активной фазы в микропорах оксида кремния, тогда как в случае катализатора, синтезированного методом пропитки, частицы активной фазы формируются в мезопорах.
Рис. 2.
Распределение пор по размерам для образцов SiO2 (а), Co/SiO2(H2O) (б), Co/SiO2(ИПС) (в), Co/SiO2(ЭГ) (г).
Микрофотографии кобальтсодержащих образцов представлены на рис. 3. Для катализаторов Co/SiO2(Н2О) и Co/SiO2(ИПС) (рис. 3а и 3б) было отмечено формирование мелких наночастиц со средним диаметром 5 и 7 нм соответственно. При этом наблюдалось равномерное распределение частиц активной фазы на поверхности носителя. Наибольшая плотность распределения частиц была в случае образца, полученного в среде субкритической воды, что указывает на высокое содержание кобальта на поверхности оксида кремния. Для катализатора, приготовленного методом пропитки (рис. 3в), наблюдалось формирование частиц со средним диаметром 20 нм. Также для него были характерны наименьшая плотность и неравномерное распределение частиц активной фазы на поверхности носителя.
Рис. 3.
ПЭМ-микрофотографии кобальтсодержащих образцов: Co/SiO2(H2O) (а), Co/SiO2(ИПС) (б), Co/SiO2(ЭГ) (в).

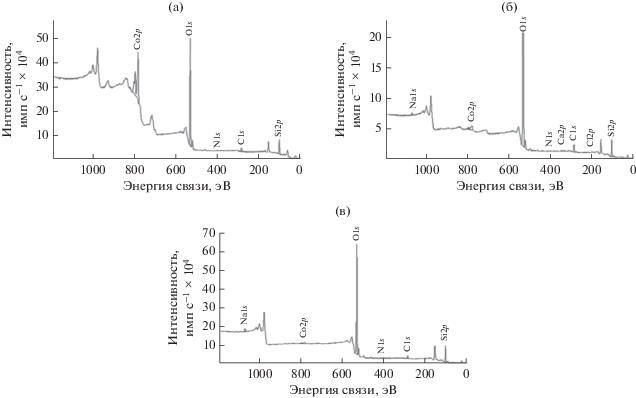
На рис. 4 представлены обзорные РФЭ-спектры образцов катализаторов. Анализ элементного состава поверхности показывает, что кобальт наиболее полно сорбируется на оксиде кремния при синтезе в среде субкритической воды (11.8 ат. %). В случае субкритического пропанола-2 и этиленгликоля кобальт на поверхности был обнаружен в незначительных количествах (1.3 и 0.5 ат. % соответственно), что подтверждается результатами ПЭМ-анализа.
Рис. 4.
Обзорные РФЭ-спектры кобальтсодержащих образцов: Co/SiO2(H2O) (а), Co/SiO2(ИПС) (б), Co/SiO2(ЭГ) (в).
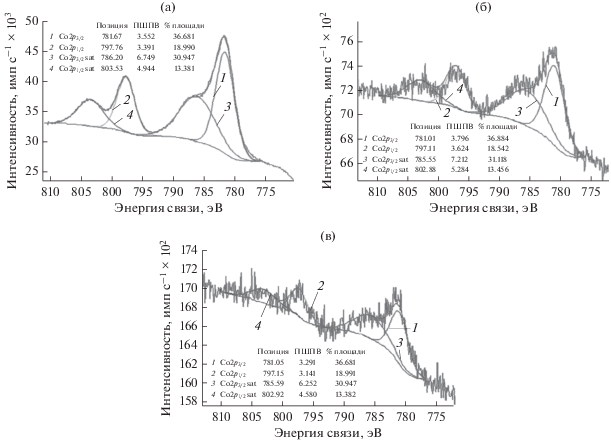
Анализ спектров высокого разрешения [36, 37] для кобальта (рис. 5) свидетельствует, что состав поверхности образцов Co/SiO2(ИПС) и Co/SiO2(ЭГ) (рис. 5б и 5в) практически идентичен и представлен в основном гидроксидом кобальта(II) (энергии связи 781.0 и 785.5 эВ). Кроме того на поверхности этих образцов также найдены оксиды кобальта(III) и кобальта(II) – 797.1 и 802.9 эВ соответственно. Для катализатора Co/SiO2(Н2О) (рис. 5а) основным поверхностным соединением также является гидроксид кобальта(II) – 786.2 и 803.5 эВ. Кроме гидроксида кобальта(II) на поверхности катализатора присутствуют гидратированные катионы кобальта(II) (781.7 эВ). На поверхности образца, синтезированного в среде субкритической воды, обнаружен смешанный оксид кобальта (Co3O4, 797.7 эВ), который проявляет каталитическую активность во многих реакциях (гидрирование оксидов углерода, деоксигенирование органических кислот и т.д.) [38]. Образование смешанного оксида Co2+/Co3+ на поверхности носителя объясняется окислительным действием молекул воды в субкритическом состоянии [39].
Рис. 5.
РФЭ-спектры высокого разрешения для кобальтсодержащих образцов: Co/SiO2(H2O) (а), Co/SiO2(ИПС) (б), Co/SiO2(ЭГ) (в).
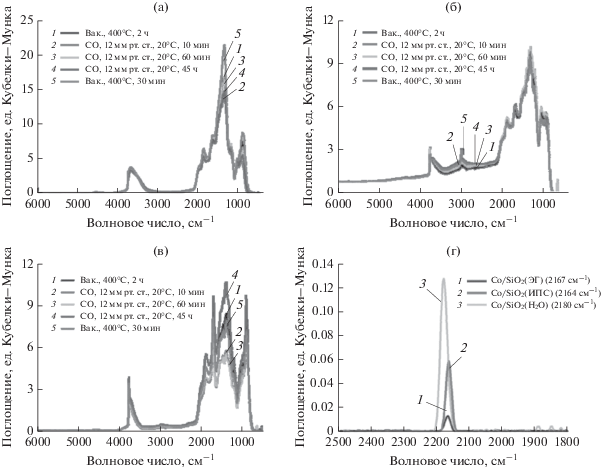
На рис. 6а–6в представлены обзорные ИК-спектры диффузного отражения СО катализаторов. Сравнение спектров показало, что для образцов, синтезированных методом пропитки и в среде субкритического пропанола-2, видны узкие полосы поглощения при 3740 см–1 с протяженным в сторону меньших частот “хвостом”, принадлежащие колебаниям изолированных Si–OH- и ОН-групп, возмущенных водородной связью. В этой же области отмечены широкие полосы колебаний спиртовых ОН-групп. В процессе адсорбции СО, особенно после длительной выдержки, в спектрах образцов появляются полосы при 3680 см–1, при этом интенсивность полосы 3740 см–1 снижается. Анализ спектров позволяет предположить, что возникновение новой полосы связано с взаимодействием СО с ОН-группами на поверхности носителя. В спектре образца, синтезированного в среде субкритической воды, видна узкая полоса при 3736 см–1 и широкая полоса с центром около 3669 см–1, принадлежащие колебаниям изолированных Si–OH- и ОН-групп, возмущенных водородной связью. В процессе адсорбции СО для вышеуказанного образца изменений в этом районе спектра не обнаружено. Анализ спектров высокого разрешения (рис. 6г) показал, что для всех образцов после адсорбции СО в спектрах появляются полосы поглощения в области 2160–2180 см–1, характеризующие валентные колебания связи С=О в молекуле монооксида углерода, адсорбированного в линейной форме на катионах Со2+ [40]. Положение этих полос практически не меняется в процессе длительной адсорбции СО, а при десорбции в вакуум при комнатной температуре они из спектра исчезают. Сопоставление спектров высокого разрешения для исследуемых образцов свидетельствует, что наибольшая доступность для молекул СО у катионов Со2+ в образце, синтезированном в среде субкритической воды. Таким образом, кобальтсодержащий катализатор, полученный в среде субкритической воды, обладает лучшими физико-химическими характеристиками по сравнению с другими исследуемыми образцами.
Рис. 6.
Обзорные ИК-спектры диффузного отражения:Co/SiO2(H2O) (а), Co/SiO2(ИПС) (б), Co/SiO2(ЭГ) (в) и спектр высокого разрешения диффузного отражения для кобальтсодержащих образцов (г).
Приготовленные образцы тестировали в жидкофазном синтезе Фишера–Тропша, который позволяет получать широкий спектр газообразных, жидких и твердых продуктов. В последнее время большой интерес исследователей вызывает проведение жидкофазного процесса, который дает возможность контроля стадии роста цепи за счет уменьшения растворимости СО, снижения образования кислородсодержащих продуктов и предотвращения реакции конверсии водяного пара. Основными металлами, используемыми в качестве активных центров катализаторов, являются металлы VIII группы, среди которых наиболее активны Ru, Co, Fe, Ni. Для увеличения площади поверхности их наносят на пористые носители, такие как силикагель. В промышленности наиболее часто применяют Co и Fe. Результаты, полученные при тестировании катализаторов, представлены на рис. 7.
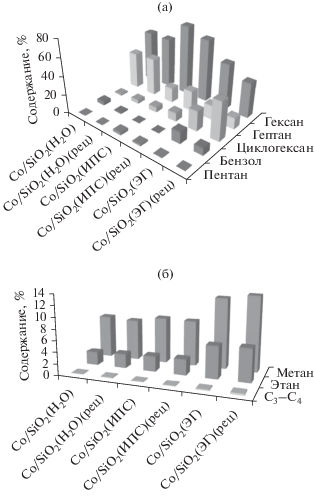
Синтезированные образцы обладают высокой эффективностью в процессе получения жидких углеводородов С5–С7 (рис. 7). Анализ состава газовой фазы показал присутствие метана и этана для всех образцов. Однако на катализаторах, полученных в субкритических условиях, количество насыщенных углеводородов С1–С2 было меньше по сравнению с образцом, приготовленным методом пропитки. Пропан и бутан в газовой фазе содержались в следовых количествах. При повторном использовании исследуемых катализаторов состав газовой фазы для образцов Co/SiO2(H2O) и Co/SiO2(ИПС) практически не изменился, а для катализатора Co/SiO2(H2O) повысилось содержание метана и этана в газовой фазе, что привело к снижению селективности образования углеводородов С5+ в его присутствии.
При анализе жидкой фазы обнаружено, что синтезированные катализаторы позволяют получать в основном линейные и циклические углеводороды состава С5–С7. Углеводороды С8+ присутствуют в следовых количествах. В случае Co/SiO2(H2O) и Co/SiO2(ИПС) селективность сдвигается в сторону образования углеводородов С6–С7. Катализатор Co/SiO2(ЭГ) проявляет достаточно высокую селективность по ароматическим и циклическим углеводородам. При повторном использовании катализатора Co/SiO2(H2O) его селективность в отношении углеводородов С6–С7 сохраняется, однако наблюдается незначительное повышение содержания гептадекана и возрастает содержание циклогексана за счет гидрирования бензола. Это можно объяснить превращением продуктов, сорбированных в порах катализатора. В случае повторного использования Co/SiO2(ИПС) наблюдается значительное увеличение содержания гептадекана, что указывает на протекание реакции роста цепи. Для катализатора, синтезированного методом пропитки, характерно повышение содержания ароматических и циклических углеводородов, что может быть следствием протекания реакции циклизации гексана в порах носителя.
Расчет молекулярно-массового распределения продуктов синтеза Фишера–Тропша показал, что в присутствии катализаторов, синтезированных в субкритической воде и пропаноле-2, коэффициент роста цепи α составляет 0.77 и 0.68 соответственно, что указывает на максимальный выход углеводородов С6 и согласуется с литературными данными о кобальтовых катализаторах [29, 31, 32]. Для образца, полученного методом пропитки, коэффициент α был равен 0.61. Такое значение свидетельствует о сдвиге продуктов реакции в сторону образования низкомолекулярных углеводородов и о высокой селективности по метану. Таким образом, катализатор, приготовленный с использованием субкритической воды, наиболее эффективен в жидкофазном синтезе Фишера–Тропша с образованием жидких углеводородов С5–С7 и наибольшим выходом углеводородов С6. Результаты тестирования катализаторов хорошо коррелируют с данными об их составе и структуре.
ЗАКЛЮЧЕНИЕ
Сравнение кобальтсодержащих катализаторов, полученных в субкритических условиях и методом пропитки, показало, что субкритический синтез – перспективная альтернатива традиционным методам, позволяющая готовить образцы с развитой внутренней поверхностью, равномерным распределением, меньшим размером и большей доступностью частиц активной фазы для реагентов. Оптимальной средой для синтеза катализаторов является субкритическая вода. Тестирование катализаторов в жидкофазном синтезе Фишера–Тропша свидетельствует, что наиболее эффективен катализатор, полученный в условиях субкритической воды.
Список литературы
Dantas A.L., Alves P.D., Arando D., Schmal M. // Appl. Catal. A: Gen. 2004. V. 277. P. 71.
Antolini E., Salgado J.R.C., Santos L.G.R.A., Garcia G., Ticianelli E.A., Pastor E., Gonzalez E.R. // J. Appl. Electrochem. 2005. V. 36. P. 355.
Luo J., Njoki P.N., Lin Y., Mott D., Wang L., Zhong C. // Langmuir. 2006. V. 22. P. 2892.
Xiong L., Manthiram A. // Electrochim. Acta. 2005. V. 50. P. 2323.
Angelucci C.A., Silva M., Nart F.C. // Electochem. 2007. V. 52. P. 7293.
Ren G., Shi H., Xing Y. // IOP Electron. J. Nanotechnol. 2007. V. 18. P. 5596.
Wang W., Zheng D., Du C., Zou Z., Zhang X., Xia B., Yang H., Akins D. // J. Power Sources. 2007. V. 167. P. 243.
Morère J., Tenorio M.J., Torralvo M.J., Pando C., Renuncio J.A.R., Cabanas A. // J. Supercritical Fluids. 2011. V. 56. P. 213.
Zhang Y., Erkey C. // J. Supercritical Fluids. 2006. V. 38. P. 252.
Tenorio M.J., Torralvo M.J., Enciso E., Pando C., Renuncio J.A.R., Cabanas A. // J. Supercritical Fluids. 2009. V. 49. P. 369.
Aymonier C., Loppinet-Serani A., Reveron H., Garrabos Y., Cansell F. // J. Supercritical Fluids. 2006. V. 38. P. 242.
Cangül B., Zhang L.C., Aindow M., Erkey C. // J. Supercritical Fluids. 2009. V. 50. P. 82.
Romang A.H., Watkins J. // J. Chem. Rev. 2010. V. 110. P. 459.
Yang J.X., Hasell T., Smith D.C., Howdle S.M. // J. Mater. Chem. 2009. V. 19. P. 8560.
Bozbag S.E., Yasar N.S., Zhang L.C., Aindow M., Erkey C. // J. Supercritical Fluids. 2011. V. 56. P. 105.
Галкин А.А., Костюк Б.Г., Кузнецова Н.Н., Туракулова А.О., Лунин В.В., Поляков М. // Кинетика и катализ. 2001. Т. 42. № 2. С. 172.
Hayashi H., Hakuta Y. // Materials. 2010. V. 3. P. 3794.
Hayashi H., Hakuta Y., Kurata Y. // J. Mater. Chem. 2004. V. 14. P. 2046.
Choi H., Veriansyah B., Kim J., Kim J.-D., Kang J.W. // J. Supercritical Fluids. 2010. V. 52. P. 285.
Chen Q.S., Prasad V., Hu W.R. // J. Cryst. Growth. 2003. V. 258. P. 181.
Lu X.M., Hanrath T., Johnston K.P., Korgel B.A. // Nano Lett. 2003. V. 3. P. 93.
Davolos M.R., Feliciano S., Pires A.M., Marques R.F.C., Jafelicci M. // J. Solid State Chem. 2003. V. 171. P. 268.
Pestov D., Levit N., Kessick R., Tepper G. // Polymer. 2003. V. 4. P. 3177.
Nieuwoudt I., Koen L. // Proc. 6thInt. Symp. Supercrit. Fluids. 2003. V. 3. P. 1871.
Галкин А.А., Туракулова А.О., Кузнецова Н.Н., Лунин В.В. // Вестн. Московского Университета. Сер. 2. Химия. 2001. Т. 42. № 5. С. 305.
Otsu J., Oshima Y. // J. Supercritical Fluids. 2005. V. 33. P. 61.
Sun Y.-P., Rollins H.W., Guduru R. // Chem. Mater. 1999. V. 11. P. 7.
Yahya R.B., Hayashi H., Nagase T., Ebina T., Onodera Y., Saitoh N. // Chem. Mater. 2001. V. 13. P. 842.
Хенрици-Оливэ Г., Оливэ С. Химия каталитического гидрирования СО. Москва: Мир, 1987. 248 с.
Bartholomew C.H. // 2003 Spring Meeting, American Institute of Chemical Engineering (AIChE). New Orleans, LA, 2003. II [83b].
Jahangiri H., Bennett J., Mahjoubi P., Wilson K., Gu S. // Catal. Sci. Technol. 2014. V. 4. P. 2210.
Oh J.-H., Bae J. W., Park S.-J., Khanna P. K., Jun K.-W. // Catal. Lett. 2009. V. 130. P. 403.
Иоффе И.И., Письмен Л.М. Инженерная химия гетерогенного катализа. Ленинград: Химия, 1972. 464 с.
Грег С., Синг К. Адсорбция, удельная поверхность, пористость. М.: Мир, 1984. 311 с.
A New Classification of Adsorption Isotherms. http://www.nigelworks.com/mdd/…/NewClass.pdf
NIST X-ray Photoelectron Spectroscopy Database, Version 3.5 (National Institute of Standards and Technology, Gaithersburg, 2003). http://srdata.nist.gov/xps/
Wagner C.D., Riggs W.M., Davis L.E., Moulder J.F., Muilenberg G.E. Handbook of X-Ray Photoelectron Spectroscopy. Minn. Perkin–Elmer Corporation. 1979. 55 344 p.
Ernst B., Libs S., Chaumette P., Kiennemann A. // Appl. Catal. A: Gen. 1999. V. 186. № 1–2. P. 145.
Stepacheva A.A., Markova M.E., Bykov A.V., Sidorov A.I., Sulman M.G., Matveeva V.G., Sulman E.M. // Reac. Kinet. Mech. Cat. 2018. V. 125. P. 213–226
Браун Д., Флойд А., Сейнзбери М. Спектроскопия органических веществ. М.: Мир, 1992. 300 с.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ