

Кинетика и катализ, 2019, T. 60, № 6, стр. 683-724
“Золотой век” гомогенно-каталитической химии алкинов: димеризация и олигомеризация алкинов
a МИРЭА – Российский технологический университет (Институт тонких химических технологий
имени М.В. Ломоносова)
119571 Москва, просп. Вернадского, 86, Россия
* E-mail: olegtemkin@mail.ru
Поступила в редакцию 25.02.2019
После доработки 14.04.2019
Принята к публикации 16.04.2019
Аннотация
Рассмотрена история развития гомогенных металлокомплексных катализаторов в химии ацетилена и его производных. Проанализированы достижения каталитической химии алкинов в создании эффективных каталитических систем (на основе комплексов металлов 3–11 и 13 групп Периодической системы элементов Д.И. Менделеева) для димеризации алкинов, позволяющих с высокой хемо-, стерео- и региоселективностью получать нужные изомеры димеров. Обсуждена природа элементарных стадий в четырех механизмах процесса димеризации и связь природы металлоорганических интермедиатов с природой катализатора (металла и лигандов), субстрата и с типом механизма, и, следовательно, с селективностью процесса димеризации.
СОДЕРЖАНИЕ
1. Введение
2. Краткая история развития каталитической химии ацетилена и его производных в растворах металлокомплексных катализаторов
3. Димеризация алкинов в растворах металлокомплексных катализаторов
3.1. Комплексы металлов 11 группы периодической системы элементов
3.2. Комплексы Ru, Os и Fe
3.3. Комплексы Rh, Ir и Co
3.4. Комплексы Ni, Pd и Pt
3.5. Комплексы металлов 4–7 групп периодической системы элементов
3.6. Комплексы металлов 3 и 13 групп периодической системы элементов.
4. Заключение
1. ВВЕДЕНИЕ
В 1991 г. была опубликована монография [1], подводящая итоги развития каталитической химии ацетилена и его производных с участием гомогенных металлокомплексных и гетерогенных катализаторов. Успехи кислотно-основного и нуклеофильного катализа обобщены в монографиях Р. Тедеши [2] и Б.А. Трофимова [3] (см. также обзоры последних десятилетий [4–6]), а некоторые проблемы “органокатализа” в химии алкинов изложены в обзоре [7]. Кинетические аспекты и механизмы старых классических и ряда новых реакций алкинов, открытых за последние четверть века, рассмотрены в монографии [8].
За последние 25–30 лет появились новые катализаторы, новые реакции, новые типы интермедиатов и новые механизмы каталитических реакций алкинов. Развитие современной химии алкинов отражено в многочисленных монографиях [8–21] и обзорах [22–45]. Наиболее популярные катализаторы в гомогенном катализе сегодня – комплексы меди, никеля, золота, рутения, палладия, родия, платины, титана и циркония. Впечатляет масштабность работ в области катализа комплексами золота в тонком органическом синтезе карбоциклов, гетероциклов и различных биологически активных соединений [9, 11, 13, 15, 18, 19, 22, 36, 44].
Возросший интерес к химии алкинов, связанный с появлением более активных катализаторов хемо- и стереоселективных реакций, протекающих в более мягких условиях, позволяет говорить о явном “ренессансе”, о “золотом веке” развития ацетиленовой химии. Вместе с тем вряд ли можно согласиться с утверждением редакторов книги [18] Б.Троста (Barry M.Trost) и Чао-Джун Ли (Chao-Jun Li), что современная каталитическая химия алкинов, возникшая за последние несколько десятков лет, “provide an alternative to classical stoichiometric alkyne chemistry”. Здесь следует внести некоторые уточнения. Во-первых, классическая химия ацетилена и его соединений, начиная с работ М.Г. Кучерова (1881 г.), А.Е. Фаворского, Ю. Ньюленда и В. Реппе, была в значительной степени каталитической, а не только стехиометрической, с использованием металлокомплексов в растворах и на поверхности металлов, оксидов металлов, кислотно-основных и нуклеофильных катализаторов [1–3, 9, 46–52]. Гомогенный металлокомплексный катализ до начала 50-х гг. прошлого столетия и развивался практически на базе химии ацетилена. Во-вторых, и в книгах, посвященных, в основном, термическим стехиометрическим реакциям ацетиленовых соединений и их металлоорганических производных, рассматривается много реакций, катализируемых кислотами Бренстеда и Льюиса, основаниями и солями металлов [48–50, 53–55].
За последние три десятилетия химия ацетилена превратилась в химию сложных ацетиленовых соединений – енинов, диалкинов, алкинолов, арил- и гетероарилацетиленов и других производных, однако впечатляющие результаты получены и с самим ацетиленом даже в форме СаС2 [45]. Значительное развитие получила химия фармацевтических хиральных препаратов и материалов со специальными оптическими, электрическими, термохимическими и механохимическими свойствами, использующая ацетиленовые субстраты. Новые реакции, гипотезы и парадигмы, возникшие в процессе развития химии ацетиленов, несомненно, заслуживают обсуждения и обобщения, как и результаты успешного развития и подтверждения “старых” гипотез о механизмах каталитических реакций. В этом обзоре будут рассмотрены вопросы, касающиеся применения металлокомплексного катализа в химии ацетиленовых соединений, с привлечением необходимой информации о нанесенных на поверхности металлокомплексах и о “микрогетерогенных” каталитических системах.
2. КРАТКАЯ ИСТОРИЯ РАЗВИТИЯ КАТАЛИТИЧЕСКОЙ ХИМИИ АЦЕТИЛЕНА И ЕГО ПРОИЗВОДНЫХ В РАСТВОРАХ МЕТАЛЛОКОМПЛЕКСНЫХ КАТАЛИЗАТОРОВ
Интересно проследить историю использования различных металлов в качестве металлокомплексных катализаторов в химии ацетилена и его производных, а также открытия новых реакций алкинов. Выделим для удобства анализа 4 этапа:
первый этап – период до начала 50-х гг.,
второй этап – 1950–1970 гг.,
третий этап – 1970–1990 гг.,
четвертый этап – 1991–2018 гг.
В монографии 1968 г. [52] была сделана попытка проследить появление различных металлов в каталитической химии ацетиленовых соединений на первом этапе в периоды до начала 50-х гг. XX-го столетия, начиная с первой работы Кучерова по гидратации ацетилена (1881 г.), и с 50-х гг. до 1967 г. Работы Кучерова, Фаворского, Ньюленда, Реппе, В. Карозерса, А.Л. Клебанского, Ю.С. Залькинда и многих других исследователей в первой половине XX-го столетия заложили основы каталитической химии ацетилена.
В период с 1925 по 1945 гг. группа химиков, руководимых Реппе в главной лаборатории “ИГ Фарбениндустри” в Людвигсхафене (Германия), разработала химию и технологию четырех типов каталитических реакций ацетилена и его производных с использованием оснόвных и металлокомплексных катализаторов. Это реакции этинилирования, винилирования, карбонилирования и циклизации ацетилена (до бензола и циклооктатетраена). Результаты работ этой лаборатории стали известны научной общественности только после Второй мировой войны в результате докладов сотрудников технической миссии США [56], отчета Реппе [47] и монографии [57], обобщившей работы Реппе по карбонилированию ацетилена. В обзоре [56] было приведено общее мнение американских химиков (110th Meeting of ACS): “выдающиеся результаты, полученные группой Реппе, станут исключительно важными для развития органической химии в США” (см. также [39]).
В одной из лекций по органической химии в МИТХТ им. М.В. Ломоносова в начале 60-х гг. академик И.Л. Кнунянц вспоминал о работе в Германии после войны в комиссии по репарациям. Однажды он встретил на железнодорожных путях химического комбината цистерну с надписью “Cyclooctatetraen” и решил, что это название скрывает какой-то секретный продукт. Он, конечно, знал, что это соединение было впервые синтезировано лауреатом Нобелевской премии Р.М. Вильштеттером в 1911 г. в результате сложнейшего 9-стадийного синтеза из алкалоида псевдопельтьерина (из коры гранатового дерева). Каково же было его удивление, когда он выяснил, что в цистерне был действительно циклооктатетраен – продукт одностадийного каталитического промышленного синтеза. История открытия реакции синтеза циклооктатетраена и написания отчета пленным Реппе [47] о своих научных результатах изложена в лекциях Ю.А. Устынюка по органической химии [58].
В первой половине ХХ в. катализаторами различных гомогенных превращений ацетилена служили, кроме кислот и оснований, в основном, соединения непереходных и постпереходных металлов – Hg(II), Zn(II), Cd(II), Bi(III), Sb(V), Cu(I), Ag(I), Fe(III, II) и Cr(III). Соединения золота использовали для катализа этинилирования карбонильных соединений (Au2C2/SiO2 [59]) и для стехиометрического окисления ацетилена до глиоксаля (AuCl3 [60]), а комплексы Pt(II) – только однажды для катализа окисления ацетилена азотной кислотой до щавелевой кислоты [52].
Превращения ацетиленовых соединений в гетерогенном катализе проводили на более разнообразном ассортименте катализаторов – на металлах и сплавах (Pt, Pd, Ni, Co, Fe, Cu), оксидах металлов (Zn, Cd, Sn, Ni, Co, Mo, Ag, V, Ti и Th), сульфидах Zn, Cd, Mo и Th, а также на нанесенных солях Co, Ni и Ce.
Металлоорганические соединения Hg(II), Cu(I) и Sb(V), образующиеся из ацетилена, стали впервые рассматривать в качестве интермедиатов каталитических реакций. Так, например, процесс хлорирования ацетилена до тетрахлорэтана в растворах SbCl5 через промежуточное соединение SbCl4(CH=CHCl) (“квазикомплексное” соединение Несмеянова) считали редким примером каталитической реакции с “ясным механизмом” [61]. Комплексы Ni(0, II) были введены Реппе в практику гомогенного катализа в синтезе акриловой кислоты и ее производных и в реакциях циклотримеризации и циклотетрамеризации ацетилена. В 1952 г. появилась работа Реппе по синтезу 5-этилпиридина из ацетилена и аммиака в растворах комплексов Ni(II) и Co(II) [52].
На втором этапе одновременно с развитием координационной и металлоорганической химии началось интенсивное развитие металлокомплексного катализа вообще и, в частности, в химии ацетилена. В каталитическую практику были введены комплексы всех металлов 8–10 групп, а также комплексы некоторых металлов 4–7 групп Периодической системы элементов (табл. 1) [52].
Таблица 1.
Комплексы металлов 4–10 групп элементов в каталитической химии алкинов
| Металл | Группа элементов | ||||||
|---|---|---|---|---|---|---|---|
| 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Валентность в комплексах |
Ti(II, III, IV) | V(III, V) | Cr(II, III, IV) Mo(0, V) W(0) |
Mn(0, II) | Fe(–II, 0) Ru(0, III) Os(II) |
Co(–I, 0, II) Rh(I, III) Ir(I) |
Ni(0, II) Pd(0, II) Pt(0, II, IV) |
Появились новые катализаторы для уже известных реакций (гидратации, гидрохлорирования, изомеризации, карбонилирования, циклизации и др.). Так, уже в конце 50-х–начале 60-х гг. были открыты первые активные нертутные каталитические системы для гидратации ацетилена и алкинов – система CuCl–ZnCl2–H2O, не уступающая ртутному катализатору по производительности в синтезе ацетальдегида, но значительно более стабильная [52, 62, 63], и RuCl3 [64]. В составах металлокомплексных катализаторов появились новые типы лигандов: PR3, AsR3, SbR3, ${\text{SnCl}}_{3}^{ - }$ и Py. После открытия реакции окисления олефинов в растворах комплексов палладия [65] (так называемое Вакер-окисление олефинов [66]) заметно возрос интерес к различным превращениям ацетилена, катализируемым комплексами палладия [52]. Было обнаружено, что в спиртовых растворах комплексов Pd(II) при карбонилировании ацетилена получается смесь продуктов окислительного, восстановительного и аддитивного моно- и дикарбонилирования (см., например, [67]), а в системе Pd(PPh3)4–CHCl3–n-BuOH в условиях существования комплекса ClPd(CHCl2)(PPh3)2 селективно образуется бутилакрилат [68–70]. Аналогичные процессы обнаружены и в растворах комплексов Co(0) и Rh(III) [52]. После открытия катализаторов Циглера–Натта реакции полимеризации олефинов их стали использовать не только в полимеризации ацетилена и алкинов, но и в реакциях гидрирования, димеризации и циклизации алкинов [52]. К новым каталитическим реакциям алкинов, несомненно, можно отнести реакции социклизации ацетилена с нитрилами, кросс-сочетания Кадье–Ходкевича, аллилирования ацетилена (конденсация Курца), некоторые реакции окислительного хлорирования [52] и метатезис алкинов [71], каталитический диеновый синтез норборнадиена с участием этинильного комплекса [Cu](C≡CH) в качестве интермедиата [72], реакции гидросилилирования алкинов [1].
С начала 50-х гг. началось и исследование механизмов гомогенно-каталитических превращений алкинов – известных и новых реакций (Ф. Больманном, А.Н. Несмеяновым, Е.А. Шиловым, Р.М. Флидом, Р. Вестиным, Д.В. Сокольским, А.А. Петровым, О.А. Чалтыкяном, И.Л. Котляревским, С.А. Вартаняном и др. [8, 52, 73]). Появилось множество работ по металлоорганической и координационной химии соединений, полученных на основе ацетилена, что сразу продвинуло понимание механизмов каталитических реакций. На втором этапе были синтезированы этинильные (алкинидные) комплексы Cu, Ag, Au, Zn, Cd, Hg, Cr, Mn, Fe, Co, Ni, Pd, Pt и Ir, установлена структура моноядерных и кластерных π-комплеков 16 металлов, синтезированы линейные β-замещенные винильные производные ряда металлов, μ2-производные М–СН=СН–М никеля, кобальта и хрома, металлациклопентадиены и другие σ-металлоорганические соединения [52]. Следует отметить появление многочисленных работ по изучению кинетики и механизмов реакций димеризации и окислительной димеризации алкинов, механизмов реакций циклизации и социклизации [52].
После работ М. Дьюара (1946–1949 гг.) о π-комплексах олефинов удалось установить соответствие гипотезы об участии π-комплексов металлов с ацетиленами в механизме каталитических реакций закономерностям реакций присоединения различных молекул к тройной связи (в работах Е.А. Шилова, Р.М. Флида, Р. Вестина, И.И. Моисеева и др.). В конце второго этапа появились попытки решения проблемы активации ацетиленов комплексами металлов (см. [74]) и разработки научных принципов подбора катализаторов [52], завершившиеся на третьем этапе [1] пониманием простой идеи – научно обоснованный выбор катализатора для любой реакции может быть реализован только при использовании процедуры выдвижения гипотез о механизмах реакции и, соответственно, о природе интермедиатов с последующим анализом гипотез. Комплекс металла в качестве предполагаемого катализатора должен обеспечить образование и превращение интермедиатов конкретного механизма. Проблемы, связанные с выдвижением гипотез о механизмах реакций, и создание компьютерных программ для генерации гипотез рассмотрены в работах [75–78].
На третьем этапе к уже известным комплексам – катализаторам превращений алкинов – добавились комплексы Sc, Y, Zr, Nb и Ta [1]. Хлоридные комплексы Au(III) в растворах оказались активными катализаторами в реакциях гидратации [79] и аминирования [80] алкинов, а комплексы Au(I, III) в растворах и на носителях – в гидрохлорировании ацетилена [15, 81]. Новые металлокомплексные катализаторы появились и в других известных реакциях – гидратации, гидроиодирования, гидроцианирования, арилирования, гидрирования, циклизации, димеризации, полимеризации, карбонилирования и гидросилилирования алкинов [1].
В это же время были открыты и новые реакции – винилирования олефинов [82] и метана [83, 84], анти-Марковниковской (анти-М) гидратации алкинов в полифункциональной систeме трех катализаторов CuCl–RSH–HCl в результате замены “жeсткого” нуклеофила (Н2О) “мягким” – RSH [85, 86] и анти-М-присоединения к терминальным алкинам карбамат-анионов в растворах комплексов рутения с участием винилиденовых интермедиатов [87]. Новые катализаторы были предложены для арилирования ацетилена в растворах комплексов Co(II), Ni(II) и Pd(II) с образованием 1,2-диарилэтанов [88], кросс-сочетания (работа Соногашира) в растворах комплексов палладия и меди(I) [89], гомогенного варианта гидродимеризации ацетилена до бутенов и гидросодимеризации ацетилена и этилена до бутенов в растворах комплекса Pd[P(OPh)3]4 [90]. Появились каталитические системы для реакций окислительного карбонилирования алкинов по связи С–Н [91, 92] и разнообразных реакций карбонилирования [93], включая колебательную реакцию окислительного карбонилирования ацетилена [94], реакции окислительного хлоркарбонилирования ацетилена до транс-1-хлоракрилатов и транс-1-хлоракриловой кислоты [95, 96]. Показано, что синтез смесей эфиров насыщенных и ненасыщенных моно- и дикислот катализируется комплексами Pd(I) и происходит в результате сопряженного процесса [97–99]. Найдены катализаторы метатезиса алкинов в гомогенных условиях [100].
Были синтезированы или выделены из реакционной среды многочисленные металлоорганические комплексы – π-комплексы 25 металлов состава МmAn (A – алкин) и потенциальные интермедиаты каталитических реакций, образующиеся из π-комплексов (схема 1 ) и из малых кластеров состава М2 и алкина (схема 2 ) [1].
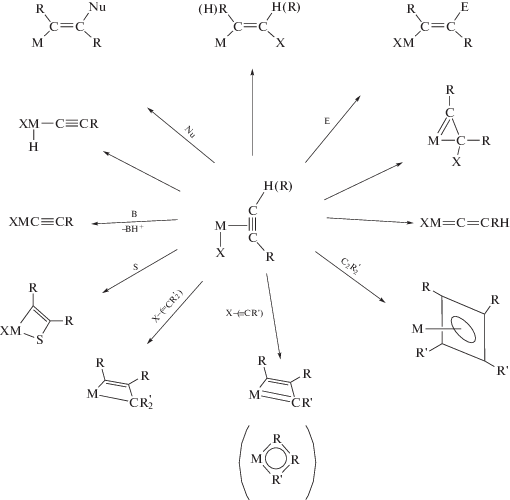
Схема 1 . Металлоорганические соединения, полученные из π-комплексов алкинов.
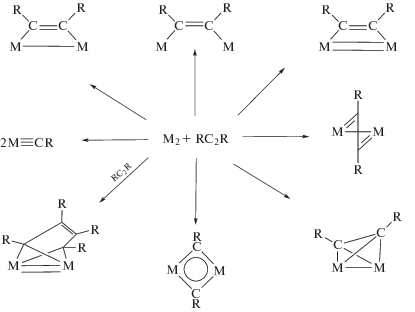
Схема 2 . Продукты реакций биядерных кластеров металлов с алкинами.
В результате систематических исследований кинетики и механизмов каталитических превращений ацетилена и его производных в растворах металлокомплексов были получены кинетические модели реакций в концентрированных растворах купрохлоридных комплексов – гидрохлорирования и гидратации ацетилена, метилацетилена и винилацетилена, анти-М-гидратации метилацетилена в системе полифункциональных катализаторов, реакции синтеза норборнодиена из ацетилена и циклопентадиена, реакции гидроцианирования ацетилена и тримеризации ацетилена [1, 8]. Исследовались также механизмы реакций карбонилирования [101], гидрирования, циклизации и полимеризации алкинов, окислительного хлорирования алкинов в водных системах полифункциональных катализаторов CuCl–CuCl2, CuCl2–HgCl2 и PdCl2–HgCl2 [8].
На четвертом этапе развития каталитической химии ацетиленовых соединений, которому и посвящен настоящий обзор, наибольший прогресс был достигнут в проведении каталитических реакций на основе комплексов Ni(II), Pt(II, IV), Ru(0, II, III, IV), Pd(0, I, II) и Au(I, III), а также на основе кластеров и наночастиц, содержащих эти металлы. Появилось множество новых лигандов. Характерной чертой последнего этапа является невероятно большое разнообразие ацетиленовых субстратов в синтезе сложнейших биоактивных соединений [9, 12–19, 22, 24, 27, 34, 35, 44].
В этот период были открыты новые реакции дикарбонилирования ацетилена и алкинов до соответствующих ангидридов насыщенных дикислот [93, 102–104], обнаружены новые колебательные реакции в каталитических системах окислительного карбонилирования алкинов [8, 104–106], реакции каталитического окисления ацетилена азотной кислотой до глиоксаля в растворах комплексов Hg, Pd, Pt, Rh, Ir и Au в присутствии катализатора нитрита натрия [107, 108], новые варианты реакций циклообразования, изомеризации, циклоизомеризации [11–15, 17–19, 24] и, в частноости, чрезвычайно важной реакции циклоизомеризации несопряженных енинов [11 (гл. 3)]. Значительный прогресс был достигнут в создании новых эффективных катализаторов реакций присоединения к алкинам соединений со связями Е–Н, Е–Е и E–E' (Е = S, Se, Te, P, B) и способов контроля селективности этих процессов [23, 28, 29, 33, 109–111]. Были найдены каталитические системы на основе комплексов палладия для исключительно активного и региоселективного процесса метоксикарбонилирования метилацетилена до метилметакрилата [112, 113], а на основе комплексов рутения для региоселективного анти-М-карбоксилирования алкинов [114, 115]. Открыта реакция окислительного иодирования ацетилена (и терминальных алкинов) с образованием (Е,Е)-1,4-дииодбутадиенов [116] и другие каталитические реакции алкинов в растворах комплексов платины(IV). Замечательными достижениями отмечено развитие региоселективных синтезов продуктов классической реакции димеризации алкинов [18]. Установлена специфическая реакционная способность комплексов золота в каталитических синтезах исключительно сложных молекулярных структур [117].
Ряд промышленно важных направлений применения С2Н2, а также перспектив его использования в синтетической органической химии, отраженных в публикациях 2013–2018 гг., обсуждены в обзоре [45].
На четвертом этапе интенсивно изучались механизмы старых и новых реакций. Была установлена важная роль в ряде процессов нанокластеров, “гигантских” кластеров и наночастиц в каталитических циклах, множественность центров катализа в “гомогенных” процессах [8, 118–120], а также достигнуто понимание важной роли стадий формирования и дезактивации активных центров в механизме каталитических реакций, т.е. возможной реализации цепных механизмов в металлокомплексном катализе [8, 118].
В задуманной серии обзоров планируется рассмотреть ряд каталитических реакций алкинов, в которых особенно ярко проявились достижения металлокомплексного катализа в ацетиленовой химии, – разработка высокоселективных процессов с разнообразием катализаторов и механизмов. Первый обзор этой серии посвящен одной из самых старых каталитических реакций – реакции димеризации и линейной олигомеризации терминальных алкинов. Димеры алкинов (енины) служат важными субстратами в синтетической химии биологически активных соединений и в химии материалов. Синтетические аспекты реакции димеризации детально описаны в обзоре С.Е. Гарсиа-Гарридо ([18 (с. 301)]). Кинетические и механистические аспекты процесса димеризации, влияние природы интермедиатов на региоселективность реакции, включая новые результаты 2014–2018 гг., рассмотрены в разделе 3 настоящего обзора. Окислительные превращения алкинов будут предметом второго обзора серии.
3. ДИМЕРИЗАЦИЯ АЛКИНОВ В РАСТВОРАХ МЕТАЛЛОКОМПЛЕКСНЫХ КАТАЛИЗАТОРОВ
Образование ениновых углеводородов в процессе димеризации терминальных алкинов можно отнести к реакциям различных типов – к реакциям олигомеризации (первый член ряда), этинилирования и винилирования (присоединение молекул НХ к тройной связи). В случае каталитической гомодимеризации терминальных алкинов могут образоваться три ениновых изомера – I (E-изомер), II (Z-изомер), III (гем-изомер) – и два кумуленовых 1,4-замещенных бутатриена IV и V (схема 3 ). В случае кросс-димеризации алкинов число хемо-, регио- и стереоизомеров возрастает до 18. В связи с этим создание каталитических систем (металл, лиганды, растворитель), обеспечивающих селективность процесса димеризации, является важнейшей задачей, решение которой для ряда систем относится к замечательным достижениям металлокомплексного катализа [18].
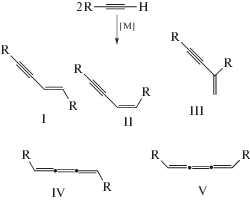
Схема 3 . Продукты гомодимеризации алкинов.
3.1. Комплексы металлов 11 группы Периодической системы элементов
Обзор катализаторов и реакций димеризации алкинов начнем с 11 группы металлов (Cu и Au). Комплексы Cu(I) являются исторически первыми катализаторами этой реакции (димеризации ацетилена) [46, 121, 122]. Их координационная и каталитическая химия детально исследована и, особенно, в химии алкинов [1, 8, 12, 14, 34, 51, 123]. Комплексы золота за последние 20–25 лет вошли как катализаторы в арсенал каталитической химии алкинов. Специфические особенности координационной химии Au(I) и Au(III) уже весьма глубоко изучены, исследованы природа и реакционная способность золотосодержащих интермедиатов каталитических реакций [10, 11, 13, 15, 18, 33, 124–127].
Фундаментальные различия физических и химических свойств Cu(I) и Au(I) связаны с особенностями электронного строения атома золота. Заполнение 4f-оболочки (так называемое лантаноидное сжатие), 4d- и 5d-оболочек и, соответственно, большое число электронов на атомных орбиталях (АО) при высоком положительном заряде ядра (79) приводят к специфическим релятивистским эффектам (РЭ), определяющим в значительной степени реакционную способность золота в степенях окисления 0, I и III [124, 125 и ссылки в них].
Очень кратко суть этого эффекта сводится к необходимости учитывать возрастание массы электронов на s-АО вследствие большой скорости вращения вокруг ядра удаленных от него электронов. Возрастание массы электрона сопровождается уменьшением боровского радиуса электрона и, соответственно, уменьшением атомного и ионного радиусов. РЭ касается всех s- и (в меньшей степени) р-электронов. Лантаноидное сжатие и сжатие АО за счет РЭ приводит к следующим последствиям [124, 125]:
1) сжатие 6s-АО стабилизирует вакантные АО, увеличивает акцепторные свойства Au(I) и Au(III), т.е. повышает Льюисову кислотность Au(I) и Au(III);
2) сильно возрастают окислительно-восстановительные потенциалы Au+/Au0 (~1.68 В), Au3+/Au0 (1.5 В), Au3+/Au1+ (~1.4 В), Au2+/Au1+ (>1.3 В) по сравнению с ионами меди (0.15 В для Cu2+/Cu1+). Аналогичная картина наблюдается в рядах Ni–Pt и Zn–Hg, поскольку РЭ существеннен и для Pt и Hg;
3) ион Au+ очень неустойчив и в отсутствие лигандов, содержащих атомы азота, серы и фосфора, легко диспропорционирует (реакция дисмутации) по реакции 3Au+ ⇄ Au3+ + $2{\text{Au}}_{{{\text{мет}}}}^{0}$ с константой равновесия, близкой к 1010;
4) заметно повышается первый потенциал ионизации атома Au (9.22 эВ) по сравнению с атомами серебра (7.57 эВ) и меди (7.72 эВ);
5) электроотрицательность Au(I) достигает значения 2.4 (1.8 для Cu(I) и 1.9 для Ag(I));
6) 5d-АО становятся более диффузными, но при этом уменьшается отталкивание электронов и понижается их энергия, что сопровождается увеличением энергии связи этих электронов с ядром и заметным снижением донорных свойств атома и ионов золота. Таким образом, способность к образованию дативных связей и к участию в реакциях окислительного присоединения заметно понижается;
7) уменьшение отталкивания 5d-электронов на диффузных АО в комплексах Au(I) сопровождается также сближением s-, p- и d-AO, что облегчает их гибридизацию и приводит к повышению устойчивости линейной геометрии комплексов LAuX и L2Au+X–.
8) наблюдается отчетливо выраженная аурофильность (образование связей Au(I)–Au(I)) по сравнению с купрофильностью;
9) высокая электрофильность Au(I) повышает прочность связей Au(I)–L, Au(I)–X и в том числе в многочисленных π-комплексах алкинов [125, 126], что делает π-комплексы алкинов способными присоединять различные нуклеофилы (Nu), включая олефины и даже алкины. π-Комплекс Au(С2Н2)+ прочнее на 10 ккал/моль аналогичного комплекса Au(С2Н4)+ ;
10) дативные связи Au+ → L менее выражены и в π-комплексах по сравнению с ионом меди, но весьма заметны в σ-комплексах, таких как Au=СНR+ и Au≡СR+, что и приводит к появлению стабильных катионных карбеновых интермедиатов, например,
11) в результате способности Au(I) к образованию π-комплексов, этинильных (алкинидных) соединений и аурофильности (связи Au(I)–Au(I)) получено огромное количество разнообразных кластеров и супрамолекулярных ассоциатов [11, 15, 125], осуществлены уникальные синтезы сложных органических молекул [117]. Все эти свойства Au(I), естественно, и привели к его использованию в катализе процессов димеризации алкинов.
Рассмотрим вначале купрокаталитическую димеризацию. Открытие реакции димеризации терминальных алкинов имеет очень долгую и поучительную историю, которая началась с работы Штрауса в 1905 г. [121]. Изучая реакцию Глязера [1, 8, 51, 52] окислительной конденсации этинильных соединений Cu(I) с различными окислителями, Штраус обнаружил, что в кипящей уксусной кислоте в отсутствие кислорода из PhC≡CCu образуется осадок состава PhC≡CCu · Cu(OAc) и углеводород С16Н12 (1,4-дифенилбутенин) по стехиометрическому уравнению
Образование димера фенилацетилена происходит в результате ацидолиза PhC≡CCu и катализа димеризации фенилацетилена ацетатом Cu(I). Позднее в работе 1959 г. [122] наблюдение Штрауса было подтверждено, и продукт димеризации алкинов был получен из исходного RC≡CCu в горячей уксусной кислоте даже при аэрировании раствора (для окисления металлической меди, возникающей при дисмутации ацетата Cu(I)) с образованием 1% диацетиленового производного.
К системе RC≡CCu–АсОН вернулись в 1997 г. [128] и было установлено, что при растворении Cu2О в кипящей уксусной кислоте и добавлении алкильных терминальных алкинов образуются Z- и E-1,4-диалкилбутенины (II и I). Процесс заканчивается появлением RC≡CCu и Cu(OAc)2. При использовании более сильной дихлоруксусной кислоты алкин не димеризуется. По аналогии со структурой тетрамерного комплекса [PhC≡CCuPMe3]4 [129] авторы полагали, что интермедиатом, ведущим к продукту внедрения фенилацетилена по связи ≡С–Cu, является димерный комплекс [Cu(PhC≡CH)2]+[Cu(C≡CPh)2 · AcOH]–, но никаких доказательств такого механизма не привели.
Более детально в течение второго и четвертого этапов была изучена димеризация ацетилена в каталитической системе, открытой Ньюлендом в 1931 г. [46], в концентрированных водных растворах CuCl–NH4Cl(KCl)–HCl [1, 8, 52, 123]. В этой системе (называемой “катализатором Ньюленда”) была получена корректная кинетическая модель с весьма обоснованным механизмом реакции димеризации ацетилена. Рассмотрим кратко основные этапы изучения катализатора Ньюленда.
1) Исследование растворимости CuCl в растворах NH4Cl и KCl в интервале 25–100°С показало, что при 80–100°С отношение [СuCl]/[NH4Cl] достигает значений 1.1–1.5 [8, 73, 123]. Этот факт однозначно указывает на образование многоядерных анионов ${\text{C}}{{{\text{u}}}_{m}}{\text{Cl}}_{n}^{{(n - m) - }}.$ Чалтыкяном [73] было замечено, что при концентрациях CuCl ниже 4–5 m и NH4Cl 6–7 m каталитическая система перестает быть гомогенной: из раствора выпадают различные ацетиленидные соединения (m – моляльная концентрация).
2) Изучение равновесий реакций комплексообразования CuCl с ионом Cl– потенциометрическим методом по Хедстрему [130–134] с использованием электронной спектроскопии [134] показало наличие в концентрированных растворах ~20 анионных комплексов ${\text{C}}{{{\text{u}}}_{m}}{\text{Cl}}_{n}^{{(n - m) - }}$ с m = 1–5 и n – m = 1–4. Например, при 25°С, [СuCl] = 4 m, [NH4Cl] = 6.5 m и катионном фоне (КФ) [NH4Cl + + NH4NО3] = 14 m мольная доля одноядерных комплексов равна ~0.4, а мольная доля только шести комплексов с m = 2–5 составляет ~0.35 [132].
Бόльшая доля моноядерных комплексов в системе CuCl–KCl–Н2О [135] и приводит, вероятно, к кристаллизации из растворов комплексов KCuCl2 и KCuCl3 при охлаждении растворов. Заметим, что системы, использованные авторами этой работы для проведения димеризации ацетилена до винилацетилена при 75°С, содержали 6.7 М CuCl, 6.4 М KCl и 8 М CuCl и 7.6 М KCl. В таких растворах содержание хлорид-иона недостаточно для образования даже комплекса ${\text{CuCl}}_{2}^{ - },$ не говоря уже о ${\text{CuCl}}_{3}^{{2 - }},$ что указывает на существование в растворе заметных количеств многоядерных анионов. Образование двухядерного комплекса было установлено даже при невысоких концентрациях CuCl для [NaCl] $ \leqslant $ 5 M и [CuCl] $ \leqslant $ 1.6 M в работе [136] методом растворимости. Из каталитической системы Ньюленда выделено много кристаллических многоядерных комплексов [см. 123, 137], например, комплекс (NH4)2Cu3Cl5 · 1/3(H2O) · 1/5(CuCl).
3) Для исследования кинетики и механизма реакции димеризации ацетилена в таких многокомпонентных средах, как катализатор Ньюленда, с очень сложной функцией закомплексованности катализатора была разработана специальная методика [8] на основе потенциометрического метода in situ, позволяющая определять активности ионов Cu+ (${{a}_{{{\text{C}}{{{\text{u}}}^{ + }}}}}$) и Cl–(${{a}_{{{\text{C}}{{{\text{l}}}^{ - }}}}}$), пропорциональные при КФ = сonst концентрациям этих ионов, и, главное, поддерживать их постоянными при варьировании ${{P}_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{2}}}}}$ и концентрации H3O+ [8, 138–141]. Концентрации ионов H3O+ определяли по результатам измерения кислотности Гаммета h0, практически также in situ, измеряя h0 в пробах раствора из реактора индикаторным методом в герметичной термостатируемой кювете [138, 140].
Исследование кинетики [138, 141–145] в проточном безградиентном реакторе [146] с измерением ${{a}_{{{\text{C}}{{{\text{u}}}^{ + }}}}}$, ${{a}_{{{\text{C}}{{{\text{l}}}^{ - }}}}}$, h0 и ${{a}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$ [130] при постоянных значениях КФ позволило получить корректные зависимости скорости реакции от ${{P}_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{2}}}}}$ и h0 $ \propto $ [H3O+] при ${{a}_{{{\text{C}}{{{\text{u}}}^{ + }}}}}$ = сonst, ${{a}_{{{\text{C}}{{{\text{l}}}^{ - }}}}}$ = сonst и уравнение (1) [138, 141]:
(1)
$W = \frac{{{{k}_{Н}}P_{{{{C}_{2}}{{H}_{2}}}}^{2}}}{{{{h}_{0}}{{а}_{{{{H}_{2}}O}}}{{а}_{{C{{l}^{ - }}}}} + K_{a}^{/}{{P}_{{{{C}_{2}}{{H}_{2}}}}}}}.$Поскольку величина константы $K_{a}^{/}$ не зависела от ${{a}_{{{\text{C}}{{{\text{u}}}^{ + }}}}}$и ${{a}_{{{\text{C}}{{{\text{l}}}^{ - }}}}}$ ([СuCl]), можно было провести анализ зависимости kH = f(${{a}_{{{\text{C}}{{{\text{u}}}^{ + }}}}}$, ${{a}_{{{\text{C}}{{{\text{l}}}^{ - }}}}}$) при варьировании [СuCl], предположив (как приближение), что в катализе участвует один активный комплекс ${\text{C}}{{{\text{u}}}_{m}}{\text{Cl}}_{n}^{{(n - m) - }}$ [142]. Анализ показал, что активными являются многоядерные комплексы со значениями m, равными 3 и 4, с преимущественным вкладом комплекса ${\text{C}}{{{\text{u}}}_{4}}{\text{Cl}}_{5}^{ - }$ [142]. Более строгий анализ [145] приводит к выводу, что значения m находятся в интервале 4.3–4.7, а в число активных комплексов входят анионы ${\text{C}}{{{\text{u}}}_{4}}{\text{Cl}}_{5}^{ - },$ ${\text{C}}{{{\text{u}}}_{4}}{\text{Cl}}_{6}^{{2 - }},$ ${\text{C}}{{{\text{u}}}_{5}}{\text{Cl}}_{6}^{ - }$ и ${\text{C}}{{{\text{u}}}_{5}}{\text{Cl}}_{7}^{{2 - }}.$ Механизм реакции димеризации ацетилена представлен реакциями (I)–(VI).
(I)
$\begin{gathered} {\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}{{({\text{O}}{{{\text{H}}}_{2}})}^{(}}{{^{{n - m}}}^{{) - }}} + {{{\text{C}}}_{2}}{{{\text{H}}}_{{2{\text{(aq)}}~}}}~~\underset{{{{k}_{{ - 1}}}}}{\overset{{{{k}_{1}}}}{\longleftrightarrow}} \\ \rightleftarrows {\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}{{({{{\text{С}}}_{2}}{{{\text{H}}}_{2}})}^{(}}{{^{{n - m}}}^{{) - }}} + {{{\text{H}}}_{2}}{\text{O}}. \\ \end{gathered} $В исходном купрохлоридном комплексе показана только одна из нескольких молекул воды, координационно связанных с атомами меди, замещение которой ацетиленом приводит к образованию π-комплекса (π1), который превращается в этинильный комплекс (σ1).
(II)
$\begin{gathered} {\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}{{({{{\text{С}}}_{2}}{{{\text{H}}}_{2}})}^{(}}^{{n - m) - }} + 2{{{\text{H}}}_{2}}{\text{O}}~~\underset{{{{k}_{{ - 2}}}}}{\overset{{{{k}_{2}}}}{\longleftrightarrow}} \\ \rightleftarrows {\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}_{{ - 1}}({{{\text{С}}}_{2}}{\text{H}}){{({{{\text{H}}}_{2}}{\text{O}})}^{(}}{{^{{n - m}}}^{{) - }}} + {{{\text{H}}}_{3}}{{{\text{O}}}^{ + }} + {\text{C}}{{{\text{l}}}^{ - }}. \\ \end{gathered} $Этинильный комплекс (σ1) через π-комплекс (π2) превращается в σ-металлоорганическое соединение (σ2) в результате внедрения π-координированного ацетилена по связи Cu–С≡ (реакция (IV)). Соединение σ2 быстро протолизуется с образованием моновинилацетилена (МВА).
(III)
$\begin{gathered} {\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}_{{ - 1}}({{{\text{С}}}_{2}}{\text{H}}){{({{{\text{H}}}_{2}}{\text{O}})}^{(}}{{^{{n - m}}}^{{) - }}} + {{{\text{C}}}_{2}}{{{\text{H}}}_{{2({\text{aq)}}}}}~\,\,\underset{{{{k}_{{ - 3}}}}}{\overset{{{{k}_{3}}}}{\longleftrightarrow}} \\ \rightleftarrows {\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}_{{ - 1}}({{{\text{С}}}_{2}}{\text{H}}){{({{{\text{C}}}_{2}}{{{\text{H}}}_{2}})}^{(}}{{^{{n - m}}}^{{) - }}} + {{{\text{H}}}_{2}}{\text{O}}, \\ \end{gathered} $(IV)
$\begin{gathered} {\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}_{{ - 1}}({{{\text{С}}}_{2}}{\text{H}}){{({{{\text{C}}}_{2}}{{{\text{H}}}_{2}})}^{(}}{{^{{n - m}}}^{{) - }}}~\xrightarrow{{{{k}_{4}}}} \\ \to {\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}_{{ - 1}}{{({{{\text{С}}}_{4}}{{{\text{H}}}_{3}})}^{(}}{{^{{n - m}}}^{{) - }}},\,\,\left( {{{\sigma }_{2}}} \right), \\ \end{gathered} $(V)
$\begin{gathered} {\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}_{{ - 1}}{{({{{\text{С}}}_{4}}{{{\text{H}}}_{3}})}^{(}}{{^{{n - m}}}^{{) - }}} + {{{\text{H}}}_{2}}{\text{O}}\,\,\overset {{\text{быстро}}} \leftrightarrows ~ \\ \rightleftarrows \,\,~{\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}_{{ - 1}}({{{\text{С}}}_{4}}{{{\text{H}}}_{3}}){{({{{\text{H}}}_{2}}{\text{O}})}^{(}}{{^{{n - m}}}^{{) - }}}, \\ \end{gathered} $(VI)
$\begin{gathered} {\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}_{{ - 1}}({{{\text{С}}}_{4}}{{{\text{H}}}_{3}}){{({{{\text{H}}}_{2}}{\text{O}})}^{(}}{{^{{n - m}}}^{{) - }}} + \\ + \,\,{{{\text{H}}}_{{\text{3}}}}{{{\text{O}}}^{ + }} + {\text{C}}{{{\text{l}}}^{ - }}~\,\xrightarrow{{{{k}_{6}}}} \\ \to {\text{C}}{{{\text{u}}}_{m}}{\text{C}}{{{\text{l}}}_{n}}{{({\text{O}}{{{\text{H}}}_{2}})}^{(}}{{^{{n - m}}}^{{) - }}} + {{{\text{С}}}_{4}}{{{\text{H}}}_{{4({\text{aq}})}}}. \\ \end{gathered} $Возможно также, что в образовании σ2 в стадии (IV) участвует молекула воды, приводя к аква-комплексу, получающемуся в стадии (V), а протолиз σ2 и присоединение Cl– (реакция (VI)) осуществляются в результате двух быстрых стадий. Используя условия квазистационарности для этой схемы механизма с одним маршрутом, получаем кинетическое уравнение скорости синтеза МВА. Оценка параметров модели и статистический анализ показали [142], что наименьшее значение дисперсии скоростей реакции получается при допущениях k–1 $ \gg $ ${{k}_{2}}{{a}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$ и ${{k}_{{ - 3}}}{{a}_{{{{{\text{H}}}_{2}}{\text{O}}}}}$ $ \gg $ k4. В результате получаем уравнение (2)
(2)
$W = \frac{{{{k}_{{{\text{эфф}}}}}a_{{{\text{C}}{{{\text{u}}}^{ + }}}}^{m}a_{{{\text{C}}{{{\text{l}}}^{ - }}}}^{n}P_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{2}}}}^{2}{{a}_{{{{{\text{H}}}_{2}}{\text{O}}}}}}}{{{{h}_{0}}a_{{{{{\text{H}}}_{2}}{\text{O}}}}^{2}{{a}_{{{\text{C}}{{{\text{l}}}^{ - }}}}} + {{K}_{{\text{a}}}}{{P}_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{2}}}}}}},$Из уравнений (1) и (2) следует, что процесс димеризации ацетилена в интервале температур 50–100°С [143] происходит без лимитирующей стадии (см. обсуждение понятия лимитирующей стадии в работе [147]). В исследованных условиях скорости протодеметаллирования [Cu]C≡CH (W–1) и стадии образования [Cu]C4H3 соизмеримы. При ${{h}_{0}}a_{{{{{\text{H}}}_{2}}{\text{O}}}}^{2}{{a}_{{{\text{C}}{{{\text{l}}}^{ - }}}}}$ $ \gg $ ${{K}_{{\text{a}}}}{{P}_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{2}}}}}$ (k–2${{a}_{{{{{\text{H}}}_{3}}{{{\text{O}}}^{ + }}}}}{{a}_{{{\text{C}}{{{\text{l}}}^{ - }}}}}$ $ \gg $ k4) лимитирующей будет стадия (IV), а при ${{h}_{0}}a_{{{{{\text{H}}}_{2}}{\text{O}}}}^{2}{{a}_{{{\text{C}}{{{\text{l}}}^{ - }}}}}$ $ \ll $ ${{K}_{{\text{a}}}}{{P}_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{2}}}}}$лимитирующей станет реакция образования σ1 (II).
Состав этинильного комплекса CumCln– 1(С2H)(n –m)– был установлен методами спектрофотометрии и потенциометрии [148]. В предположении преимущественного вклада только одного такого “желтого” комплекса в каталитический процесс наименьшая дисперсия значений функции F, связанной с интегральным оптическим поглощением “желтых” комплексов в интервале 350–420 нм, наблюдается при значениях m = 4.3 и n = 5.0. Исследования показали также, что ацетиленид меди Cu2C2 хорошо растворяется в концентрированных растворах СuCl, но существует только в слабокислых растворах (рН > 5 и h0 < 0.2). Поэтому в условиях синтеза МВА (h0 в интервале 0.2–0.5 в растворе СuCl (6.6 m) и NH4Cl (8.84 m) при оптимальной температуре 80°С [143]) его концентрация неизмеримо мала по сравнению с этинильным комплексом.
Полезная информация о природе второго металлоорганического интермедиата [Cu]C4H3 (σ2), который рассматривается как продукт внедрения ацетилена по связи [Cu]–C≡ в π-комплексе, получена при изучении влияния концентраций хлоридов различных металлов на скорость процесса в системе CuCl–NH4Cl–HCl–Н2О [144]. Обнаружено, что хлориды металлов, различающихся влиянием на скорость реакции, подразделяются на три группы:
– ZnCl2, CdCl2 и BiCl3 увеличивают скорость реакции за счет увеличения ${{a}_{{{\text{C}}{{{\text{u}}}^{{\text{ + }}}}}}}$ и, следовательно, концентрации активного купрохлоридного аниона и комплекса σ1;
– BеCl2, MgCl2, CaCl2, SrCl2, BaCl2, FeCl2, CoCl2, MnCl2 и CrCl3 уменьшают скорость реакции, увеличивая ${{a}_{{{\text{C}}{{{\text{l}}}^{ - }}}}}$ и снижая ${{a}_{{{\text{C}}{{{\text{u}}}^{{\text{ + }}}}}}}$ по аналогии с действиями NH4Cl и KCl;
– HgCl2, SnCl2 и CuCl2 резко понижают скорость реакции димеризации. Действие хлоридов SnCl2 и HgCl2 связано с увеличением кислотности раствора в результате гидролиза SnCl2 и в результате образования стабильных к протолизу ртутьорганических соединений, которые появляются в процессе переноса органических групп по реакции
при R = C≡CH, CH=CHC≡CH и C≡CCН=CH2.
Как было показано, основным источником, повышающим h0 до 2 при добавлении сулемы, служит образование в растворе соединения ClHgCH=CHC≡CH, которое при действии НCl (конц.) разлагается с получением МВА. Добавление CuCl2 приводит к превращению предполагаемого промежуточного [Cu]C4H3 (σ2) в 2-хлорвинилацетилен (ХВА) – CH2=C(Cl)C≡CH. Исследование кинетики конкурентных реакций образования МВА и ХВА [149, 150] позволило получить информацию о стадиях механизма после необратимой стадии образования [Cu]C4H3. Показано также, что в присутствии CuCl2 в стационарных условиях ([CuCl] = 12 m, [NH4Cl] = 12 m, [CuCl2] = 10–3–10–2 М (при непрерывной регенерации CuCl2 анодным окислением CuCl) при установленной линейной зависимости ${{a}_{{{\text{C}}{{{\text{u}}}^{{\text{ + }}}}}}}$ = const[Cu(II)]Σ удалось получить уравнения скоростей образования МВА (3) и ХВА (4) в условиях ${{a}_{{{\text{C}}{{{\text{u}}}^{{\text{ + }}}}}}}$ = const и ${{a}_{{{\text{C}}{{{\text{l}}}^{ - }}}}}$ = const, cоответствующие механизму синтеза МВА ((I)–(VII)) и схеме 4 превращения общего интермедиата [Cu]C4H3 (σ2).
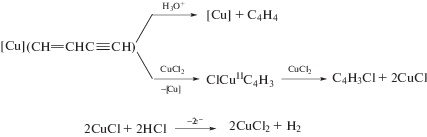
Схема 4. Механизм превращения интермедиата [Cu]C4H3 (σ2) в системе [CuCl] = 12 m, [NH4Cl] = 12 m, [CuCl2] = 10–3–10–2 М.
(3)
${{W}_{{{\text{МВА}}}}} = \frac{{P_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{2}}}}^{2}{{h}_{0}}}}{{({{k}_{{\text{I}}}}{{P}_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{2}}}}} + {{k}_{{{\text{II}}}}}{{h}_{0}})({{k}_{{{\text{III}}}}}[{\text{Cu}}{{{({\text{II}})}}_{\sum }}] + {{h}_{0}})}},$(4)
${{W}_{{{\text{ХВА}}}}} = \frac{{{{k}_{{{\text{III}}}}}P_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{2}}}}^{2}[{\text{Cu}}{{{({\text{II}})}}_{\sum }}]}}{{({{k}_{{\text{I}}}}{{P}_{{{{{\text{C}}}_{2}}{{{\text{H}}}_{2}}}}} + {{k}_{{{\text{II}}}}}{{h}_{0}})({{k}_{{{\text{III}}}}}[{\text{Cu}}{{{({\text{II}})}}_{\sum }}] + {{h}_{0}})}}.$Из уравнений (3) и (4) следует, что WХВА/WМВА = = kIII[Cu((II)Σ]/h0, а сумма скоростей WХВА + WМВА равна скорости образования МВА в отсутствие CuCl2. Механизм превращения [Cu]C4H3 в ХВА обсуждается в монографии [8].
4) Твердые π-комплексы CuCl и Cu2SO4 c ацетиленом и CuCl с МВА, а также CuC≡CH и твердые “желтые” ацетиленидные комплексы, описанные в ходе первых двух этапов развития химии ацетилена в работах М. Бертло, Р. Шавастелона, В. Маншо, Дж. Остерлофа, О.А. Чалтыкяна, Р. Вестина, А.Л. Клебанского, И.М. Долгопольского, Л.Г. Цюриха, А.А. Гинзбурга, И.К. Христича и Р. Наста, приведены в монографии [52].
Заметим, что разнообразная металлоорганическая и координационная химия, относящаяся к каталитической системе Ньюленда, была развита Львовской группой кристаллохимиков (М.Г. Мыськив и сотр.) [137, 151–160 ] и в работах других исследователей [123, 129, 161–165] в основном в период 1990–1999 гг. (четвертый этап). Эти исследования позволили получить представление о структуре многоядерных π-комплексов ацетилена, МВА, дивинилацетилена (ДВА), этинильных комплексов с фрагментами CuC≡CH, CuC≡CR и Cu2C2, которые позволяют представить структурный (кристаллохимический) аспект механизма димеризации ацетилена, детально обсужденный в работах [8, 123].
5) На схеме 5 представлены структуры четырехядерных по атому меди возможных интермедиатов процесса, построенные на основании фрагментов структур соответствующих кристаллических комплексов Cu(I), т.е. кристаллохимическая реконструкция механизма. В соответствии с кинетикой димеризации образование π-комплекса ацетилена В из А на стадии (I) происходит в результате разрыва хлоридного мостика или замещения координированной молекулы воды (заштрихована) с перемещением мостикового Cl– на место молекулы воды (без вытеснения хлорид-иона). Координационный узел в комплексе В аналогичен по строению одному из π-комплексов [123], например, NH4Cu3Cl4(C2H2) [152] или NH4Cu8Cl9(C2H2)4 [154 ] . При образовании этинильного интермедиата С (σ1) ион хлора может уходить в раствор одновременно с Н3О+ (стадия (II)) или выделяться в следующей квазиравновесной стадии без изменения вида кинетического уравнения.
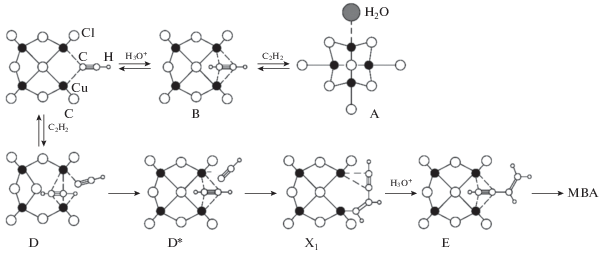
Схема 5. Кристаллохимический аспект механизма димеризации [123].
Из огромного разнообразия структур этинильных комплексов [123] интермедиат С можно сопоставить с комплексом (PyH)4Cu9Cl12(C≡CH) [123, 160] (рис. 1).
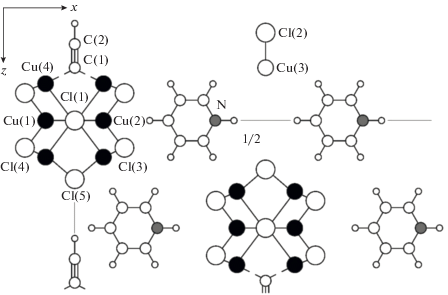
Более богатая координационная химия обнаружена в случае этинильных производных пропаргилового спирта (NH4)2Cu4Cl5(C≡CCH2OH) · H2O (рис. 2) [123, 156].
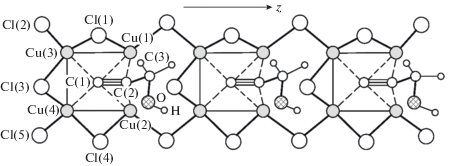
Отметим, что в растворе катализатора Ньюленда в зависимости от рН существуют этинильные и симметричные “желтые” ацетиленидные комплексы $({\text{С}}{{{\text{u}}}_{2}}{{{\text{C}}}_{2}}){\text{C}}{{{\text{u}}}_{p}}{\text{Cl}}_{q}^{{(q--p)--}}.$ Повышение кислотности (рН < 1.5) приводит к разрушению ацетиленидного комплекса и к увеличению до определенного предела концентрации интермедиата С.
Внедрение ацетилена из π-комплекса по связи Cu–C≡ в интермедиате D (схема 5 ) происходит с образованием фрагмента [Cu](CH=CHC≡CH) в Х1 (стадия (IV) механизма), вероятно, через интермедиат D*. Механизм этой стадии, отвечающей за стереохимию продуктов димеризации терминальных алкинов, обсуждается в обзоре [123]. Стадия протолиза интермедиата Х1 (стадия (VI)) с образованием Е не вызывает вопросов и давно известна в химии σ-металлоорганических соединений Cu(I) [166].
Кинетические модели реакций тримеризации ацетилена (синтез гексадиен-1,3-ина-5) и содимеризации ацетилена и МВА (синтез гексадиен-1,5-ина-3, ДВА) также были получены описанным выше методом [167, 168].
6) Кроме известных ранее типов координации алкинидных лигандов RC≡C– с атомами меди (VI, VII, VIII, IX), в случае купрохлоридных комплексов обнаружены еще 2 структуры – Х и ХI [123].
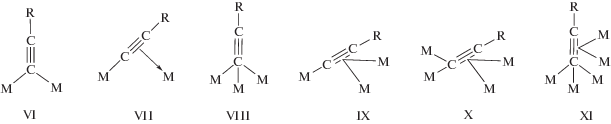
Весьма интересные варианты координации обнаружены и при взаимодействии Сu2C2 с купрохлоридными анионами [158].
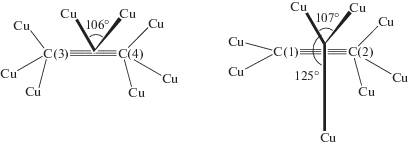
π,6 – Координационные узлы в структуре комплекса (NH4)8Cu29Cl29(C≡C)4 · 7H2O.
Полезные для понимания механизма синтеза МВА дополнительные результаты были получены недавно [169, 170] при анализе 1Н и 13С ЯМР-спектров в купрохлоридных растворах при пропускании ацетилена. При пропускании ацетилена в раствор 7.0 М CuCl и 6.66 М KCl при 50°С отмечено появление сигналов, соответствующих π-комплексам ацетилена, МВА и ДВА (табл. 2), и оценено изменение концентраций π-комплексов по времени реакции. Обнаружен также быстрый H/D-обмен ацетилена в растворах D2O с образованием HC≡CD, DC≡CD и дейтерированного МВА с заметным кинетическим изотопным эффектом (КИЭ), показавшим, что скорость расходования π-комплексов С2Н2 и самого ацетилена (в газовой фазе) в ~3 раза ниже в D2O. Эти наблюдения вполне ожидаемы в рамках кинетически и спектрофотометрически изученного механизма димеризации и тримеризации ацетилена (см. выше) с кинетически обратимой стадией (VIII)
(VIII)
$[{\text{Cu}}]({{{\text{C}}}_{2}}{{{\text{H}}}_{2}}) + {{{\text{Н}}}_{2}}{\text{О}} \rightleftarrows [{\text{Cu}}]{{({{{\text{C}}}_{2}}{\text{H}})}^{--}} + {{{\text{Н}}}_{3}}{{{\text{О}}}^{ + }}.$Таблица 2.
Значения химических сдвигов в π-комплексах алкинов
| Метод ЯМР | Химические сдвиги, м. д. | ||
|---|---|---|---|
| [Cu](CH≡CH) | [Cu](MBA) | [Cu](ДВА) | |
| 1H | 5.46 | 5.00 | 5.05 |
| 13С | 76.5 | 89.5 | 93.7 |
Кинетические результаты работ [169, 170] по определению первого порядка реакции (VIII) по концентрации π-комплекса С2Н2 и по концентрации ацетилена в газовой фазе, полученные в нестационарных условиях по значениям интенсивностей химических сдвигов при отсутствии контроля за изменением кислотности, вызывают большие сомнения. Так, например, при обратимости реакции (VIII) не может наблюдаться первый порядок по концентрации π-комплекса (в координатах ln(C0/C)–t) даже при практически постоянной концентрации Н3О+, а в случае кинетической необратимости этой стадии не должен наблюдаться быстрый H/D-обмен, если обмен вообще протекает через образование σ-этинильного комплекса Cu(C2H) и его протолиз. Так, например, полученные ранее результаты [171] по кинетике изотопного обмена в АсОН показали, что скорость образования RC≡CCu в ~100 раз меньше скорости изотопного H/D-обмена. Возможные механизмы изотопного обмена обсуждаются в работе [1]. Не ясна также роль диффузионных ограничений при определении порядка реакции по газообразному ацетилену.
Повышение стабильности действия катализатора Ньюленда в водных [172] и неводных [173] растворах регулированием кислотности растворов (добавлением НCl по ходу процесса) давно применяется на практике в водных системах, поскольку концентрация ацетиленидных “желтых” комплексов и [Cu](C2H)– и их растворимость зависят от концентрации кислоты НCl, которая расходуется на побочные процессы гидрохлорирования ацетилена, МВА и тримеров ацетилена и на окислительное хлорирование всех алкинов из-за присутствия следов кислорода и, следовательно, CuCl2.
Теперь рассмотрим аурокатпалитическую димеризацию. В результате изучения активности комплексов Au(I) в реакции димеризации алкинов в различных растворителях и условиях [174] было установлено, что димеризация эфира н‑BuO(CH2)3C≡CH в кипящем толуоле приводит к образованию изомеров III и II в соотношении (4.5–20) : 1. Это соотношение зависит от природы лиганда и природы основания, добавляемого в каталитическую систему, с выходом гем-изомера, достигающим 91–96%. В качестве катализаторов использовали растворимые комплексы LAuNTf2 (${\text{NTf}}_{2}^{ - }$ – некоординирующийся анион), а L – карбеновый (NНС) и фосфиновые лиганды.
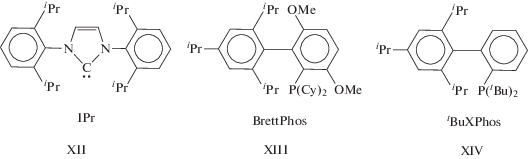
Лиганды в комплексах Au(I).
Наилучшие результаты были получены с лигандом XIV с добавками твердого NaOAc.
Изучена димеризация 9 алифатических алкинов с различными заместителями в алкильных группах. Предполагаемый механизм реакций аналогичен механизму в растворах комплексов Cu(I), который далее будем называть этинильным. В результате цис-внедрения связанного в π-комплексе алкина по связи [Au]–C≡ по Марковникову (или взаимодействия [Au]–C≡CR с π-комплексом [Au](RC≡CH)+ (вследствие аурофильности)) образуется преимущественно интермедиат RC≡C–C(R)=CHAuL. Замещение AuL+ протоном (вероятно, уксусной кислоты) приводит к изомеру III. Образование побочного продукта – изомера II (анти-М-внедрение) – может свидетельствовать и о возможном антиприсоединении аниона RC≡C– к алкину. Учитывая способность Au+ стабилизировать карбеновые и винилиденовые лиганды, можно также предполагать и участие винилиденового лиганда в механизме димеризации с образованием димера II. Превращение RC≡CAu(L)(=C=CHR)– может, вероятно, привести к изомеру II. Этот механизм, однако, требует образования трижды координированного атома золота, не характерного для Au+.
Уже на третьем этапе развития химии алкинов были найдены катализаторы весьма селективных процессов димеризации алкинов на основе комплексов Ni(II), Rh(I), Cr(IV), Sc(III), Y(III) и Ti(IV) [1]. За период 1990–2018 гг. (четвертый этап) появились новые металлокомплексные катализаторы димеризации терминальных алкинов и новые интермедиаты и механизмы реакции (см. также [18]).
3.2. Комплексы Ru, Os и Fe
Начнем с наиболее детально изученных процессов димеризации алкинов, катализируемых комплексами металлов 8 группы Периодической системы элементов и, в первую очередь, с катализа комплексами Ru.
Исследования селективности гомодимеризации алкинов и активности комплексов Ru с разнообразными моно- и полидентатными лигандами, включая циклопентадиенильные и карбеновые комплексы (табл. 3) показали, что в растворах комплексов Ru(II, 0) в качестве прекурсоров активных катализаторов селективно (≥95%) образуются изомеры I и II (схема 6 ), соотношение Е/Z которых определяется стерическими и электронными свойствами лигандов и заместителей в алкинах.
Таблица 3.
Катализаторы – комплексы рутения
| Номер кат-ра | Комплекс-катализатор1 | Лиганд | R в алкине | Селективность, % | Лит. ссылка |
|---|---|---|---|---|---|
| 1 | Ru(CO)L3(H)2 | PPh3 | tBu | IV (E) = 98 | 175 |
| 2 | RuL4(H)2 | PPh3 | tBu | IV (E) = 35, III = 43 | 175 |
| 3 | RuL3(OAc)H | PPh3 | tBu | IV (E) = 6, III = 50 | 175 |
| 4 | Ru(PP3)(Ph)H | PPh3 | tBu | I/II (E/Z) > 95/5 | 176 |
| Ph | I/II = 95/5 | ||||
| n-Bu | I/II = 5/95 | ||||
| 5 | Ru(NP3)(Ph)H | PPh3 | Ph | I/II > 95/5 | 176 |
| n-Bu | I/II < 5/95 | ||||
| 6 | Ru(SiP3)(Ph)H | PPh3 | Ph | I/II = 71/29 | 176 |
| n-Bu | I/II = 50/50 | ||||
| 7 | Ru(PP3)(H2)(H)BPh4 | PPh3 | Ph | II = 92 | 177 |
| 8 | Ru(PP3)(N2)(H)BPh4 | PPh3 | Ph | II = 92 | 177 |
| 9 | Ru(PP3)(C≡CSiMe3)BPh4 | PPh3 | SiMe3 | II = 95 | 177 |
| 10 | Ru(PP3)(C≡CPh)2 | PPh3 | Ph | II = 95 | 178 |
| Ru(PP3)(H)2 | |||||
| 11 | Ru(COD)(COT) + PR3 | PiPr3 | tBu | V(Z) = 78 | 179, 180 |
| 12 | Ru(Tp)L2Cl | PPh3 | Ph | I = 90 | 181 |
| SiMe3 | II = 82, III = 15 | ||||
| tBu | II = 98 | ||||
| 13 | Ru(Tp)L2(Py)Cl | PPh3 | Ph | I = 92 | 181 |
| 14 | Ru(Tp)L2H | PPh3 | Ph | I = 91 | 181 |
| 15а | Cp*RuL(H)3 | PPh3 | tBu | I/II = 95/5 | 182, 183 |
| SiMe3 | I/II = 98/2 | ||||
| Ph | I/II = 33/67 | ||||
| PhCH2 | III = 73 | ||||
| 15б | PCy3 | tBu | II/I/III = 17/35/48 | 182, 183 | |
| SiMe3 | I/II = 95/5 | ||||
| Ph | I/II = 10/90 | ||||
| PhCH2 | V > 95 | ||||
| 15в | PMe3 | tBu | II/I/III = 33/39/12 | 182, 183 | |
| SiMe3 | II/I/III = 10/28/62 | ||||
| Ph | I/II = 90/10 | ||||
| PhCH2 | II/I/III = 14/62/24 | ||||
| Cp*RuL(C≡CPh) | Ph | I/II = 80/20 | 184 | ||
| 16 | Cp*RuL(=C=CHPh)Cl Прекурсор + Et3N |
PPh3 | CO2Me | I + II = 100 | 185 |
| 17 | RuCl2(p-cymene)(L) | NHC | tBu | I + II = 67, III = 33 | 186 |
| Ph | I + II = 90, III = 10 | ||||
| 1-C8H14 | I + II = 18, III = 82 | ||||
| 18 | [RuCl2(p-cymene)]2 | – | Ar | I > 98 | 187 |
| 19 | RuCl2(=C=CHPh)L2 | PiPr3 | Ar | II = 80–89 | 188 |
| SiMe3 | III = 85 | ||||
| 20 | Ru(NP3)(CH3CN)(H)OTf | PiPr3 | Ar | I > 95 | 189 |
| Alkyl | |||||
| 21a | RuH(X)(PiPr3)2 | X = Cl | Ph | II = 76, I = 7, III = 17 | 190 |
| SiMe3 | I = 73.5, II = 21 | ||||
| 21б | X = N(SiMe3)2 | Ph | II = 78.8, I = 13.8 | ||
| tBu | II = 85.6 | ||||
| SiMe3 | II = 67.5, V = 32.5 | ||||
| 22 | Ru(CO)2Cl($\widehat {{\text{C N}}}$) | – | Ar | I ~ 100 | 191 |
1 Обозначения лигандов: 4. PP3 = P(CH2CH2CH2PMe2)3; 5, 20. NP3 = N(CH2CH2PPh2)3; 6. SiP3 = Si(Me)(CH2PMe3)3; 7–10. PP3 = = P(CH2CH2PPh2)3; 11. COD = циклооктадиен-1,5; COT = циклооктатриен; 12–14. Тр = трис-(пиразолилборат); 15, 16. Cp* = = C5Me5; 17. NHC = триазол-5-илиден  ; 22. $\widehat {{\text{C N}}}$
; 22. $\widehat {{\text{C N}}}$ 
Схема 6 . Продукты димеризации алкинов в каталитических системах на основе комплексов рутения.
Гем-изомер III является редким продуктом в селективной гомодимеризации – он образуется в растворах комплексов 15 (табл. 3, R = PhCH2) и 17 (табл. 3, R = н-гексил). Редкими изомерами являются и кумулены IV и V (табл. 3, схема 6 ). Особенно заметно влияние на селективность процесса геометрии координационной сферы атома рутения, т.е. степени его экранирования.
Наиболее подробно вклады стерических эффектов и природы лиганда в регио- и стереоселективность димеризации прослежены в случае весьма “закрытого” комплекса 15 (η5-С5M5)RuLH(H2) [182, 183]. При L = PPh3 (15a) и больших R (tBu и SiMe3) изомер I образуется с селективностью 95–98%. В случае L = PMe3 (15в) с теми же заместителями R образуется смесь изомеров I–III, и только при R = Ph получается изомер II с выходом 90%.
Изомер V (>95%) получается в растворах комплекса 15б (L = PCy3) и R = PhCH2. Реакции кросс-димеризации алкинов рассмотрим после анализа механизмов реакции, катализируемых комплексами рутения.
Общие представления о механизмах димеризации алкинов можно сформулировать в результате исследований димеризации ацетилена и алкинов в растворах комплексов Сu(I) (раздел 3.1), Ni(II) и Ni(0) [1], а также на основе винилиденовой гипотезы, предложенной Ямазаки в первой работе по катализу димеризации алкинов комплексами Ru(II) [175].
А. Этинильный механизм
B. Винильный механизм
Последовательность образования интермедиатов может быть и другой – сначала образуется этинильная группа, а затем винильная (алкенильная), но ключевой интермедиат должен содержать обе группы.
С. Винилиденовый механизм
Винилиденовая группа образуется различными путями:
1) H[M](C≡CR) $ \to $ [M](=C=CHR).
2) [M](η2-HC≡CR) $\xrightarrow[{ - B{{Н}^{ + }}}]{{B{{H}^{ + }}}}$ [M](=C=CHR).
3) [M](η2-HC≡CR) $ \to $ [M](=C=CHR).
Различные варианты распада интермедиата [M]-C4 в рамках этого механизма могут привести стерео- и региоселективно к изомерам I, II, IV и V.
Анализ работ по синтезу енинов в растворах комплексов Ru(II, IV) [175–191], в которых часто приводятся результаты исследований отдельных сторон механизма димеризации, а также работ, посвященных именно механизмам реакций [192–200], катализируемых комплексами рутения, позволяет выявить основные проблемы и сложившиеся парадигмы в этой области процессов димеризации. Наиболее обоснованным и разработанным механизмом катализа комплексами Ru является механизм С [176–178, 181, 192, 195, 197, 200]. Прокомментируем кратко несколько исследований.
В работе Добсона и др. [192] были изучены реакции комплекса HRu(O2CCF3)(CO)(PPh3)2 с терминальными алкинами и с диарилацетиленами с образованием винильных производных. Также был синтезирован гипотетический модельный интермедиат димеризации алкинов с 1,4-дифенилбут-1-ен-3-ин-2-ильным лигандом из гидридного комплекса HRu(O2CCF3)(CO)(PPh3)2 и 1,4-дифенилбутадиина, т.е. [M]-C4 или σ-С4. Структура этого комплекса Ru(С4НPh2)(O2CCF3)(CO)(PPh3)2 содержит фрагмент [Ru]–C(=CHPh)–C≡CPh (XII, σ-С4).
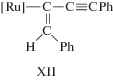
В этой работе отмечается, что такой интермедиат не может образоваться при внедрении алкина по связи [Ru]–C≡CPh, т.е. в рамках механизма А. На основании описанных в литературе в 1968–1972 гг. (см. обзор [200]) винилиденовых комплексов Fe и Pt было предположено, что интермедиат XII может получиться в результате образования винилиденового комплекса из алкина и последующего сочетания винилиденовой группы с этинильным лигандом (нуклеофильная атака группы RC≡C– по α-атому винилиденовой группы). В случае образования изомера III винилиденовый механизм С может быть исключен.
Комплекс с фрагментом XII был обнаружен в работах [176, 178, 194] среди интермедиатов, образующихся непосредственно из алкина и комплекса рутения в ходе димеризации алкинов. В работах [177, 193] синтезированы комплексы также с фрагментом [M]-C4, но со структурой XIII, отличающейся от комплекса XII.
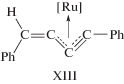
Переход комплекса XII в комплекс XIII становится понятным с учетом двух приведенных ниже мезомеров несимметричного η3-аллильного комплекса XIII.
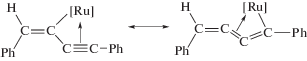
Таким образом, интермедиаты типа XII и XIII рассматривают как ключевые в винилиденовых механизмах, ведущих к димерам I, II, IV и V [179, 180, 190, 194, 197, 198].
Винилиденовые комплексы рутения были, по-видимому, впервые получены из алкинов в 1979 г. в работе Брюса и Уоллиса [201], изучены (в ряде случаев структурно) в работах [176–178, 195] и детально описаны в обзорах [115, 180, 200–203] и в монографии [204]. В этих обзорах обсуждаются и возможные механизмы образования винилиденовых лигандов. Установлено также образование моно- и диэтинильных комплексов из алкинов и из винилиденовых комплексов [177, 178, 181, 184] по реакции (IX)
(IX)
$\left[ {{\text{Ru}}} \right]\left( { = {\kern 1pt} {\text{C}}{\kern 1pt} = {\kern 1pt} {\text{CHR}}} \right)\xrightarrow{{\text{B}}}~\left[ {{\text{Ru}}} \right]\left( {{\text{C}}{\kern 1pt} \equiv {\kern 1pt} {\text{CR}}} \right) + {\text{В}}{{{\text{Н}}}^{ + }}.$В работе [195] была изучена кинетика изомеризации π-комплекса метилацетилена в винилиденовый комплекс
(X)
$\begin{gathered} {\text{CpRu}}{{{\text{L}}}_{2}}{{({{\eta }^{2}}{\text{ - MeC}}{\kern 1pt} \equiv {\kern 1pt} {\text{CH}})}^{{\text{ + }}}}{\text{PF}}_{6}^{ - }~\xrightarrow[{{\text{MeCN}}}]{{15 - 40^\circ {\text{C}}}} \\ \to {\text{CpRu}}{{{\text{L}}}_{2}}{{\left( { = {\kern 1pt} {\text{C}}{\kern 1pt} = {\kern 1pt} {\text{CHMe}}} \right)}^{ + }}{\text{PF}}_{6}^{ - }. \\ \end{gathered} $Реакция (X) оказалась мономолекулярной (первый порядок по π-комплексу) с $\Delta {{H}^{ \ne }}$ = 23.4 ккал/моль и $\Delta {{S}^{ \ne }}$ = 3.9 ± 0.9 кал моль–1 град–1. При 80–110°С винилиденовый комплекс превращается с выделением метилацетилена в комплекс CpRuL2(MeCN)+${\text{PF}}_{6}^{ - }$. Интересно, что превращение π-комплекса ацетилена в винилиденовый комплекс происходит только при 60–90°С.
В работе [197] был проведен квантово-химический анализ мономолекулярной трансформации π-комплекса ацетилена с Ru(II) в винилиденовый комплекс с анализом различных конформаций π-комплекса RuCl2(PH3)2(η2-C2H2) (XIV) и комплекса RuCl2(PH3)2(=C=CH2) (XV). Сравнение результатов расчета термической изомеризации
(XI)
${\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} = {\kern 1pt} {\text{С}} \to {\text{НС}}{\kern 1pt} \equiv {\kern 1pt} {\text{СН}},$∆Н0 = –48.7 ккал/моль и ∆Ea =1.07 ккал/моль (MP2/6-31G*) с результатами экспериментов ∆Н0 = –47.4 ± 4 и ∆Ea = 2 ккал/моль дало хорошее соответствие [197]. Расчеты авторов этой работы по той же методике с различными наборами базисов дали значения ∆Н0 = – 51.7 ккал/моль и ∆Ea = = 0.1 ккал/моль. Для перехода комплекса XIV в комплекс XV
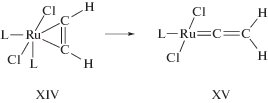
получено ∆Н0$ \cong $ 0, а величина барьера переходного состояния TS2 реакции XIV → XV равна 34.5 ккал/моль. Переходное состояние TS2 (а) образуется через интермедиат (б)
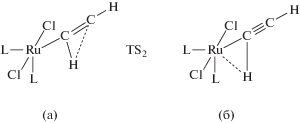
с агостиковой связью
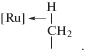
Оценки вероятности перехода π-комплекса RuCl2(PH3)2(η2-C2H2) (XIV) в продукт окислительного присоединения С2Н2 к Ru(II) с образованием этинильного октаэдрического комплекса Н[Ru](C≡CH) показали, что энергия такого комплекса намного выше энергии TS2 и что в данной системе такой переход даже термодинамически невероятен.
Здесь уместно обсудить и вопрос о степени окисления (СТО) Ru в винилиденовых комплексах, поскольку карбеновые лиганды часто рассматривают как нейтральные лиганды (синглетные карбены), предоставляющие атому металла σ-пару электронов. Такой подход справедлив для карбеновых комплексов Фишера. В случае карбеновых комплексов Шрока (М=СН2 и М=С=СНR) по всем формальным правилам карбен образует σ- и π-связи с металлом, т.е. повышает степень окисления металла на 2 ед., в качестве лиганда с зарядом –2 (общие подходы к определению СТО см. в [205]). Таким образом, степень окисления рутения в винилиденовом комплексе становится равной IV, т.е. повышается на 2 ед. по сравнению с исходным π-комплексом, в котором она равна II, как в работе [197]. Аналогичная ситуация наблюдается и в имидах [M2+](=NR2–), нитридах [M3+](≡N3–) и фосфидах [M3+](≡P3–) переходных металлов. Заметим также, что связи Ru=С= (1.76 и 1.84 Å [202]) и 1.8 Å [181]) существенно короче связей Ru–С≡ (2.08, 2.05 Å). Длина связи Ru–С в [Ru]–С4 находится в интервале 2.07–2.48 Å [177, 192, 193]. Длина связи Ru=С=, полученная в результате оптимизации геометрии в работе [197], составляет 1.63–1.66 Å (в зависимости от конформации комплекса), а длина связи Ru–С≡ в этинильном комплексе – –1.95 Å.
Образование кумуленовых продуктов димеризации алкинов происходит в результате цепочки превращений интермедиатов XII и XIII со сдвигом атома Ru из положения 2 (XII) в положение 4 (через XIII) (схема 7 , где А – алкин).
Схема 7. Механизм синтеза кумуленовых изомеров IV и V.
Еще в работе 1991 г. [179] было показано, что кумулен Z–H3SiCH=C=C=CHSiH3 обладает энергией на 17–19 ккал/моль выше соответствующих енинов I и II, т.е. термодинамически возможна также изомеризация кумуленов IV и V в изомеры I и II. Такая возможность была подтверждена экспериментально [190] с комплексом HRuCl(PiPr3)2. Таким образом, некоторые стадии схемы 7 могут быть кинетически обратимы.
В связи с возможной реализацией этинильного механизма димеризации (А) интересно отметить, что процесс может начинаться с образования винилиденового комплекса, который по реакции (IX) превращается в этинильный комплекс [181]. Механизм димеризации в таком случае представлен на схеме 8 .
Схема 8. Механизм А в катализе комплексами рутения.
Отметим, что последняя стадия деметаллирования интермедиата [Ru](σ-C4) с образованием продукта I в этом механизме происходит в результате метатезиса σ-связей [Ru]–C и Н–С≡, однако в некоторых каталитических системах на основе Ru источник атома Н остается неясным. В работе [187] проблема деметаллирования в рамках механизма С решалась в стадии протолиза интермедиата [Ru](σ-C4) в системе, содержащей АсОН.
По кросс-димеризации алкинов в случае комплексов Ru очень мало примеров селективных реакций. В реакции присоединения RC≡CH к интернальным алкинам R1C≡CR2 при катализе комплексом Cp*Ru(C≡CPh)(PPh3) (16, табл. 3) из винилиденового прекурсора [185] получен продукт PhC≡C–C(R1)C=CHR2 с выходом 44–90%. Высокая хемоселективность достигается только с R1 = tBu, iBu, SiMe3 и R2 = CO2Et, COMe. В реакции tBuC≡CH с MeС≡СMe селективность составляет 100%. При изучении этой реакции выделен интересный интермедиат механизма А с агостиковой связью [Ru](Н–СН2–) (XVI).
Описана кросс-димеризация алкинов [188] ArC≡CH c Me3SiC≡CH, катализируемая винилиденовым комплексом RuCl2L2(=C=CHPh). В случае p‑TolC≡CH и L = PiPr3 в присутствии аминов образуется, в основном, изомер II – Me3SiC≡CCH=CH(p-Tol) – с выходом от 82 до 93% (в зависимости от природы амина). Содержание гем-изомера III варьируется в пределах 7–14%. Образование изомера II рассматривают как результат реализации механизма С, а изомеров I и III – механизма А.
Комплексы осмия(II) в реакциях с алкинами исследовали Добсон и сотр. [192], показавшие, что комплексы Os(O2CCF3)2(CO)L2, OsH(O2CCF3)(CO)L2 и Os{C(C≡CPh)=CHPh)}(O2CCF3)(CO)L2 являются катализаторами олигомеризации фенилацетилена с образованием олигомеров С64Н48 и С90Н80. Гидридный комплекс осмия катализирует образование изомерных винильных продуктов присоединения фенилацетилена к толану и метилфенилацетилену.
Различные гидридные комплексы осмия (H[Os], H2[Os], H3[Os] и H4[Os]) в реакциях с алкинами образуют разнообразные металлоорганические соединения [206 и ссылки в ней], содержащие алкенильный, алкилкарбиновый (алкилидиновый), винилиденовый и алкинидный лиганды соответственно. Комплекс HOs(=C=CHPh)(η2-O2CCH3)L2 катализирует димеризацию PhC2H до смеси изомеров I и II. В работе [207] детально изучен механизм димеризации с участием комплекса HOs(PP3)(N2)BPh4 (PP3 = P(CH2CH2PPh3)3, XVII). Аналогично комплексам рутения (см. выше) комплекс XVII в реакции с Me3SiC≡CH образует через винилиденовый комплекс H[Os](=C=CHSiMe3) интермедиат типа ХIII (Os[η3–Me3SiC3=CHSiMe3] [BPh4], XVIII) и 1 экв. стирола. В избытке PhC2H протекает каталитический процесс образования изомера II с выходом ~90%. Исследованием 1H, 31P (1H) ЯМР in situ и моделированием интермедиатов и их реакций установлена последовательность превращений фенилацетилена в присутствии гидридного комплекса осмия:
Показано, что каталитический цикл начинается и заканчивается образованием [Os](C≡CR)+:
– формирование активного центра
– каталитический цикл
Любопытно, что еще в работе 1985 г. [208] было установлено, что окисление комплекса Os(C≡CPh)L2 (L = PMe3) ионом серебра (AgPF6) также дает продукт Os(σ-C4)L4PF6 в результате окислительного сочетания этинильных групп с отрывом атома Н от растворителя и с восстановлением Ag+ до Ag0.
В случае простых гидридных комплексов HOsCl(CO)(PiPr3)2 в присутствии Et2NH из алкинов были получены и бутатриены IV и V, стабильные при R = tBu и Cy [209]. В случае R = Ph полученные бутатриены полимеризуются с образованием олигомеров [–CH(Ph)C≡CC(H)Ph)–]n. При R = Me3Si бутатриен изомеризуется в енин II. Показано, что роль амина состоит в формировании активного в процессе интермедиата:
Изучение различных пинцерных комплексов осмия показывает [210 и ссылки в ней], что комплексы Os являются обещающей альтернативой другим металлокомплексным катализаторам. Так, пинцерный комплекс с РОР-лигандом –H4Os[POP] (XIX), где РОР = [Xant(PiPr3)2] (XX),
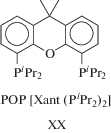
является активным катализатором димеризации алкинов до изомера II с выходом 94% (R = Ph, TOF = 100 ч–1) и 90% (R = tBu, TOF = 30 ч–1). Формирование активного катализатора происходит из XIX с образованием двух молекул олефина и выделением молекулы Н2:
Каталитический цикл представлен на схеме 9 [210].
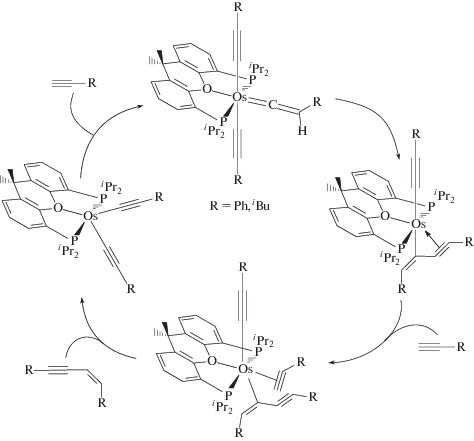
Схема 9. Каталитический цикл с прекурсором H4Os[POP] (XIX) (схема воспроизводится с разрешения из Inorg. Chem. 2013. V. 52. P. 6199 [210]. Copyright (2013) Am. Chem. Soc.).
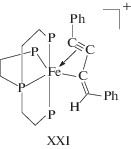
Комплексы Fe хотя и редко используются в работах по катализу димеризации алкинов, но и в этой области получены интересные результаты [211–216]. Комплексы FeCl2L и HFeClL, HFe(H2)L+ c L = PP3 и [P(CH2CH2PMe2)3] мало активны в димеризации алкинов [211]. Z-изомер (II) из PhC2H образуется селективно, но медленно – со скоростью 1 оборот катализатора в сутки.
Однако в этой работе получены и изучены 4 изомера промежуточных комплексов (PP3)Fe(η3-PhC3C=CHPh)+${\text{BPh}}_{4}^{ - }$ с преимущественным образованием изомера XXI.
В работах Даша и сотр. [212, 213] исследовались простые системы на основе комплексов FeCl3L (L – диметилэтилендиамин (ДМЭДА)) в присутствии KOtBu и других оснований. На примере 17 арилацетиленов показано образование смеси изомеров I и II с преимущественным синтезом изомера I (78–89%). Изучение влияния различных аминов в качестве лигандов и радикальных ингибиторов (ТЕМРО) [213] привели к созданию оригинального механизма димеризации, включающего участие арилвинильных радикалов и изменение степени окисления атома железа в ходе процесса (Fe(II) → Fe(III) → Fe(IV) → Fe(II)). Роль KOtBu сводится к образованию анион-радикала из ArC2H и арилвинильных радикалов из анион-радикала:
Полученные интермедиаты взаимодействуют, например, с Fe(III) с образованием интермедиата [Fe(IV)](C≡CAr)(CН=CHAr), распадающегося до Fe(II) и изомера I.
В работе Мильштейна и сотр. [214] исследовали пинцерный комплекс HFe(BH4)(iPr-PNP) и установили селективное образование изомера II в гомодимеризации фенилацетилена (94%) и других арилалкинов (13 примеров). Показано также весьма селективное кросс-сочетание ArC2H и Me3SiC2H (Z-изомер II с выходом 79–99%) без каких-либо добавок. Активный катализатор [Fe](BH4)(C≡CAr) формируется из исходного комплекса с выделением Н2 и с сохранением лиганда BH4 в координационной сфере атома Fe в ходе каталитического процесса.
Интересный аминокарбеновый лиганд XXII был использован в работе [215] в присутствии KOtBu.
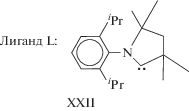
Комплекс Fe(CО)4L с этим лигандом катализирует димеризацию фенилацеилена с высокой селективностью по отношению к изомеру I (82–84%) и с высоким числом оборотов катализатора (6500). В статье обсуждается механизм изомеризации II → I через металлкарбеновый интермедиат, а также выполненный квантово-химический анализ механизма, однако по неясной причине рассматривается транс-присоединение этинильного фрагмента к π-комплексу алкина.
В работе 2017 г. [216] исследован катализ димеризации алкинов пентаметилциклопентадиенильными комплексами Fe–Ср*Fe(HL)Cl и Ср*Fe(L), где HL = XXIII, и установлена исключительно высокая селективность в отношении гем-изомера III
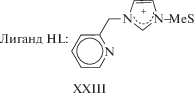
(>99%) в случае комплекса Ср*Fe(L). Селективность кросс-димеризации пропаргилового спирта с арилалкинами также высока с этим комплексом. Изомер III ArC≡CC(CH2OH)=CH2 образуется с выходом 90–94%. Как и в случае других металлов, природа лиганда является существенным фактором, определяющим селективность процесса.
В заключение этого раздела заметим, что интерес к катализу различных реакций комплексами железа в органическом синтезе возрос невероятно быстро. Так, в обзоре 2015 г. [217] библиография включает 1651 ссылку.
3.3. Комплексы Rh, Ir и Co
Комплексы Rh(I) начали использовать в реакции димеризации (и олигомеризации) алкинов еще на втором и третьем этапах развития химии ацетилена. По-видимому, первыми Уилкинсон и сотр. [218] провели реакцию димеризации алкинов с комплексом Rh(I) (с так называемым комплексом Уилкинсона RhCl(PPh3)3). Оказалось, что фенилацетилен быстро полимеризуется, а замещенные α-гидроксипропаргильные амины димеризуются с образованием изомера I (73%) и π‑комплекса I с RhCl(PPh3)2. Авторы предположили этинильный механизм димеризации А с образованием продукта окислительного присоединения RC≡CH к Rh(I) в качестве интермедиата.
В работе [219] изучали активность катионного комплекса цис-Rh(diphos)(CO)2BPh4 в неселективной олигомеризации МеС2Н в СН2Cl2. При 60оС было получено 50% димеров (I/III = 3), 8% линейных тримеров и 42% изомерных триметилбензолов. Аналогичные димеры образуются из α- и β-гидроксизамещенных терминальных алкинов. При 80°С в случае β-гидроксипроизводных получены изомеры в процентном соотношении III/I, равном от (55/45) до (75/25) [220]. Кинетические измерения показали второй порядок по концентрации алкина для этих субстратов. Показано также, что добавки лигандов AsPh3 и PPh3 к комплексу Rh(C8H14)2Cl влияют на соотношение тримеров и димеров, которое при [L]/[Rh] = 6 и варьировании отношения [AsPh3]/[PPh3] меняется от 1/19 до 6.7/1 [220].
В работе 1990 г. Виноградов и сотр. показали [221], что комплекс RhCl(PМе3)3 в ацетоне приводит к образованию суммы изомеров I и III (95–98%), причем, в отличие от алкильных алкинов, фенилацетилен не димеризуется. При анализе 1Н и 31Р (1Н) ЯМР-спектров в реакции пентина-1 (–65°С) было установлено образование π-комплекса, который при –10оС превращается в π-комплексные изомеры Rh(PМе3)3(RC≡CH$)_{2}^{ + }$Cl– и в смесь этинильных комплексов НRh(PМе3)3(C≡CR)(RC≡CH)+Cl– (а, b) с отношением изомеров комплексов (а)/(b) ≈ 1.5, что близко к отношению концентраций изомерных димеров I/III. Повышение температуры до 25оС приводит к образованию этинилвинильных интермедиатов (c, d).
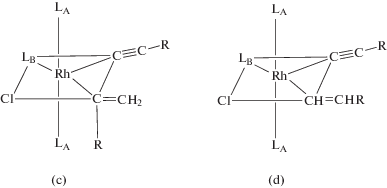
Первый изомер (с) приводит продукту III, а комплекс (d) – к продукту I. Полученные результаты подтвердили реализацию винильного механизма димеризации В. В работе [222] также подтверждено цис-внедрение алкина по связи Rh–Н (по Марковникову и против правила Марковникова) и показано, что в случае димерного комплекса (RhClL2)2 при 25–50оС получаются изомеры I и III. При этом при R = Ph получено 100% изомера III, при R = tBu – 100% изомера I, а при R = = n-Pr – 37% изомера I и 63% изомера III.
При использовании π-аллильного комплекса родия Rh(η3-C3H5)(PiPr3)2 [223] были получены доказательства механизма димеризации С через образование этинилвинилиденового интермедиата. В работе 2005 г. [224] эти же авторы продолжили изучение интермедиатов и закономерностей димеризации алкинов и показали, что этинилвинилиденовый комплекс в реакции с СО превращается в интермедиат типа XII (σ-C4):
Полученный E-изомер при действии кислот образует изомерные димеры (R = Ph):
Изомеризация E-изомера типа XII при действии сильной кислоты приводит к кумуленовому продукту [Rh](RC=C=C=CHR), превращающемуся в димер IV. В работе также детально изучены превращения этинилвинилиденового комплекса в продукт XII (E, Z) и далее в димеры I, II и в бутатриены в ходе термического и фотохимического процессов. Интересные превращения обнаружены в случае незамещенного ацетилена:
Окислительное присоединение MeI к комплексу RhCl(CO)L2 приводит к стимулированию реакции димеризации с активными комплексами Rh(III) [225]. Наряду с небольшим количеством алкина RC≡CMe (ацетон, тетрагидрофуран, СН2Cl2), который количественно образуется в избытке MeI, в ходе димеризации в присутствии метилата натрия по механизму А образуются продукты I и III (80/20, в %). При этом природа продуктов и селективность димеризации существенно зависят от природы растворителя. В ацетонитриле из фенилацетилена и метилиодида получается только метилфенилацетилен, а в метаноле с добавкой К2СО3 из алкинов в присутствии метилиодида получается продукт II (80–99%). Образование этого изомера авторы рассматривают как результат реализации механизма С.
Изучение реакции присоединения алкинов к диэфирам ацетилендикарбоновой кислоты [226] в растворах пинцерных комплексов Rh(III)(NCN) (OAc)2(H2O), где NCN – бис-(оксазолинил)фенил, позволило получить Z-изомер продукта димеризации с выходом 98–100%. Показано два пути образования этинильного интермедиата в рамках механизма А:
При использовании H2 этинильный лиганд образуется в результате метатезиса σ-связей H–[Rh] и Н–С≡:
Весьма редкий продукт димеризации III получается селективно (90%) в случае N-замещенных пропаргиламинов RNHCH2C≡CH в растворах комплексов RhClL3 и [RhCl(COD)]2 в присутствии dppf (dppf – 1,1-бис-(дифенилфосфино)ферроцен) [227]. В случае 8-оксихинолинатных комплексов Rh(I) с COD и в присутствии PPh3 из арилалкинов образуются изомеры III и I в процентном соотношении 56/8 [228]. Катализ димеризации алкинов обнаружен также и в растворах карбеновых комплексов Rh(I) – [Rh(NHC)(Cl)(COE)]2 и Rh(NHC)(Cl)(COE)(Py) (XXIV), где СОЕ – циклооктен, а NHC – 1,3-бис-(2,6-диизопропилфенил)имидазол-2 [229]. Комплекс XXIV, например, катализирует образование из фенилацетилена в С6D6 при 40°С 82% III, 4% тримеров и 10% 1,2,4-Ph3С6Н3. При избытке Py этот комплекс приводит к 92% III и 6% тримеров. В случае R = tBu, Me3Si при конверсии алкинов 90–100% получается продукт III с выходом 84–98%. Синтезированы интермедиаты процесса и получены ЯМР-спектры π-комплексов с PhCH2C≡CH и с продуктом I (tBu), а также этинильного комплекса Rh[NHC](Cl)(H)(C≡CSiMe3)Py2. Авторы считают комплекс XXIV активным универсальным катализатором, работающим в рамках механизма А в течение 3–6 ч, при 100%-ной конверсии с селективностью 92–95%.
Комплексы Ir и Co использовались лишь в нескольких работах по катализу реакции димеризации. Комплекс Ir (η2-SNC5H4)(PPh3)2 [230] по механизму В катализирует димеризацию алкинов с образованием изомера I (94–100% для R = Ph , tBu и n-Bu) через интермедиат НIr(η2-SNC5H4)(С≡СR)L2.
Детальное экспериментальное и квантово-химическое исследование активности гидридных пинцерных комплексов иридия Ir(РСР)(Н)2 (РСР – (2,6-СН2РtBu2)2С6Н3) и возможных интермедиатов процесса позволило авторам работы [231] сделать интересные выводы. Реакция гидридного комплекса с норборненом (NВЕ) приводит к норборнану и к π-комплексу Ir(РСР)(NВЕ), который и является активным катализатором димеризации PhC2H с образованием продукта I в рамках механизма В
Приведем некоторые любопытные наблюдения и заключения авторов этого исследования.
1) Гидрид этинильный комплекс на 29 ккал/моль стабильнее фенильного аналога H[Ir]Ph.
2) Внедрение алкина по связи H–[Ir] происходит быстро и дает продукт 1,2-внедрения [Ir]–С(Ph)=CH2, но этот процесс обратим.
3) Продукт 1,2-внедрения в результате β-элиминирования H–[Ir] превращается в π-комплекс и затем более медленно в продукт 2,1-внедрения [Ir](C≡CPh)(CH=CHPh), который быстро в ходе восстановительного элиминирования дает продукт I.
4) Затруднения в образовании связей С–С являются общим свойством интермедиатов типа [Ir](C≡CR)(С(R)=CH2) и возникают из-за экранирования Ir заместителями лиганда РСР.
5) Компьютерные расчеты показали, что винильная группа в переходном состоянии стадии (3) должна повернуться на 90° для достижения нужной ориентации. Это вращение затрудняется существенным экранированием заместителями в случае α-заместителя в винильной группе. Интересно, что, хотя восстановительное элиминирование с образованием связи С–С уменьшает напряженность в координационной сфере металла, стерические препятствия ингибируют сам процесс элиминирования димера.
Заметим, что наблюдаемые эффекты не требуют обратимости стадии внедрения алкина по связи М–Н. Продукты 2,1- и 1,2-внедрения могут возникать и параллельно. При этом продукт 2,1-внедрения будет образовываться медленно, но быстро превращаться в димер, а продукт 1,2-внедрения будет возникать быстро, но медленно превращаться в продукт димеризации. При этом обратимость стадии внедрения, конечно, полезна, поскольку будет препятствовать накоплению ненужного интермедиата (см. кинетику асимметрического гидрирования олефинов [8]).
Исследуя реакцию гидросилилирования алкинов в растворах комплексов Со(I), авторы работы [232] установили, что в случае фенилацетилена комплекс СоСl(РМе3)3 катализирует реакцию димеризации с образованием изомера I (78%) и цикломеризацию алкина до 1,3,5-Ph3С6Н3 (19%), а комплекс СоСl(РМе3)2(СО)2 в основном – образование изомеров I (76%) и II (13%). Реакции димеризации и циклообразования катализируют также комплексы CoBr2(dppe) в присутствии восстановителей Zn0 и Mg0 [233]. Выход димера I из фенилацетилена составляет 47%, а циклического тримера – 41%.
В заключении раздела заметим, что эта триада металлов участвует в катализе димеризации алкинов с преимущественным образованием изомеров I, III, IV и линейных тримеров в комплексах с различным лигандным окружением и с металлами в степенях окисления I и III.
3.4. Комплексы Ni, Pd и Pt
Комплексы Ni(0, II) исследовали в реакциях димеризации и олигомеризации алкинов [1, 52] на втором и третьем этапах развития химии ацетиленовых соединений [234–237], начиная с работ Реппе по карбонилированию и циклизации ацетилена и алкинов [1, 47, 56].
Из сравнения реакционной способности металлов 10 группы Периодической системы элементов, проведенного в обзоре [43], следует, что энергии активации стадии восстановительного элиминирования в реакции (XII)
(XII)
${{{\text{L}}}_{2}}{\text{M}}{{\left( {{\text{C}}{{{\text{H}}}_{{\text{3}}}}} \right)}_{2}} \to {{{\text{L}}}_{2}}{\text{M}} + {\text{C}}{{{\text{H}}}_{3}}{\text{C}}{{{\text{H}}}_{3}}$Рассмотренные выше свойства комплексов никеля и приводят к высокой активности комплексов Ni(CO)2(PPh3)2 в реакциях линейной и циклической олигомеризации терминальных алкинов [234, 235] и к полимеризации несопряженных терминальных алкинов [236]. Среди димеров [235] в случае алкильных заместителей С2–С5 были обнаружены изомеры I и III, а среди линейных тримеров – 3 разветвленных изомера, структуры которых были определены спектроскопическими и химическими методами.
В работе [237], подводящей итоги серии исследований реакций алкинов в циглеровской системе NiX2–AliBu3, приведена интересная информация о механизме образования Е-2,4-диалкил-1,3-бутадиена из алкинов одновременно с триалкилбензолами (после гидролиза алюминийорганических интермедиатов). Показано образование алкилгидридных комплексов HNi(iBu)Ln и ${\text{NiL}}_{n}^{0}$ из комплексов Ni(II) с ацетилацетонатным и салицилальдиминатным лигандами. На основании изотопных экспериментов с RC≡CD и H2O/D2O авторы делают вывод, что, помимо винильного механизма образования связи С–С в диене, получающемся после гидролиза интермедиатов iBu2AlC(R)=CH–C(R)=CH2 и RCH=CH–C(R)=CHAlIBu2, часть диена образуется после гидролиза циклических продуктов (XXV), полученных окислительным присоединением алкинов к комплексу ${\text{NiL}}_{n}^{0}$ и соответствующих циклических алюминийорганических соединений.
В случае комплекса Ni(CO)2(PPh3)2 при больших соотношениях AliBu3/Ni = 60 в результате димеризации гексина-1, кроме диенов, получен изомер III с выходом 17% в смеси с изомерными трибутилбензолами.
Селективный синтез димера I был проведен в очень активной каталитической системе Ni(COD)–PtBu3–C6D6 [238] при комнатной температуре. Выход продукта за 5 мин достигал (в зависимости от природы заместителя в алкине) 88–98%. Некоторые алкины побочно превращались в тримеры и олигомеры. На этом катализаторе получен первый пример димеризации Bu3Sn(C≡CH) с выходом 88% изомера I. Предполагаемый механизм димеризации В состоит в цис-внедрении алкина по связи H–Ni(C≡CR) c образованием интермедиата (RCH=CH)Ni(PtBu3)(C≡CR). В отсутствие растворителя полученный димер может реагировать с арилбромидом (в присутствии комплексов Pd(0) в диметилформамиде, приводя к продуктам реакции Мигита–Стилле (кросс-сочетание с участием SnR4).
Комплексы палладия в качестве катализаторов димеризации алкинов в отличие от никелевых катализаторов изучали, в основном, на четвертом этапе развития химии ацетиленовых соединений, однако разнообразие типов комплексов и используемых алкинов, возможности регулирования регио- и стереоселективности реакции привлекли к палладиевым катализаторам заслуженное внимание специалистов. Заметим, что катализ комплексами палладия в химии алкинов вообще весьма развит. Так, в обзоре 2014 г. [41] приведено более 600 ссылок на статьи по этой тематике.
В работе, опубликованной еще в 1984 г. [239], было показано, что ацетат палладия в таких растворителях, как CH3CN, C6H6 и Et3N, в присутствии фосфонильных лигандов (например, iPr2(Ph)P=O) катализирует образование димеров III и тримеров пропаргиловых спиртов, причем отношение димер/тример значительно возрастает при добавлении CuI и при увеличении числа заместителей у γ-атома углерода, связанного с ОН-группой. В оптимальных условиях это соотношение достигает 99/1.
Неселективная кросс-димеризация происходит также с образованием 4 возможных изомеров геминальных димеров. Предполагается образование промежуточного комплекса RC≡CPdH, но без каких-либо оснований. Интересно, что в случае ацетилена в этой системе образуется только моновинилацетилен.
Систематические исследования каталитических свойств комплексов Pd(OAc)2 с фосфиновыми лигандами в реакциях гомодимеризации и кросс-сочетания (присоединения терминальных алкинов к дизамещенным ацетиленам) было предпринято Тростом с соавт. [240]. В качестве фосфина использовали стерически нагруженный трис-(2,6-диметоксифенил)фосфин (ТДМФФ).
В гомодимеризации различных алкинов с алкильными, арильными, алкилгидроксильными заместителями, а также с несопряженными олефиновыми группами были получены геминальные изомеры III с выходами до 90%. В кросс-сочетании донорных алкинов с алкинами, имеющими электроноакцепторные заместители (EWG),
получаются енины с выходом 70–95%, свидетельствующие о важной роли заместителя EWG (СО2СН3, SO2Ph) в стабилизации карбаниона, связанного с Pd(II). Такая стабилизация, вероятно, осуществляется и в гомодимеризации TMSC≡CH с образованием изомера I.
При анализе различных гипотез авторы остановили свой выбор на этинильном механизме A (схема 10 , А – алкин). Отметим, что во всех случаях происходит нуклеофильное присоединение RC≡C– по правилу Марковникова (кроме TMSC≡CH) с предполагаемым образованием Pd(IV) (см. обзор [241]).
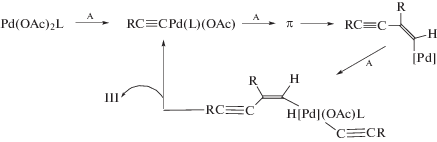
Схема 10. Механизм А гомодимеризации в растворах комплексов палладия (А – алкин).
Депалладирование енинового интермедиата [Pd](σ-C4) может также осуществляться выделяющейся в первой стадии АсОН или непостедственно алкином как синхронная реакция электрофильного замещения с нуклеофильным содействием (SEi) (так называемый метатезис σ-связей). Авторы проверили активацию алкинов и комплексом Pd(0) Pd2(dba)3 · CHCl3 c ТДМФФ (1 : 1) и выяснили, что реакция сочетания протекает медленно и неполностью. Окисление Pd(0) аллилацетатом в указанной системе приводит к ускорению процесса и к 94%-ной конверсии алкина.
В работе [242] были, однако, найдены условия селективной димеризации арилалкинов с образованием изомера I и показано, что комплексы PdL4 и Pd2(dba)3 · CHCl3 с лигандом P(o-Tol)3 образуют также изомер III, однако тот же комплекс Pd2(dba)3 с лигандом ТДМФФ, наряду с III, приводит к продукту I c выходом 70% (PhC2H) и 91% (4‑NCC6H4C2H). Лучший выход линейного Е-изомера I получается при соотношении Pd/L = 1/5. В отличие от работы [240], в которой аллилацетат использовали для окисления Pd(0) до Pd(II), в работе [242] применили π-аллильный комплекс Pd(II) для его восстановления диэтиламином до Pd(0) и аллиламина в присутствии 5-кратного избытка ТДМФФ с образованием бидентатного комплекса
и получили 93% изомера I. Любопытно, что введение орто-заместителей в фенильное кольцо ArC≡CH независимо от их природы (Ме, МеО, F) приводит к полной остановке процесса димеризации. По мнению авторов [242], процессу региоселективного внедрения ArC≡CH по связи ArC≡C–Pd способствует агостиковое взаимодействие орто-Н-арильной группы с Pd в интермедиате ArC≡CPd(L)H(η2-ArC≡CH). Ими же было показано, что обратимого внедрения Pd(II) по о-С–Н(D)-связи с образованием Pd(IV) не происходит. Агостиковое взаимодействие, по мнению авторов, рационально объясняет превосходную региоселективность в изученной системе.
Карбеновые комплексы PdI2 (NHCR2 с R = Me, tBu и 2-этилфенил) в растворе Et3N в присутствии PPh3 были использованы в димеризации фенилацетилена [243]. Комплекс PdI2(NHCR2)(PPh3) с R = 2-этилфенил катализирует образование димера I с выходом 95% на сумму димеров. Добавки CuI приводят к появлению 1,4-дифенилдиина, который получается количественно в присутствии 5 мол. % CuI. Другой способ синтеза карбеновых лигандов in situ предложен в работе [244]. В реакции Pd(OAc)2 с хлоридом имидазолия (R = Cy, Tol, Mes, iPr)
в диметилацетамиде в присутствии Cs2CO3 при 80°С гепт-1-ин образует 90% изомера I и небольшое количество изомеров II и III. Близкие результаты получили в диметилформамиде, тетрагидрофуране и в диоксане. Любопытно, что К2CO3 меняет региоселективность димеризации и приводит к образованию димера III с выходом 89%. Система активна в случае различных алкинов. Влияние природы катиона в карбонатах на региоселективность в работе не объясняется. В реакциях ArC≡CH в присутствии К2CO3 соотношение изомеров I/III меняется от 70/26 до 60/34 (в %).
Весьма универсальный катализатор для синтеза изомеров I из очень широкого круга алкинов (22 алкина) предложен Геворгяном и соавт. [245] на основе бис-карбенового комплекса Pd(IPr)2 (Pd(0) с лигандом IPr):
Этот комплекс в присутствии ТДМФФ в толуоле при 60°С приводит к образованию изомера I с входом 51–94%. При этом отсутствие орто-Н в арилах не препятствует каталитической реакции в отличие от каталитической системы в работе [242]. Предполагается винильный механизм В, включающий стадию гидропалладирования алкина по Марковникову с учетом гидридного характера атома Н в связи Pd–Н. Механизм подтверждается и расчетами (методом теории функционала плотности (DFT)). Заметим, что преимущественное гидропалладирование алкинов в случае гидридэтинильных комплексов Pd(II) и Pt(II) было показано в реакции этих комплексов с RCO2C≡CCO2R [246, 247]. Даже при 100°С в диоксане не было обнаружено продуктов карбометаллирования [246]. В продолжение работы [245] было расширено исследование каталитических свойств предложенной системы [248] в связи с наблюдениями [244] о сильном влиянии добавок и природы оснований на региоселективность димеризации. Показано, что в исследованных системах в толуоле при варьировании координационной сферы комплексов Pd(0) добавки карбонатов цезия и калия не меняют региоселективности процесса (изомер I), однако использование карбоксилат-анионов резко меняет природу продукта – образуется изомер III с выходом 100% в случае пиволата цезия. Авторы предложили рациональное объяснение этого влиянием оснований на региоселективность димеризации. Основание, которое отщепляет протон от интермедиата RC≡C[Pd]H, переводит процесс с гидропалладирования алкина (М) с образованием продукта I (механизм В) на карбометаллирование алкина в анионном интермедиате RC≡C[Pd](η2-HC≡CR)– (анти-М) с образованием изомера III (механизм А). Эта гипотеза была подтверждена расчетами методом DFT.
Альтернативная региоселективность гидропалладирования в винильном механизме димеризации обнаружена в синтезе винил-этинильного интермедиата PhC≡CPd(L)2–[C(Ph)=CH2] в реакции Pd(PEt3)4 с PhC≡CH в присутствии кислоты Ph2P(O)OH при 25°С в С6D6 с определением структуры интермедиата методом рентгеноструктурного анализа (РСА) [249]. Комплекс палладия Pd2(dba)3 в этой же системе в присутствии лиганда dppe весьма селективно катализирует образование изомера III с выходом 40–93% в зависимости от заместителей в алкине. Интересно, что и в случае кросс-сочетания PhC≡CH с PhC≡CR в присутствии Ph2P(O)OH атом палладия присоединяется к атому углерода, имеющему наибольший отрицательный эффективный заряд, с образованием интермедиатов (R = P(O)(OEt)2 и COCH3):
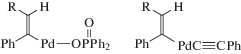
Возможны следующие причины влияния кислоты Бренстеда на региоселективность образования алкенильной группы: (а) менее выраженный гидридный характер Н-атома в комплексе НPdLn+, полученном в результате протонирования комплекса Pd(0), и (б) внешнесферное протонирование кислотой НА π-комплекса (схема 11 ).
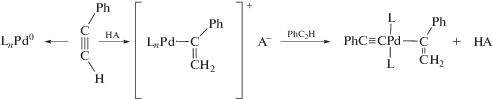
Схема 11. Механизм В с внешнесферным протонированием π-комплекса.
В работе [250] убедительно показано, что в реакции Ph2C2 с RCO2H и M(РEt3)4 (M = Ni, Pd, Pt) в C6D6 при 25оС получается исключительно цис-алкенильный продукт. При этом PhC≡CH в случае палладия образует продукт только анти-М-типа.
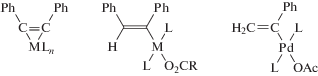
Исследование комплексов Pt(0) показало, что гидрометаллирование происходит в результате реакции π-комплекса с кислотой RCO2H, т.е. по варианту (б). Так, образующийся в реакции PtL4 c AcOH гидридный комплекс Pt(II) не взаимодействует с Ph2C2 с образованием алкенильного комплекса в течение 20 ч при 25°С и еще в течение 10 ч при 60–100°С. При этом π-комплекс Pt(0) реагирует с AcOH при 25°С. Вероятно, и в случае Pd(0) осуществляется аналогичный механизм образования алкенильных интермедиатов. Синтез цис-алкенильного комплекса в случае Ph2C2 обусловлен, вероятнее всего, электорофильным замещением протоном металла в π-комплексе со структурой металлациклопропена [1].
3.5. Комплексы металлов 4–7 групп Периодической системы элементов
Только отдельные представители металлов 4–7 групп элементов протестированы в качестве катализаторов димеризации алкинов [251–255].
Комплекс Re2(CO)10 в присутствии Bu4NF в толуоле при 80°С катализирует образование изомера I с выходом 50% [251]. Использование комплекса ReBr(CO)3(THF)2 приводит к селективному синтезу продукта I с выходом 98%. Различные арилалкины с электронодонорными и электроноакцепторными заместителями димеризуются с высокими выходами. В случае этилпропиолата в тех же условиях получается 1,3,5-изомер циклотримера с выходом 60%.
Еще на втором этапе развития химии ацетиленовых соединений было показано [252], что в системе Cr(tBu)4–ZnEt2 алкилацетилены димеризуются с образованием изомера III с почти количественным выходом при 30оС при соотношении ZnEt2/Cr(tBu)4 = 3. При использовании AlEt3 вместо ZnEt2 образуются только триалкилбензолы.
Циглеровские системы на основе пентаметилциклопентадиенильных комплексов Ti(IV) также оказались активны в гомо- и кросс-димеризации алкинов [253]. Различные алкины димеризуются с образованием изомера III с региоселективностью >99% и выходом 92–99% в системе ${\text{Cp}}_{2}^{*}{\text{TiC}}{{{\text{l}}}_{2}}$–iPrMgBr при 30оС в Et2О. Интересно, что незамещенный лиганд η5-С5Н5 приводит к олигомеризации алкинов. Различные комбинации RC≡CH и R/C≡CH изучены в кросс-димеризации. Наиболее региоселективно димер R/C≡CС(R)=СН2 (92%) образуется при R/ = Me3Si и R = n-Bu. Восстановленный изопропилмагнийбромидом ${\text{Cp}}_{2}^{*}{\text{TiC}}{{{\text{l}}}_{2}}$ приводит к образованию комплекса ${\text{Cp}}_{2}^{*}{\text{TiH}}$, превращающегося в реакции с RC≡CH в комплекс ${\text{Cp}}_{2}^{*}{\text{TiC}} \equiv {\text{C}}{{{\text{R}}}^{/}}$ (по различным механизмам). Этот комплекс, по мнению авторов [253], и является активным катализатором процесса в рамках этинильного механизма с Марковниковским вариантом присоединения нуклеофила R/C≡C– к алкину в π-комплексе Ti(III). Синтезированный этинильный комплекс (через LiC≡CR/) успешно катализирует процесс димеризации. Показано также, что внедрение RC≡CH по связи Ti–C≡CR происходит как цис-внедрение.
Изомер III получен и в растворах катионного комплекса Zr(IV)–${\text{Cp}}_{2}^{*}$ZrMe+A– (A = B[4-C6H4F] (XXVI)) [254]. Реакция комплекса XXVI с 5–10-кратным избытком RC≡CH (R = tBu, Pr) при ‒30°С в C6D5Br приводит к получению металлоорганического продукта XXVII
наряду с олигомерами алкинов. В случае R = Me3Si комплекс XXVII реагирует далее при 25оС с избытком алкина с образованием нового продукта XXVIII (выход 50%) путем внедрения третьей молекулы RC≡CH по связи Zr–СН=. Показано, что комплекс XXVII так же, как и XXVI, катализирует димеризацию и тримеризацию алкинов в течение 5 мин при 25оС. Число оборотов катализатора в случае t BuС≡СН равно 1300 при селективности по изомеру III более 99.5% с TOF = 15 600 ч–1. В случае R = Ме образуется 72.5% тримера. Предполагается, что образование димера III из интермедиата XXVII или тримера из интермедиата XXVIII происходит в результате метатезиса σ-связей в реакции с алкином с образованием активного катализатора [Zr](C≡CR)+. Интересно, что внедрение третьей молекулы алкина по связи Zr–СН= в XXVI происходит как анти-М-присоединение.
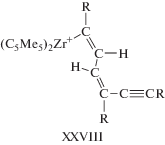
Как и в других каталитических системах, лигандное окружение атома циркония существенно влияет на региоселективность димеризации. Так, Z-изомер II из фенилацетилена получается с выходом 92% в растворах комплекса XXIX
с лигандом бис-уреатом, который в присутствии анилина в толуоле превращается в двухядерный мостиковый амидо-имидо-димер, являющийся катализатором димеризации [255]. На основании определения структуры димерного комплекса (Z‑изомер II) методами РСА и 1Н ЯМР-спектроскопии предложено объяснение региоселективности реакции с участием двух атомов циркония в интермедиате и транс-присоединения нуклеофильной группы RC≡C– к π-комплексному фрагменту [Zr](НC≡CR) против правила Марковникова (анти-М, схема 12 ).
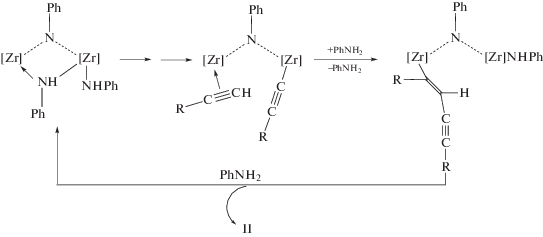
Схема 12. Механизм димеризации A с исходным комплексом XXVIII [255].
3.6. Комплексы металлов 3 и 13 групп Периодической системы элементов
В классической работе Беркоу и соавт. [256], посвященной изучению метатезиса σ-связей в реакциях ${\text{Cp}}_{2}^{*}$ScR (R = H, алкил, арил, винил, алкенил и др.) с H2, C6H6, RCH=CH2 и RC≡CH, было показано, что ${\text{Cp}}_{2}^{*}$ScCH3 реагирует с пропином в течение нескольких минут при Т < 0°C с образованием комплекса ${\text{Cp}}_{2}^{*}$ScC≡CCH3, который при 25°С катализирует селективную димеризацию пропина с получением изомера III. Предложенный механизм димеризации включает внедрение пропина по связи Sc–С (по Марковникову) с образованием ${\text{Cp}}_{2}^{*}$Sc[CH=C(CH3)C≡CCH3] и с протолизом этого алкенильного интермедиата пропином по механизму метатезиса s-связей, т.е. без предварительной координации алкина скандием в степени окисления (III). В отличие от Zr и Hf, легко внедряющих пропин по связи М–Н, ${\text{Cp}}_{2}^{*}$ScH быстро образует в реакции с пропином пропинильный комплекс и Н2. В реакциях этих же комплексов скандия с ацетиленом [257] обнаружено образование при –78°С ацетиленидных соединений ${\text{Cp}}_{2}^{*}$ScC≡CH и ${\text{Cp}}_{2}^{*}$ScC≡CSc${\text{Cp}}_{2}^{*}$. Симметричный продукт синтезируется при повышенной концентрации комплекса ${\text{Cp}}_{2}^{*}$ScC≡CH по реакции
Катализ димеризации алкинов аналогичными комплексами иттрия (Y) в неполярных растворителях протекает также региоселективно с образованием изомера III [258]. Так, в реакции ${\text{Cp}}_{2}^{*}$YCH(SiMe2)2 c пропином в бензоле при 25°С образуются ${\text{Cp}}_{2}^{*}$YC≡ССН3 и 2-метил-1-пентен-3-ин (изомер III), а с фенилацетиленом получается аналогичный димер с выходом 89%. Величина TOF в случае пропина равна 5400 ч–1.
Каталитические свойства иттрия часто сравнивают со свойствами других редкоземельных металлов – лантанидов и актинидов [259–263]. Так, например, в [259] реакции фенилацетилена с комплексами ${\text{Cp}}_{2}^{*}$МCH(SiMe2)2 (М = Y, Ln, Ce) сопровождаются образованием олигомеров алкина и комплексов ${\text{Cp}}_{2}^{*}$МC≡СPh]n, однако регио- и хемосективности процесса зависят от природы R в алкине и от природы металла. Так, в случае Y и RC≡CH реакция проходит исключительно до димера III (R = tBu, Pr) или до смеси димеров III и I (R = SiMe3, Ph). В случае Ln и Ce при R = SiMe3 и Ph также появляются продукты III и I, но вместе с тримерами.
Изучение ИК- и 1Н ЯМР-спектров олигомерных этинильных комплексов Се [259] привело к выводу об их димерной природе с мостиковыми этинильными лигандами и со значительным вкладом π-системы этинильного лиганда в координацию с металлом. Также показано, что скорости димеризации tBuC≡CH в растворах исходного комплекса ${\text{Cp}}_{2}^{*}$СеCH(SiMe2)2 и этинильного комплекса близки.
Еще один оригинальный механизм образования 1,4-енинов (D), ранее не встречавшийся, был обнаружен при изучении свойств этинильных комплексов Sm [260, 261], Ce и Nd [261]. В отличие от рассмотренной выше димерной структуры с мостиковыми этинильными лигандами [${\text{Cp}}_{2}^{*}$Ln(μ-C≡CR)]2 [259], методом РСА было показано, что любые процессы, приводящие к комплексу ${\text{Cp}}_{2}^{*}$Sm(C≡CR)(THF), дают в результате комплекс [${\text{Cp}}_{2}^{*}$Sm]2(μ-η2,η2-RC=C=C=CR) (XXX). При действии двух молекул фенилацетилена на комплекс XXX получается PhCH=CHC≡CPh. Вероятно, в комплексе XXX после метатезиса связей Sm–C и Н–С≡ с образованием XXXI и ${\text{Cp}}_{2}^{*}$SmC≡CPh происходит 1,3-сдвиг [Sm] в рамках π-аллильного комплекса XXXI с получением комплекса XXXII
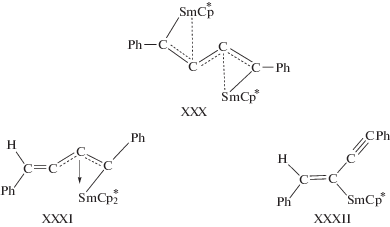
Протолиз комплекса XXXII вторым алкином и приводит к продукту реакции по уравнению реакции (XIV)
(XIV)
$\begin{gathered} {\text{XXX}}~ + {\text{ }}2{\text{PhC}}{\kern 1pt} \equiv {\kern 1pt} {\text{CH}}~\,\, \to \\ \to {\text{PhCH}}{\kern 1pt} = {\kern 1pt} {\text{CHC}}{\kern 1pt} \equiv {\kern 1pt} {\text{CPh\;}} + 2{\text{Ср}}_{2}^{*}{\text{SmC}}{\kern 1pt} \equiv {\kern 1pt} {\text{CPh}}. \\ \end{gathered} $Синтез интермедиата XXX напоминает реакцию окислительной димеризации алкинов Глязера–Залькинда [51], один из механизмов которой включает димеризацию этинильных комплексов RC≡CMXn через бициклическое σ-металлоорганическое соединение типа XXXIII [1]:
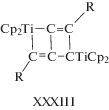
В случае окислителя Cu(II) и катализа комплексами Cu(I) механизм окислительной димеризации алкина был представлен cхемой 13 [1].
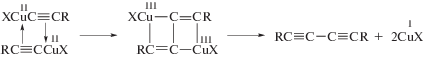
Cхема 13. Вероятный механизм окислительной димеризации алкинов.
Распад аналогичного бициклического комплекса в реакция комплексов ${\text{Cp}}_{2}^{*}$LnC≡CPh (Ln = Sm, Ce, Nd) может привести и к 1,4-енину по схеме 14 .
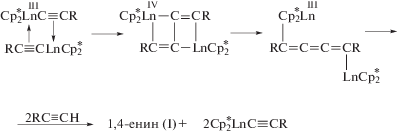
Схема 14. Возможный механизм димеризации алкинов D с участием двух этинильных интермедиатов.
При изучении каталитических свойств моноциклопентадиенильных комплексов (Y, Yb и Lu) в реакции димеризации алкинов было показано [262], что эти комплексы приводят к очень высокой селективности в образовании изомера II из алкинов с арильными и алкильными заместителями. Особенно активны комплексы Lu XXXIV
которые при конверсии 99% обеспечивают селективность 100% по II (80°С, C6D6). Получены убедительные доказательства, что реальным катализатором в этом случае является димерный мостиковый этинильный комплекс XXXV, изученный методом РСА.
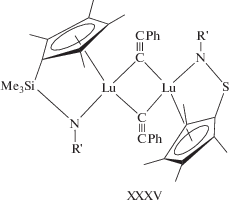
Предполагается, что π-связанный алкин внедряется по связи Lu–С≡ с последующим метатезисом σ-связей. В мини-обзоре катализа комплексами Sc, Y, La, Ce, Yb, Lu и Pr [263] показано, что в зависимости от лигандного окружения атома металла в степени окисления (III) и природы заместителей в алкине возможно образование и изомеров I и III со 100%-ной региоселективностью, а также небольших количеств олигомеров. Димерные этинильные комплексы Lu при охлаждении растворов до комнатной температуры осаждаются из растворов и легко отделяются от продуктов реакции [263]. Эти же авторы показали возможность полимеризации 1,4-диэтинильных ароматических соединений в растворах одноядерных комплексов Lu и Pr с образованием полимеров с молекулярной массой (Мn), равной 5300–16 000 Д, с выходом 95–99% и селективностью 95–100%.
Роль стерически затрудненных лигандов в управлении стерео- и региоселективностью показана на примере комплексов иттрия [264]. Если при использовании комплекса ${\text{Cp}}_{2}^{*}$YCH(SiMe2)2 в реакции с гетероароматическими алкинами образуются преимущественно изомеры I и III [259], то при использовании очень сложного макроциклического имидного лиганда с 4 атомами азота (L) комплекс Y(L)CH(SiMe2)2 образует изомер II с селективностью 100%.
В заключении этого раздела рассмотрим два металла 3 группы элементов, относящиеся к 6d-металлам группы актинидов (An) – Th и U [265–268], и непереходный металл 13 группы элементов – Al [269].
Синтезированные комплексы Th и U [265] ${\text{Cp}}_{2}^{*}$Аn(μ-C≡CR)2 обладают примерно одинаковой каталитической активностью в олигомеризации алкинов. При 80°С tBuC≡CH образует изомер III с выходом 98–100%, а Me3SiC≡CH регио- и стереоселективно превращается в тример Е,Е‑1,4,6-трис-(триметилсилил-1,3-гесадиен-5-ин) с выходом 90–95%. В качестве первой стадии механизма тримеризации Me3SiC≡CH рассматривается стадия внедрения алкина по связи [Аn]–C≡ по уравнениям первого порядка по алкину и по [Аn]. Медленная стадия представляет собой метатезис σ‑связей с выделением продуктов из алкенил-этинильных интермедиатов. Изучение 1Н ЯМР-спектров и стадий обрыва процесса в присутствии Н2О и D2O [265] натолкнули исследователей на вывод о существовании интермедиатов, приводящих к енину III и тримеру ${\text{Cp}}_{2}^{*}$Th(C(R)=CHCR=CRC≡CR)2 (XXXVI).
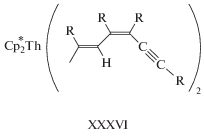
В работе [266] установлено, что селективность в образовании димера III или тримера легко регулируется добавками аминов RNH2 (R = Me, Et, 2-6-диметилбензол, tBu) и Me2NH, не участвующих в стехиометрии реакции димеризации. Роль аминов, по данным [266], заключается в образовании активных амидных комплексов ${\text{Cp}}_{2}^{*}$Th(NHR)2 и в участии аминов в стадии протолиза связи М–СН= в инермедиате (что подтверждено изотопными исследованиями с парой RNH2/RND2), приводящем к димеру или тримеру с регенерацией амидного лиганда в активном комплексе. Кинетическое уравнение в случае тория имеет вид (5) и объясняется образованием неактивного диамид-аминного компекса ${\text{Cp}}_{2}^{*}$Th(NHR)2(RNH2).
(5)
$W~ = k~{{[{\text{Th}}]}_{\Sigma }}{{\left[ {{\text{алкин}}} \right]} \mathord{\left/ {\vphantom {{\left[ {{\text{алкин}}} \right]} {\left[ {{\text{амин}}} \right]}}} \right. \kern-0em} {\left[ {{\text{амин}}} \right]}}.$Размер и индуктивные свойства заместителя R, а также природа заместителя R/ в R/С≡СН существенно влияют на хемоселективность процесса. Однако разделить стерические эффекты R в амине и влияние индуктивных эффектов на реакционную способность связи N–H в амине пока не удалось.
В ходе детального изучения каталитических свойств урансодержащего комплекса [(Et2N)3U]+ ${\text{BPh}}_{4}^{ - }$ в реакции димеризации [267] установлено преимущественное образование изомера III, однако при объемных заместителях в алкине наблюдается образование изомера II.
Исследование механизма реакции показало, что первая стадия образования (Et2N)2UС≡СR+(А1) и Et2NН является слабо обратимой с последующим образованием π-комплекса урана (В1) (Et2N)2U(С≡СR)(η2-RС≡СН)+${\text{BPh}}_{4}^{ - }$ (R = tBu). Была изучена кинетика димеризации tBuС≡СН до изомера III и установлен первый порядок по [U]Σ и переменный порядок (от 1 до 0) по алкину. В предположении, что образование интермедиата (А1) из [(Et2N)3U]+BPh4 и алкина практически смещено вправо, предложен механизм реакции, включающий двухстадийный цикл с интермедиатами А1 и (Et2N)2U–С(Н)=С(R)С≡СR+ (С1) и с неучаствующим в цикле комплексом (В1) (см. обозначения в [267]).
К сожалению, выведенное в статье кинетическое уравнение (А11) не соответствует принятым допущениям. В условиях квазистационарности стадия (1) образования π-комплекса (В1) из А1 всегда равновесна (d[B1]/dt ≈ 0), а для цикла из стадий (2) и (3) получается уравнение (6) скорости реакции W:
(6)
$W~ = {{k}_{2}}{{k}_{3}}{{[{\text{S}}]}^{2}}{{[{{{\text{A}}}_{1}}]} \mathord{\left/ {\vphantom {{[{{{\text{A}}}_{1}}]} {({{k}_{{--2}}}~\,\, + {{k}_{3}}[{\text{S}}])}}} \right. \kern-0em} {({{k}_{{--2}}}~\,\, + {{k}_{3}}[{\text{S}}])}},$При допущении k3[S] $ \gg $ k–2 получим W = k2[S][A1] и [U]Σ = [A1] + [B1] = [A1](1 + K1[S]). При этом
(7)
$W~ = {{{{k}_{2}}[{\text{S}}]{{{[{\text{U}}]}}_{{\Sigma ~}}}} \mathord{\left/ {\vphantom {{{{k}_{2}}[{\text{S}}]{{{[{\text{U}}]}}_{{\Sigma ~}}}} {(1{\text{ }} + {{K}_{1}}[S])}}} \right. \kern-0em} {(1{\text{ }} + {{K}_{1}}[S])}}$Изучение катализа димеризации димерными мостиковыми циклопентадиенильными комплексами тория [268] с более открытой координационной сферой атома тория показало, что процесс димеризации с образованием изомеров III происходит существенно быстрее и более селективно по сравнению с комплексом ${\text{Cp}}_{2}^{*}$ThМе2. Так, комплекс Me2Si${\text{Cp}}_{2}^{{//}}$ThBu2 (Cp// = C5Me4) активнее немостикового комплекса в ~200 раз (для R = Bu) и в ~105 раз (для R = iPr). Предполагается, что ускорение реакции связано с облегчением стадии деметаллирования интермедиата типа С1 (см. выше) алкином. В случае мостиковых Cp-комплексов катализатором является комплекс Me2Si${\text{Cp}}_{2}^{{//}}$Th(С≡СR)2, образование которого происходит количественно из прекурсора в необратимой стадии.
Эти же авторы показали [269], что соединения непереходного металла алюминия (13 группа элементов) в форме метилалюмоксана (МАО) в кипящем бензоле (С6H12, тетрагидрофуране, Et2O) и без растворителя селективно катализируют димеризацию различных алкинов (кроме Me3SiС≡СН) с образованием изомера III (93–99%). Предполагается, что алкин региоселективно внедряется по связи [Al]–C≡ этинильного соединения [Al](C≡CR) (А), образующегося в результате метатезиса σ-связей по реакции (XV)
(XV)
$\left[ {{\text{Al}}} \right]{\kern 1pt} --{\kern 1pt} {\text{C}}{{{\text{H}}}_{3}} + {\text{RC}}{\kern 1pt} \equiv {\kern 1pt} {\text{CH}} \to \left[ {{\text{Al}}} \right]{\kern 1pt} --{\kern 1pt} {\text{C}}{\kern 1pt} \equiv {\kern 1pt} {\text{CR}} + {\text{C}}{{{\text{H}}}_{4}}.$Стадия деметаллирования интермедиата В с образованием продукта также является результатом метатезиса σ-связей с участием алкина (схема 15 ). Частота оборотов катализатора при этом невелика (не более 5 ч–1).
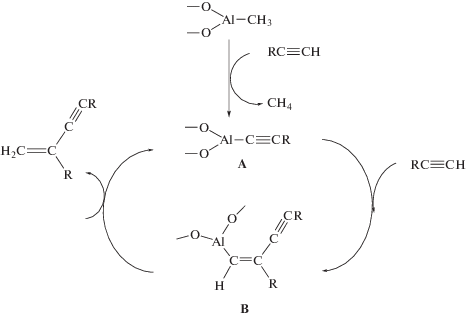
Cхема 15. Механизм димеризации в растворах МАО (схема воспроизводится с разрешения из Org. Lett. 2000. V. 2. № 6. Р. 737 [269]. Copyright (2000) Am. Chem. Soc.).
4. ЗАКЛЮЧЕНИЕ
Обзор катализаторов, механизмов и интермедиатов реакции димеризации алкинов позволяет сделать несколько обобщений. Если рассматривать успехи в области гомодимеризации алкинов, можно отметить, что для любого терминального алкина в настоящее время можно подобрать систему М–L, которая обеспечит хемо-, регио- и стереоселективность синтеза одного или нескольких возможных изомеров (схема 4 ): I (E), II (Z-), III (гем-изомер), IV и V (1,4-замещенные бутатриены).
Селективность димеризации определяется тремя контролирующими факторами – природой металла и электронными и стерическими свойствами лигандов (катализатор), природой заместителей в алкине (субстрат) и, конечно, механизмом реакции, реализуемым в реакционной среде (механизм). При этом даже на одном и том же исходном комплексе металла могут возникать различные механизмы (см., например, катализ комплексами рутения [188]). В растворах различных комплексов с добавками кислот или оснований обнаружено протекание димеризации по механизмам А, В, С (раздел 3.2) и D (раздел 3.6). Некоторые металлы в зависимости от степени окисления и набора лигандов являются катализаторами, реализующими различные механизмы, например, Rh (A, B и C), Ru (С и А), Pd (A и B), Ir (B и C).
На примере катализа комплексами Ru (табл. 3) видно, как трудно выделить и объяснить факторы, определяющие селективность процесса димеризации. Так, например, для одного и того же комплекса Ru(II) для алкина с R = Ph соотношение изомеров I/II = 95/5, а для R = n-Bu – 5/95. Для R = tBu катализ комплексами Ru(II) и Ru(0) приводит к изомерам IV и V соответственно. Очевидный факт состоит в том, что изомер III не может образоваться по механизму С. Простое объяснение стереоселективности получения продуктов I и II приведено в случае механизма С – это различная геометрия винилиденэтинильного комплекса Ru(II) и влияние на нее стерических характеристик лигандов и заместителей в алкине [18, 182, 183].
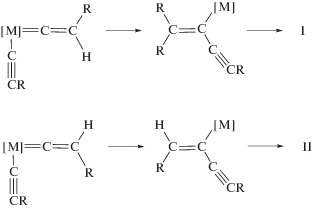
Интермедиаты различных механизмов в настоящее время изучены весьма детально: in situ, в соединениях, выделенных из реакционной среды (метод РСА), на модельных соединениях. Интермедиаты процесса димеризации приведены в табл. 4. Суммируя данные табл. 4, можно сделать следующие выводы:
Таблица 4.
Интермедиаты, механизмы и металлы в катализаторах димеризации алкинов
| Номер интермедиата |
Интермедиат | Тип механизма |
Металлы [лит. ссылка] |
|---|---|---|---|
| 1 | [M](C≡CR) | A | Cu(I) [8, 123], Au(I) [174, 270], Ru(II) [181, 192], Os(II) [207], Pd [248], Pt [246], Ti(III) [253], Sc(III) [256], Al(III) [269], Th(IV) [265] |
| 2 | [M](C≡CR)(RC≡CH) | Rh(I) [221], U(IV) [267] | |
| 3 | [M](CH(R)=CR(H)C≡CR) | Rh(III) [225], Pd(II) [240, 242, 248], Sc(III) [256], Al(III) [269], Th(IV) [265], U(IV) [267] | |
| 4 | 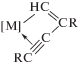 |
Zr(IV) [254, 255] | |
| 1 | [M](C≡CR) | B | Ir(I) [231] |
| 5 | [M][CH(R)=CR(H)] | Pd(0) [242, 248], Pt(II) [246] | |
| 6 | $\left. \begin{gathered} {\text{[M](C}} \equiv {\text{CR)(CH = CHR)}} \hfill \\ {\text{[M](C}} \equiv {\text{CR)(CR = C}}{{{\text{H}}}_{2}}) \hfill \\ \end{gathered} \right\}$ | Rh(I) [232], Ir(I) [231], Pd(II) [249], Ni(0) [237] | |
| 7 | [M](=C=CHR) | С | Ru(II) [175], Os(II) [207, 209, 210] |
| 8 | [M](C≡CR)(CH=CHR) | Ir(I) [231] | |
| 9 | [M](C(=CHR)C≡CR) | Ru(II) [176, 178, 193], Rh(I) [223, 224] | |
| 10 | 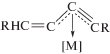 |
Ru(II) [177, 193], Os(II) [207], Sm(III) [259, 261] |
|
| 11 | [M](RC=C=C=CHR) | Rh(I) [226] | |
| 12 | [M]2(RC=C=C=CR) | D | Sm(III), Ce(III), Nd(III) [259, 261] |
1) Этинильные (алкинидные) комплексы [M](C≡CR) (1) участвуют в качестве интермедиатов, образующихся на различных стадиях процесса, во всех 4 механизмах димеризации (А, В, С и D) для металлов 3–11 и 13 групп Периодической системы элементов. Известны моно- и бис-этинильные комплексы [M](C≡CR)2 [210].
Механизмы образования этинильных лигандов из алкинов:
– замещение иона водорода ионом металла
– окислительное присоединение алкина к [M] с повышением СТО металла на 2 ед.
При наличии в исходном комплексе таких лигандов, как Н или R, образование этинильных групп может происходить в результате метатезиса σ-сязей с выделением Н2 или RH (например, в [269]).
2) Соединения (2) и (3) – типичные интермедиаты этинильного механизма А, образование которых доказано во многих исследованиях (табл. 4). Комплекс (3) приводит к димерам I и III (цис-внедрение алкина по связи металл–этинил) и к димеру II (транс-присоединение этинильной группы к π-комплексу). Комплекс (4) превращается в димер III. Региоселективность реакции внедрения (М или анти-М) зависит от распределения электронной плотности во фрагменте [M]–C≡ и в π-комплексе (2) и, в большой степени, от стерических факторов.
Образование изомеров I (Е) и II (Z) при катализе димеризации ацетатом Cu(I) в АсОН в работе [128] в рамках этинильного механизма А можно понять, если в реакции участвуют многоядерные комплексы Cu(I) (по аналогии со схемой 5 ). Транс-присоединение этинильной группы к π-комплексу вероятно и в случае двухядерных комплексов Zr (схема 12 , [255]) и Lu (комплекс XXXIV, [262]). Для объяснения образования интермедиата (3) в случае Cu(I) (анти-М) можно привлечь стерические препятствия в случае алкилацетиленов и возможное изменение распределения электронной плотности за счет вклада мезомера с винилиденовым карбанионом, меняющего нуклеофильные свойства группы RC≡C–, становящейся более “мягким” нуклеофилом
Распределение электронной плотности в исходном алкине, кроме случаев с электроноакцепторными заместителями (EWG), не является фактором, определяющим региоселективность присоединения нуклеофила к алкину в π-комплексе, поскольку в π-комплексе это распределение может значительно измениться. К тому же сама атака нуклеофила меняет распределение эффективных зарядов на атомах углерода в π-комплексе и тем сильнее, чем мягче нуклеофил. Так, на очень простой модельной реакции присоединения мягкого нуклеофила Н– к пропину расчеты ab initio поверхности потенциальной энергии показали [271, 272], что различие энергий активации реакции присоединения по Марковникову (СН3СН=СН–, М) и против правила Марковникова (СН3С–=СН2, анти-М) невелико и составляет 1.7 [271] или 1.2 ккал/моль [272] соответственно, причем более быстрой оказывается анти-М-реакция. Заметим, что термины “по Марковникову, М” и “против правила Марковникова, анти-М” здесь (и выше) применяются в классическом смысле: электроположительный конец E молекулы ENu в случае неактивированных терминальных алкинов присоединяется к наиболее гидрогенизированному атому углерода (1, М), а нуклеофильная группа Nu – к наименее гидрогенизированному атому (2, анти-М). Детальное обсуждение проблем, связанных с применением правила Марковникова, см. в обзоре [273].
3) Комплексы (5) и изомерные соединения (6) – интермедиаты винильного механизма В. Порядок появления винильного фрагмента в интермедиате (6) может быть различным. Механизмы образования винильного лиганда:
– цис-внедрение алкина по связи [M]-Н с различной региоселективностью;
– внешнесферное протонирование π-комплекса, повышающее СТО металла на 2 ед. [1, 249, 250];
– электрофильное замещение металла в π-комплексах, приближающихся по всем характеристикам к металлациклопропеновой структуре [1].
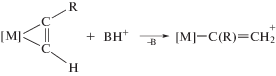
Для доказательства механизма транс-присоединения Н+(D+) к алкину в π-комплексе необходимо быть уверенным в том, что, во-первых, в π‒комплексе MLn(RC≡CH) нет свободных координационных вакансий для образования связи [M]–Н(D) и, во-вторых, образующийся цис-алкенильный лиганд не изомеризуется в транс-изомер [1].
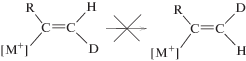
Образование енинов в результате внедрения алкина по связи [M]–алкенильный лиганд не установлено, вероятно, вследствие редко встречающейся стадии β-элиминирования [M]–Н из такого σ-бутадиенильного интермедиата (на примере реакции ацетилена):
Вместе с тем в работе [231] показано, что в реакции PhC2H с гидридным комплексом иридия бысторо образуется анти-М-продукт 1,2-цис-внедрения [Ir][C(Ph)=CH2], но процесс обратим (!) и в результате β-Н-элиминирования [Ir]–Н π-комплекс иридия медленно превращается в продукт 2,1-цис-внедрения [Ir](CН=CHPh) с образованием изомерного комплекса (6) (табл. 4) и конечного димера I. В работе Беркоу и сотр. [274] изотопными методами также была доказана стадия β-Н-элиминирования в случае алкенильных комплексов циркония(IV) с образованием π-комплексов алкинов. Так, комплекс ${\text{Cp}}_{2}^{*}$Zr(CH=CHCH3)2 при комнатной температуре превращается в цирконациклопентен, который при нагревании изомеризуется по реакции
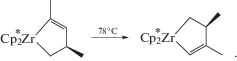
Механизм β-Н-элиминирования из алкенильных комплексов можно, вероятно, представить стадиями образования и превращения η2-винильных (алкенильных) комплексов [275].
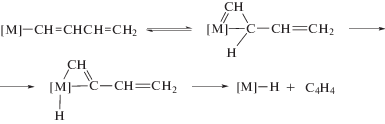
Влияние оснований на перевод винильного механизма димеризации алкинов на этинильный детально обсуждается в работе [248].
4) Винилиденовые комплексы металлов (7) изучены весьма подробно [115, 200–202, 204, 276–278]. Многократно доказана реализация винилиденового механизма димеризации алкинов С (табл. 4). Установлена природа продуктов взаимодействия винилиденового лиганда с этинильной группой с образованием различных по структуре интермедиатов (8) и (9)–(11) с фрагментом [M](σ-С4), приводящих к енинам I и II и к кумуленам IV и V. Рассматривают 3 основных механизма образования винилиденового лиганда из алкинов и π-комплексов:
– 1,3-сдвиг гидрида
– 1,2-сдвиг атома водорода в алкине
– внешнесферное протонирование этинильного лиганда
Механизм изомеризации π-комплекса в винилиденовый комплекс обсуждается в работе Морокумы и др. [197] и в последующих квантово-химических и экспериментальных исследованиях комплексов Mn(I) [279], Ru(II) [280] и Rh(I) [281–284], которые цитируются здесь без детального анализа. Интересное экспериментальное наблюдение, изученное также теоретически, сделано в работе [284], где показана положительная роль иона Cl– или метанола в процессе переноса атома Н ([M]–Н) на этинильный лиганд (1,3-сдвиг атома Н) с образованием винилиденового комплекса рутения. Очень интересный механизм образования винилиденового лиганда обнаружен в работе [285]. В реакции комплекса RuX(H)(H2)L2 с PhC≡CD получен комплекс RuX(D)(=C=CHPh). Для объяснения этого изотопного обмена авторы предложили механизм с образованием η1-винильного интермедиата по Марковникову с последующим α-элиминированием [M]–D и образованием винилиденового лиганда:
Расчеты аb initio (B3LYP) показали экзотермичность всех стадий этого процесса. Гидридный комплекс осмия OsH3ClL2 реагирует с алкином также, но в качестве интермедиата образуется η2-винильный комплекс с последующим α-элиминированием [Os]–Н и также с образованием винилиденового лиганда. Образование η2-винильных комплексов из алкинов особенно характерно для катионных комплексов осмия [286].
Известно также превращение винилиденовой группы в этинильный лиганд при действии оснований на винилиденовый комплекс [177, 181]. Таким образом, винилиденовый комплекс может стать прекурсором в этинильном механизме А димеризации [181, 207]. На основании изучения кинетики превращений винилиден → алкин в растворах катионных комплексов рутения обсуждаются и другие варианты изомеризации винилидена [287]. Принимается во внимание вклад мезомерной структуры с α-карбкатионным центром, облегчающей 2,1-Н-сдвиг:
Заметим, что превращение винилиден → алкин обнаружено даже для дизамещенных винилиденовых лигандов Ph(R)C=C= с образованием PhC≡CR в катионных комплексах Ru и Fe, причем это превращение обратимо (толуол + PR3 или CН3CN) [288].
5) Механизм деметаллирования интермедиатов [M](σ-С4) (3), (4), (9)–(11), судя по приведенным в обзоре результатам, весьма разнообразен:
– Н[M]С4 → НС4 + [M],
– [M]С4 + RC≡CH → M](C≡CR) + НС4,
– [M]С4 + НХ → [M]Х + НС4.
Механизм с участием алкинов (метатезис σ-связей) часто встречается в катализе комплексами различных металлов (Ru, Rh, Sc, Sm, Th, U, Al). Исследование кинетики и механизма димеризации при катализе комплексом [RuCl(μ-Cl)(η6-п-кумол)]2 в уксусной кислоте подтвердило роль АсОН в стадии деметаллирования [289].
6) Механизм димеризации алкинов D, в котором фрагмент С4(μ2-η2, η2-С4) в комплексе (12) образуется из двух этинильных комплексов [M](C≡CR), пока обнаружен только в катализе комплексами лантанидов, хотя уже упоминавшийся возможный бициклический интермедиат в растворах комплексов Cu(ОАс)2 в аминах также способен привести через интермедиат (12) к кумуленам и изомерам I и II
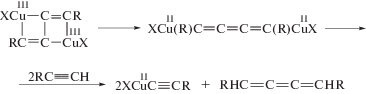
в результате метатезиса σ-связей с алкинами (или ацидолиза). При изучении реакции димеризации, катализируемой комплексами Fe(III), получена информация о возможности реализации радикального механизма синтеза енинов [213].
Разнообразие механизмов одной и той же реакции при варьировании металла-катализатора и координационной сферы каталитического комплекса является характерной чертой современного металлокомплексного катализа.
Список литературы
Темкин О.Н., Шестаков Г.К., Трегер Ю.А. Ацетилен. Химия. Механизмы реакций. Технология. М.: Химия, 1991.
Tedeschi R.J. Acetylene-Based Chemicals from Coal and Other Natural Resources. N.Y.: Dekker, 1982.
Трофимов Б.А. Гетероатомные производные ацетилена. М.: Наука, 1981.
Trofimov B.A. Acetylene and its derivatives in reactions with nucleophiles: recent advances and current trends // Curr. Org. Chem. 2002. V. 6. № 13. P. 1121.
Трофимов Б.А., Гусарова Н.К. Ацетилен: новые возможности классических реакций // Успехи химии. 2007. Т. 76. № 6. С. 550.
Трофимов Б.А., Шмидт Е.Ю. Реакции ацетиленов в суперосновных средах – итоги последних лет // Успехи химии. 2014. Т. 83. № 7. С. 600.
Salvio R., Moliterno M., Bella M. Alkynes in organocatalysis // Asian J. Org. Chem. 2014. V. 3. P. 340.
Temkin O.N. Homogeneous Catalysis with Metal Complexes: Kinetic Aspects and Mechanisms. Chichester: Wiley, 2012.
Acetylene Chemistry. Chemistry, Biology and Material Science / Eds. Diederich F., Tykwinski R.R., Stang R.J. Weinheim: Wiley–VCH, 2005.
Catalysis by Gold. Catal. Sci. Ser. V. 6 / Eds. Bond G.C., Louis C., Thompson D.T. London: Imperial College Press, 2006.
Modern Gold Supramolecular Chemistry. Gold-Metal Interactions and Applications / Ed. Laguna A. Weinheim: Wiley–VCH, 2009.
Click Chemistry for Biotechnology and Materials Science / Ed. Lahann J. Hoboken: John Wiley & Sons Ltd., 2009. 432 p.
Gold Chemistry: Applications and Future Directions in the Life Sciences / Ed. Mohr F. Weinheim: Wiley–VCH, 2009. 408 p.
Click Tiazoles. Topics in Heterocyclic Chemistry. V. 28 / Ed. Kosmrlj J. Berlin, Heidelberg: Springer–Verlag, 2012.
Modern Gold Catalyzed Synthesis / Eds. Hashmi A.S.K., Toste F.D. Weinheim: Wiley–VCH, 2012. 402 p.
Transition Metals Catalyzed Carbonylation Reactions / Eds. Beller M., Wu X.-F. Berlin, Heidelberg, N.Y., Dordrecht, London: Springer–Verlag, 2013. 228 p.
Maretina I.A., Ionin B.I. Alkynes in Cycloadditions / Consulting Ed. Tebby J.C. Chichester: Wiley, 2014. 312 p.
Modern Alkyne Chemistry. Catalytic and Atom-Economic Transformations / Eds. Trost B.M., Li Ch.-J. Weinheim: Wiley–VCH, 2015. 424 p.
Au-Catalyzed Synthesis and Functionalization of Heterocycles. Topics in Heterocyclic Chemistry. V. 46 / Ed. Bandini M. Springer, 2016. 289 p.
Lei A., Shi W., Liu C., Liu W., Zhang H., He C. Oxidative Cross-Coupling Reactions Weinheim: Wiley–VCH, 2017. 229 p.
Lu W., Zhou L. Oxidation of C–H Bonds. Hoboken: Wiley, 2017. 508 p.
Schore N.E. Transition metal-mediated cycloaddition reactiones of alkynes in organic synthesis // Chem. Rev. 1988. V. 88. P. 1081.
Beletskaya I., Moberg C. Element-element addition to alkynes catalyzed by the group 10 metals // Chem. Rev. 1999. V. 99. P. 3435.
Chung Y.K. Transition metal alkyne complexes: the Pauson–Khand reaction // Coord. Chem. Rev. 1999. V. 188. P. 297.
Kiss G. Palladium-catalyzed Reppe carbonylation // Chem. Rev. 2001. V. 101. № 11. P. 3435.
Temkin O., Bruk L. Oxidative Carbonylation-Homogeneous. Encyclopedia of Catalysis / Ed. Horvath I. N.Y.: Wiley, 2003. V. 5. P. 394.
Negishi E.-I., Anastasia L. Palladium-catalyzed alkynylation // Chem. Rev. 2003. V. 103. P. 1979.
Beller M., Seayad J., Tillack A., Jiao H. Catalytic Markovnikov and anti-Markovnikov functionalization of alkenes and alkynes: recent developments and trends // Angew. Chem. Int. Ed. 2004. V. 43. P. 3368.
Alonso F., Beletskaya I.P., Yus M. Transition-metal-catalyzed addition of heteroatom-hydrogen bond to alkynes // Chem. Rev. 2004. V. 104. P. 3079.
Fűrstner A., Davies P.W. Alkyne metathesis // Chem. Commun. 2005. P. 2307.
Gabriele B., Salerno G., Costa M. Oxidative carbonylation // Top. Organomet. Chem. 2006. V. 18. P. 232.
Hintermann L., Labonne A. Catalytic hydration of alkynes and its application in synthesis // Synthesis. 2007. № 8. P. 1121.
Beletskaya I.P., Ananikov V.P. Addition reactions of E–E and E–H bonds to triple bond of alkyne catalyzed by Pd, Pt and Ni complexes (E = S, Se) // Pure Appl. Chem. 2007. V. 79. P. 1041.
Meldal M., Tornøe C.W. Cu-catalyzed azide–alkyne cycloaddition // Chem. Rev. 2008. V. 108. P. 2952.
Hein C.D., Liu X.-M., Wang D. Click chemistry a powerful tool for pharmaceutical sciences // Pharm. Res. 2008. V. 25. № 10. P. 2216.
Hashmi A.S.K. Homogeneous gold catalysis beyond assumptions and proposals – characterized intermediates // Angew. Chem. Int. Ed. 2010. V. 49. P. 5232.
Hintermann L. Recent developments in metal-catalyzed additions of oxygen nucleophiles to alkenes and alkynes // Top. Organomet. Chem. 2010. V. 31. P. 123.
Abbiati G., Beccalli E.M., Rossi E. Groups 9 and 10 metals-catalyzed O–H bond addition to unsaturated molecules // Top. Organomet. Chem. 2013. V. 43. P. 231.
Schobert H. Production of acetylene and acetylene-based chemicals from coal // Chem. Rev. 2014. V. 114. № 3. P. 1743.
Trotus I.-T., Zimmermann T., Schüth F. Catalytic reactions of acetylene: a feedstock for the chemical industry revisited // Chem. Rev. 2014. V. 114. № 3. P. 1761.
Chinchilla R., Nájera C. Chemicals from alkynes with palladium catalysts // Chem. Rev. 2014. V. 114. № 3. P. 1783.
Quintero-Duque S., Dyballa K.M., Fleischer I. Metal-catalyzed carbonylation of alkynes: key aspects and recent development // Tetrahedron Lett. 2015. V. 56. № 21. P. 2634.
Ananikov V.P. Nickel: the “spirited horse” of transition metal catalysis // ACS Catal. 2015. № 5. P.1964.
Huple D.B., Ghorpade S., Liu R.-S. Recent advances in gold-catalyzed N- and O- functionalizations of alkynes with nitrones, nitroso, nitro and nitroxy species // Adv. Synth. Catal. 2016. V. 358. P. 1348.
Voronin V.V., Ledovskaya V.S., Bogachenkov A.S., Rodygin K.S., Ananikov V.P. // Molecules. 2018. V. 23. P. 2442.
Nieuwland J.A., Vogt R.R. The Chemistry of Acetylene. N.Y.: Reinhold, 1945.
Reppe W. Neue Entwiclungen auf dem Gebiete der Chemie des Acetylene und Kohlen Oxydes. Berlin–Gőttingen–Heidelberg: Springer–Verlag, 1949.
Johnson A.W. The Chemistry of Acetylenic Compounds. London: Edward Arnold and Co. V. 1. 1946; V. 2. 1950.
Raphael R.A. Acetylenic Compounds in Organic Synthesis. L.: Butterwort, 1955. 219 p.
Ziegenbein W. Einführungen der Ăthinyl- und Alkynyl-Gruppe in Organische Verbindungen. Berlin: Verlag Chemie, 1963.
Котляревский И.Л., Шварцберг М.С., Фишер Л.Б. Реакции ацетиленовых соединений. Новосибирск: Наука СО АН СССР, 1967. 354 с.
Темкин О.Н., Флид Р.М. Каталитические превращения ацетиленовых соединений в растворах комплексов металлов. М.: Наука, 1968. 212 с.
Rudledge T.F. Acetylenic Compounds: Preparation and Substitution Reaction. N.Y., Amsterdam, London: Reinhold Publ. Comp., 1968.
Химия ацетиленовых соединений / Под ред. Вийе Г.Г. М.: Химия, 1973.
Shostakovskii M.F., Bogdanova A.V. The Chemistry of Diacetylenes. N.Y.: J. Wiley, 1974.
Hanford W.E., Fuller D. L. Acetylene chemistry // Ind. Eng. Chem. 1948. V. 40. P. 1171.
Copenhaver J., Bigelow M. Acetylene and Carbon Monoxide Chemistry. N.Y.: Reinhold Publ., 1949.
Устынюк Ю.А. Лекции по органической химии. Ч. 2. Химия углеводородов. Сер. Мир химии. М.: Техносфера, 2016. 372 с.
Walker L.F., Landergan T.E. Pat. 2487006 USA, 1949; C.A. (1950) V. 44. 5380.
Kindler K. // Chem. Ber. 1921. B. 54. S. 647.
Фогель И.Г. Ацетилен, его свойства, изготовление и применение / Под ред. и с доп. Залькинда Ю. Ленинград: ОНТИ Госхимтехиздат, 1934. 352 с.
Флид Р.М., Темкин О.Н. // А. с. CCCP. № 117494, 1958.
Флид Р.М., Темкин О.Н. // Журн. физ. химии. 1961. V. 35. P. 452.
Halpern I., James B. R., Kemp A.L.W. // J. Amer. Chem. Soc. 1961. V. 83. P. 4097.
Моисеев И.И. π-Комплексы олефинов в жидкофазном окислении. М.: Наука, 1970. 270 с.
Jira R. Acetaldehyde from ethylene – a retrospective on the discovery of the Wacker process // Angew. Chem. Int. Ed. 2009. V. 48. P. 9034.
Lines C.B., Long R. // Preprints Division of Petrol. Chem. Inc. Am. Chem. Soc. 1969. V. 14. № 2. P. 159.
Калия О.Л., Кирченкова Г.С., Темкин О.Н., Флид Р.М. // Кинетика и катализ. 1969. Т. 10. № 5. С. 1186.
Калия О.Л., Темкин О.Н., Флид Р.М., Кимельфельд Я.М., Кирченкова Г.С., Смирнова Е.М. // Изв. АН СССР. Сер. хим. 1969. С. 2854.
Кимельфельд Я.М., Смирнова Е.М., Першикова Н.И., Калия О.Л., Темкин О.Н. // Журн. структур. химии. 1971. Т. 12. С. 1097.
Penella F., Banks R.L., Bailey G.C. // J. Chem. Soc. Chem. Commun. 1968. P. 1548.
Могилянский А.И., Коган Л.М., Бондаренко И.Е., Кулешова Л.С., Темкин О.Н. // Ученые записки МИТХТ им. М.В. Ломоносова. 1970. Т. 1. № 2. С. 10.
Chaltykian O.A. Copper-Catalyzed Reactions. Consultants Bureau. N.Y., 1966.
Шмулевич Л.А. Развитие химии ацетилена. Автореф. дис. … канд. хим. наук. М.: Ин-т истории естествознания и техники АН СССР, 1969.
Темкин О.Н., Брук Л.Г., Зейгарник А.В. // Кинетика и катализ. 1993. Т. 34. № 3. С. 445.
Zeigarnik A.V., Bruk L.G., Temkin O.N., Likholobov V.F., Maier L.I. // Russ. Chem. Rev. 1996. V. 65. P. 117.
Temkin O.N., Zeigarnik A.V., Bonchev D. Chemical Reaction Networks. A Graph-Theoretical Approach. Boca Raton: CRC Press, 1996. 286 p.
Zeigarnik A.V., Valdes-Perez R.E., Temkin O.N., Bruk L.G., Shalgunov S.I. // Organometallics. 1997. V. 16. P. 3114.
Norman R.O.C., Parr W.J.E., Thomas C.B. // J. Chem. Soc. Perkin Trans. 1976. P. 1983.
Fukuda Y., Utimoto K., Nozaki H. // Heterocycles. 1987. V. 25. P. 297.
Hutchings G.J. // J. Catal. 1985. V. 96. P. 292.
Chiusoli G.P. // J. Organomet. Chem. 1986. V. 300. P. 57.
Григорян Э.А., Гюлумян Х.Р., Гуртовая Е.И., Ениколопян Н.С., Тер-Казарян М.А. // ДАН СССР. 1981. Т. 257. № 2. С. 364.
Носкова Н.Ф., Сокольский Д.В., Изтелеулова М.Б., Гафарова Н.А. // ДАН СССР. 1982. Т. 262. № 1. С. 113.
Шестаков Г.К., Темкин О.Н., Всесвятская Н.Ю., Степанов А.М. // Журн. орган. химии. 1979. Т. 15. № 2. С. 245.
Всесвятская Н.Ю., Шестаков Г.К., Темкин О.Н. // Кинетика и катализ. 1986. Т. 27. С. 1330.
Sasaki Y., Dixneuf P.H. // J. Chem. Soc. Chem. Commun. 1986. P. 790.
Чухаджян Г.А., Абрамян Ж.И., Куколев В.П., Геворгян Г.А., Тонян Г.М., Мелконян Л.Н. // Армян. хим. журн. 1978. Т. 31. № 8. С. 607.
Sonogashira K., Tohda Y., Hagihara N. // Tetrahedron Lett. 1975. P. 4467.
Шестаков Г.К., Васильев А.М., Тищенко Л.М., Темкин О.Н., Флид Р.М. // Кинетика и катализ. 1974. Т. 15. № 4. С. 1070.
Heck R.F. // J. Amer. Chem. Soc. 1972. V. 94. P. 2712.
Tsuji J., Takahashi M., Takahashi T. // Tetrahedron Lett. 1980. V. 21. P. 849.
Брук Л.Г., Темкин О.Н. // Хим. пром-сть. 1993. С. 57.
Шуляковский Г.М., Темкин О.Н., Быканова Н.В., Ныркова А.Н. Химическая кинетика в катализе. Кинетические модели жидкофазных реакций / Под ред. Кипермана С.Л. Черноголовка: ИОХ АН СССР, ИХФ АН СССР, 1985. С. 112.
Брайловский С.М., Кабалина Г.А., Шестакова В.С., Темкин О.Н. // Журн. орган. химии. 1977. Т. 13. С. 1158.
Шестакова В.С., Брайловский С.М., Темкин О.Н. // Кинетика и катализ. 1978. Т. 19. С. 1585.
Брук Л.Г., Всесвятская Н.Ю., Стромнова Т.А., Алексеева Н.Ф., Темкин О.Н. // Журн. орган. химии. 1978. Т. 14. С. 473.
Темкин О.Н., Мехрякова Н.Г., Брук Л.Г., Калия О.Л., Прудников А.Ю. // Кинетика и катализ. 1979. Т. 20. С. 629.
Темкин О.Н., Брук Л.Г. // Успехи химии.1983. Т. 52. С. 206.
Mortreux A., Blanchard M. // Chem. Commun. 1974. P. 786.
Калия О.Л., Темкин О.Н., Мехрякова Н.Г., Флид Р.М. // Докл. АН СССР. 1971. Т. 199. № 6. С. 1321.
Bruk L.G., Temkin O.N. // Proc. 9th Soviet-Japan Seminar on Catalysis. Novosibirsk: Institute of catalysis Acad. Sci. USSR, 1990. P. 83.
Bruk L.G., Oshanina I.V., Kozlova A.P., Vorontsov E.V., Temkin O.N. // J. Mol. Catal. 1995. V. 104. P. 9.
Темкин О.Н., Брук Л.Г. // Кинетика и катализ. 2003. Т. 44. № 5. С. 661.
Malashkevich A.V., Bruk L.G., Temkin O.N. // J. Phys. Chem. A. 1997. V. 101. № 52. P. 9825.
Gorodsky S.N., Bruk L.G., Istomina A.E., Kurdiukov A.V., Temkin O.N. // Top. Catal. 2009. V. 52. P. 557.
Козлов А.И., Брайловский С.М., Темкин О.Н. // Кинетика и катализ. 1994. Т. 35. № 4. С. 551.
Козлов А.И., Брайловский С.М., Темкин О.Н. // Кинетика и катализ. 1995. Т. 36. № 2. С. 225.
Ananikov V.P., Gayduk K.A., Orlov N.V., Beletskaya I.P., Khrustalev V.N., Antipin M.Yu. // Chem. Eur. J. 2010. V. 16. P. 2063.
Khemchyan L.L., Ivanova J.V., Zalesskiy S.S., Ananikov V.P., Beletskaya I.P. // Adv. Synth. Catal. 2014. V. 356. P. 771.
Zalesskiy S.S., Khrustalev V.N., Kostukovich A.Yu., Ananikov V.P. // Organometallics. 2015. V. 34. P. 5214.
Drent E., Arnoldy D., Budzelar P.H.M. // J. Organomet. Chem. 1993. V. 455. P. 247.
Drent E., Arnoldy D., Budzelar P.H.M. // J. Organomet. Chem. 1994. V. 475. P. 57.
Doucet H., Hofer J., Bruneau C., Dixneuf P.H. // J. Chem. Soc. Chem. Commun. 1993. P. 850.
Bruneau C., Dixneuf P.H. // Angew. Chem. Int. Ed. 2006. V. 45. P. 2176.
Митченко С.А., Шубин А.А. Металлоорганические производные ацидокомплексов платины: синтез, реакционная способность и дизайн каталитических реакций. Донецк: ДонГУЭТ. 2004. 217 с.
Dorel R., Echavarren A. // Chem. Rev. 2015. V. 115. P. 9028.
Schmidt A., Halaiqa A., Smirnov V.V. // Synlett. 2006. № 18. P. 2861.
Eremin D.B., Ananikov V.P. Understanding Active Species in Catalytic Transformations: from Molecular Catalysis to Nanoparticles, Leaching, “Cocktails” of Catalysts and Dynamic Systems // Coord. Chem. Rev. 2017. V. 346. P. 2.
Schmidt A.F., Kurokhtina A.A., Larina E.V. // Mendeleev Commun. 2017. V. 27. P. 213.
Strauss F. // J. Liebig. Annalen Chem. 1905. B. 342. № 5. S. 190.
Akhtar M., Richards T.A., Weedon B.C.L. // J. Chem. Soc. 1959. P. 933.
Мыхаличко Б.М., Темкин О.Н., Мыськив М.Г. // Успехи химии. 2000. Т. 69. № 11. С. 1042.
Gorin D.J., Toste F.D. // Nature. 2007. V. 446. P. 395.
Soriano E., Marco-Contelles J. // Top. Curr. Chem. 2011. V. 32. P. 1.
Zuccaccia D., Belpassi L., Rocchigiani L., Tarantelli F., Macchioni A. // Inorg. Chem. 2010. V. 49. P. 3080.
Das A., Dash C., Celik M.A., Yousufuddin M., Frenking G., Dias R. // Organometallics. 2013. V. 32. P. 3135.
Balcioglu N., Uraz I., Bozkurt C., Sevin F. // Polyhedron. 1997. V. 16. № 2. P. 327.
Cornfield P.W.R., Shearer H.M.M. // Acta Crystal. Chem. 1966. V. 21. P. 957.
Темкин О.Н., Флид Р.М., Сухова Т.Г., Чепайкин Е.Г., Тихонов Г.Ф. // Журн. прикл. химии. 1968. Т. 41. С. 633.
Сухова Т.Г., Темкин О.Н., Флид Р.М., Калия Т.К. // Журн. неорган. химии. 1968. Т. 13. № 8. С. 2073.
Сухова Т.Г., Борщ Н.Я., Темкин О.Н., Флид Р.M. // Журн. неорган. химии. 1969. Т. 14. № 3. С. 694.
Сухова Т.Г., Темкин О.Н., Флид Р.М. // Журн. неорган. химии. 1969. Т. 14. № 4. С. 928.
Сухова Т.Г., Темкин О.Н., Флид Р.М. // Журн. неорган. химии. 1970. Т. 15. № 7. С. 1849.
Nishiwaki K.-ichiro, Kobayashi M., Takeuchi T., Matuoto K., Osakada K. // J. Mol. Catal. A. Chem. 2001. V. 175. P. 73.
Ahrland S., Rawstorne J. // Acta Chem. Scand. 1970. V. 24. P. 157.
Мыхаличко Б.М., Аксельруд Л.Г., Давыдов В.Н. // Координац. химия. 1997. Т. 42. № 3. С. 410.
Тихонов Г.Ф., Темкин О.Н., Флид Р.М. // Кинетика и катализ. 1967. Т. 8. № 3. С. 520.
Тихонов Г.Ф., Темкин О.Н., Флид Р.М. // Журн. физ. химии. 1966. Т. 11. № 12. С. 3075.
Тихонов Г.Ф., Шестаков Г.К., Темкин О.Н., Флид Р.М. // Завод. лаб. 1967. Т. 33. № 2. С. 150.
Темкин О.Н., Тихонов Г.Ф., Флид Р.М., Галеев Ю.Р. // Кинетика и катализ. 1967. Т. 8. № 6. С. 1236.
Темкин О.Н., Флид Р.М., Шестаков Г.К., Ермакова А., Тихонов Г.Ф., Яровая Л.Н., Михальченко В.Г. // Кинетика и катализ. 1969. Т. 10. № 6. С. 1231.
Шестаков Г.К., Тихонов Г.Ф., Темкин О.Н., Флид Р.М. // Кинетика и катализ. 1970. Т. 11. № 3. С. 575.
Шестаков Г.К., Тихонов Г.Ф., Темкин О.Н., Флид Р.М., Гершензон И.Ш., Брайловский С.М., Должникова С.И. // Кинетика и катализ. 1970. Т. 11. № 4. С. 875.
Темкин О.Н., Шестаков Г.К., Козлова Н.Ю. // Кинетика и катализ. 1990. Т. 31. № 4. С. 850.
Тихонов Г.Ф., Шестаков Г.К., Темкин О.Н., Флид Р.М. // Кинетика и катализ. 1966. Т. 7. № 5. С. 614.
Темкин О.Н. О различных взаимосвязях кинетики и термодинамики. Saarbrücken: Lambert Acad. Publ., 2016. 119 p.
Темкин О.Н., Сухова Т.Г., Шестаков Г.К., Тихонов Г.Ф., Флид Р.М., Чепайкин Е.Г., Новикова Г.М., Борщ Н.Я. // Кинетика и катализ. 1969. Т. 10. № 5. С. 1004.
Брайловский С.М., Темкин О.Н., Флид Р.М., Белова Н.Г. // Журн. физ. химии. 1970. Т. 44. № 4. С. 1112.
Брайловский С.М., Темкин О.Н., Флид Р.М. // Кинетика и катализ. 1971. Т. 12. № 5. С. 1152.
Осечкин С.И., Мыськив М.Г., Завалий П.Ю., Соболев А.Н. // Металлоорган. химия. 1991. Т. 4. № 5. С. 997.
Мыхаличко Б.М., Мыськив М.Г., Аксельруд Л.Г. // Координац. химия. 1993. Т. 19. № 9. С. 722.
Мыськив М.Г., Мыхаличко Б.М. // Журн. структур. химии. 1994. Т. 35. № 5. С. 120.
Мыхаличко Б.М., Мыськив М.Г. // Журн. неорган. химии. 1999. Т. 44. № 3. С. 411.
Мыхаличко Б.М., Мыськив М.Г. // Координац. химии. 1998. Т. 24. № 12. С. 939.
Мыхаличко Б.М. // Журн. неорган. химии. 1998. Т. 43. № 10. С. 1639.
Мыхаличко Б.М. // Координац. химия. 1999. Т. 25. № 5. С. 359.
Мыхаличко Б.М., Мыськив М.Г., Давыдов В.Н. // Журн. неорган. химии. 1999. Т. 44. № 1. С. 46.
Мыськив М.Г., Завалий П.Ю., Мыхаличко Б.М., Фундаменский В.С. // Координац. химия. 1988. Т. 14. № 12. С. 1619.
Мыхаличко Б.М., Мыськив М.Г. // Координац. химия. 1999. Т. 25. № 5. С. 533.
Gamasa M.P., Gimtno J., Lastra E. // J. Organomet. Chem. 1988. V. 346. P. 277.
Reger D.L., Huff M.F. // Organometallics. 1990. V. 9. P. 2807.
Osakoda K., Takizawa T., Yamamoto T. // Organometallics. 1995. V. 14. P. 3531.
Reger D.L., Collins J.E., Huff M.F. // Organometallics. 1995. V. 14. P. 5475.
Lang H., Weinmann M. // Synlett. 1995. P. 1.
Normant J.E., Alexakis A. // Synthesis. 1981. P. 841.
Вартанян В.С., Шестаков Г.К., Темкин О.Н. // Армян. хим. журн. 1979. Т. 32. № 4. С. 259.
Вартанян В.С., Шестаков Г.К., Темкин О.Н. // Армян. хим. журн. 1979. Т. 32. № 4. С. 264.
Tachiyama T., Yoshida M., Aoyagi T., Fukuzumi S. // Appl. Organomet. Chem. 2008. V. 22. P. 205.
Tachiyama T., Yoshida M., Aoyagi T., Fukuzumi S. // J. Phys. Org. Chem. 2008. V. 21. P. 510.
Hefner J.G., Zizelman P.M., Durfee L.D., Lewandos G.S. // J. Organomet. Chem. 1984. V. 260. P. 369.
Lieu J., Zuo Y., Han M., Wang Z., Wang D. // J. Nat. Gas Chem. 2012. V. 21. P. 495.
Liu J., Zuo Y., Han M., Wang Z. // J. Chem. Technol. Biotechnol. 2012. Online library. Wiley com. https://doi.org/10.1002/jctb.3860
Sun S., Kroll J., Luo Y., Zhang L. // Synlett. 2012. V. 23. P. 54.
Yamazaki H.J. // J. Chem. Soc. Chem. Commun. 1976. V. 21. P. 841.
Dahlberg L., Frosin K.-M., Kerstan S., Werner D. // J. Organomet. Chem. 1991. V. 407. P. 115.
Bianchini C., Perruzzini M., Zanobini F., Frediani P., Albinati A. // J. Am. Chem. Soc. 1991. V. 113. P. 5453.
Bianchini C., Frediani P., Masi D., Perruzzini M., Zanobini F. // Organometalliks. 1994. V. 13. P. 4616.
Wakatsuki Y., Yamazaki H. // J. Am. Chem. Soc. 1991. V. 113. P. 9604.
Wakatsuki Y., Yamazaki H. // J. Organomet. Chem. 1995. V. 500. P. 349.
Slugovc C., Mereiter K., Zobetz E., Schmid R., Kirchner K. // Organometallics. 1996. V. 15. P. 5275.
Yi C.S., Liu N. // Organometallics. 1996. V. 15. P. 3968.
Yi C.S., Liu N. // Synlett. 1999. № 3. P. 281.
Yi C.S., Liu N. // Organometallics. 1997. V. 16. P. 3910.
Yi C.S., Liu N. // Organometallics. 1998. V. 17. P. 3158.
Melis K., Samulkiewicz P., Rynkowski J., Verpoort F. // Tetrahedron Lett. 2002. V. 43. P. 2713.
Bassetti M., Pasquini C., Raneri A., Rosato D. // J. Org. Chem. 2007. V. 72. P. 4558.
Katayama H., Yari H., Tanaka M., Ozawa F. // Chem. Commun. 2005. P. 4336.
Chen X., Xue P., Sung H.H.Y., Wiliams I.D., Peruzzini M., Bianchini C. Yia G. // Organometallics. 2005. V. 24. P. 4330.
Lee J.-H., Caulton K.G. // J. Organomet. Chem. 2008. V. 693. P. 1664.
Hijazi A., Parkhomenko K., Djukic J.-P., Chemmri A. // Adv. Synth. Catal. 2008. V. 350. P. 1493.
Dobson A., Moore D.S., Robinson S.D., Hursthouse M.B., New L. // Polyhedron. 1985. V. 4. P. 1119.
Jia G., Rheingold A.L., Meek D.W. // Organometallics. 1989. V. 8. P. 1378.
Wakatsuki Y., Satoh M., Yamazaki H. // Chem. Lett. 1989. P. 1585.
Bullock R.M. // J. Chem. Soc. Chem. Commun. 1989. P. 165.
Tenorio M.J., Puerta M.C., Valegra P. // J. Chem. Soc. Chem. Commun. 1991. P. 1750.
Wakatsuki Y., Koga N., Yamazaki H., Morokama K. // J. Am. Chem. Soc. 1994. V. 116. P. 8105.
Yi C.S., Liu N. // Organometallics. 1997. V. 16. P. 3729.
Lee H.M., Yao J., Jia G. // Organometallics. 1997. V. 16. P. 3927.
Bruneau C., Dixneuf P.H. // Acc. Chem. Res. 1999. V. 32. P. 311.
Bruce M., Wallis R.C. // Austr. J. Chem. 1979. V. 32. P. 1471.
Bruce M. // Chem. Rev. 1991. V. 91. P. 197.
Naota T., Takaya H., Murahashi S.-I. // Chem. Rev. 1998. V. 98. P. 2599.
Metal Vinylidens and Allenylidens in Catalysis: From Reactivity to Applications / Eds. Bruneau Ch., Dixneuf P.H. Wiley–VCH, 2008.
Темкин О.Н., Брук Л.Г. // Рос. хим. журн. 2014. Т. 58. № 5–6. С. 90.
Esteruelas M.A., Oro L.A., Ruiz N. // Organometallics. 1994. V. 13. P. 1507.
Barbaro P., Bianchini C., Peruzzini M., Polo A. // Inorg. Chem. Acta. 1994. V. 220. P. 5.
Gotzig J., Otto H., Werner H. // J. Organomet. Chem. 1985. V. 257. P. 247.
Esteruelas M.A., Herrero J., Lopez A.M., Olivan M. // Organometallics. 2001. V. 20. P. 3202.
Alos J., Bolaño T., Esteruelas M.A., Olivan M., Oñate E., Valencia M. // Inorg. Chem. 2013. V. 52. P. 6199.
Field L.D., Messerle B.A., Smernik R.J., Hambley T.W., Turner P. // J. Chem. Soc. Dalton Trans. 1999. P. 2557.
Midya G.C., Paladhi S., Dhara K., Dash J. // Chem. Commun. 2011. V. 47. P. 6698.
Midya G.C., Parasar B., Dhara K., Dash J. // Org. Biomol. Chem. 2014. V. 12. P. 1812.
Rivada-Wheelahan O., Chakraborty S., Shimon L.J.W., Ben-David Y., Milstein D. // Angew. Chem. Int. Ed. 2016. V. 55. P. 6942.
Bhunia M., Sahoo S., Vijakumar G., Adhikari D., Mandal S. // Organometallics. 2016. V. 35. P. 3775.
Liang Q., Osten K.M., Song D. // Angew. Chem. Int. Ed. 2017. V. 56. P. 1.
Bauer I., Knölker H.-J. // Chem. Rev. 2015. V. 115. P. 3170.
Singer H., Wilkinson G. // J. Chem. Soc. A. 1968. P. 849.
Albano P., Aresta M. // J. Organomet. Chem. 1980. V. 190. P. 243.
Schāfer H.-A., Marcy R., Rüping T. Singer H. // J. Organomet. Chem. 1982. V. 240. P. 17.
Kovaler I.P., Yerdakov K.V., Strelenko Yu.A., Vinogradov M.G., Nikishin G.I. // J. Organomet. Chem. 1990. V. 386. P. 139.
Boese W.T., Goldman A.S. // Organometallics. 1991. V. 10. P. 782.
Schāfer M., Wolf J., Werner H. // J. Chem. Soc. Chem. Commun. 1991. P. 1341.
Schāfer M., Wolf J., Werner H. // J. Chem. Soc. Dalton Trans. 2005. P. 1468.
Lee C.-C., Lin Y.-C., Liu Y.-H., Wang Y. // Organometallics. 2005. V. 24. P. 136.
Ito J.-ichi, Kitase M., Nishiyama H. // Organometallics. 2007. V. 26. P. 6412.
Peng H.M., Zhao J., Li X. // Adv. Synth. Catal. 2009. V. 351. P. 1371.
Mochizuki K., Sakai K., Kochi T., Kakiuchi F. // Synthesis. 2013. V. 45. P. 2088.
Rubio-Perez L., Azpiroz R., Di Giuseppe A., Polo V., Castarlenas R., Perez-Torrente J.J., Oro L.A. // Chem. Eur. J. 2013. V. 19. P. 15304.
Ogata K., Toyota A. // J. Organomet. Chem. 2007. V. 692. P. 4139.
Ghosh R., Zhang X., Achord P., Emye T.J., Krogh-Jespersen K., Goldman A.S. // J. Am. Chem. Soc. 2007. V. 129. P. 853.
Field L.D., Ward A.J. // J. Organomet. Chem. 2003. V. 681. P. 91.
Hilt G., Hess W., Vogler T., Hengst C. // J. Organomet. Chem. 2005. V. 690. P. 5170.
Meriwether L.S., Colthup E.C., Kennerly G.W., Reusch R.N. // J. Org. Chem. 1961. V. 26. P. 5155.
Meriwether L.S., Colthup E.C., Kennerly G.W., Reusch R.N. // J. Org. Chem. 1961. V. 26. P. 5163.
Colthup E.C., Meriwether L.S. // J. Org. Chem. 1961. V. 26. P. 5
Giacomelli G., Caporusso A.M., Lardici L. // J. Org. Chem. 1979. V. 44. P. 231.
Ogoshi S., Ueta M., Oka M., Kurosawa H. // Chem. Commun. 2004. № 23. P. 2732.
Sabourin E.T. // J. Mol. Catal. 1984. V. 26. P. 363.
Trost B.M., Sorum M.T., Chan C., Harms A.E., Rühter G. // J. Am. Chem. Soc. 1997. V. 119. P. 698.
Canty A.J. // Acc. Chem. Res. 1992. V. 25. P. 83.
Herrmann W.A., Böhm V.P.W., Gstöttmayr G.V.K., Grosche M., Reisinger C.-C., Weskamp T. // J. Organomet. Chem. 2001. V. 617–628. P. 616.
Rubina M., Gevorgyan V. // J. Organomet. Chem. 2001. V. 123. P. 11107.
Yang C., Nolan S.P. // J. Org. Chem. 2002. V. 67. P. 591.
Jahier C., Zatolochnaya O.V., Zvyagintsev N.V., Ananikov V.P., Gevorgyan V. // Org. Lett. 2012. V. 14. P. 2846.
Tohda Y., Sonogashira K., Hagihara N. // J. Organom. Chem. 1976. V. 110. C. 53.
Yasuda T., Kai Y., Yasuoka N., Kasai N. // Bull. Chem. Soc. Jpn. 1977. V. 50. № 11. P. 2888.
Zatolochnaya O.V., Gordeev E.G., Jahier C., Ananikov V.P., Gevorgyan V. // Chem. Eur. J. 2014. V. 20. P. 9578.
Chen T., Guo C., Goto M., Han L.-B. // Chem. Commun. 2013. V. 49. P. 7498.
Chen R., Chen T., Zhao Y., Qiu R., Zhou Y., Yin S., Wang X., Goto M., Han L.-B. // J. Am. Chem. Soc. 2011. V. 133. P. 17037.
Kawata A., Kuninobu Y., Takai K. // Chem. Lett. 2009. V. 38. P. 836.
Hagihara N., Tamura M., Yamazaki H., Fujiwara M. // Bull. Chem. Soc. Jpn. 1961. V. 34. P. 892.
Akita N., Yasuda H., Nakamura A. // Bull. Chem. Soc. Jpn. 1984. V. 57. P. 480.
Horton A.D. // J. Chem. Soc. Chem. Commun. 1992. P. 185.
Platel R.H., Schafer L.L. // Chem. Commun. 2012. V. 48. P. 10609.
Thompson M.E., Baxter S.M., Bulls A.R., Burger B.J., Nolan M.C., Santarsiero B.D., Schaefer W.P., Bercaw J.E. // J. Am. Chem. Soc. 1987. V. 109. P. 203.
St. Clair M., Schaefer W.P., Bercaw J.E. // Organometallics. 1991. V. 10. P. 525.
Den Haan K.H., Wielstra Y., Teuben J.H. // Organometallics. 1987. V. 6. P. 2053.
Heeres H.J., Teuben J.H. // Organometallics. 1991. V. 10. P. 1980.
Evans W.J., Keyer R.A., Ziller J.W. // Organometallics. 1990. V. 9. P. 2628.
Evans W.J., Keyer R.A., Ziller J.W. // Organometallics.1993. V. 12. P. 2618.
Nishiura M., Hou Z., Wakatsuki Y., Yamaki T., Miyamoto T. // J. Am. Chem. Soc. 2003. V. 125. P. 1184.
Nishiura M., Hou Z. // J. Mol. Catal. A. Chem. 2004. V. 213. P. 101.
Ge S., Quiroga Narambuena V.F., Hessen B. // Organometallics. 2007. V. 26. P. 6508.
Haskel A., Straub T., Dash A.K. Eisen M.S. // J. Am. Chem. Soc. 1999. V. 121. P. 3014.
Haskel A., Wang J.Q., Straub T., Neyroud T.G., Eisen M.S. // J. Am. Chem. Soc. 1999. V. 121. P. 3025.
Dash A.K., Wang J.S., Berthet J.C., Ephritikhine M., Eisen M.S. // J. Organomet. Chem. 2000. V. 604. P. 83.
Dash A.K., Gourevich I., Wang J.Q., Wang J., Kapon M., Eisen M. // Organometallics. 2001. V. 20. P. 5084.
Dash A., Eisen M.S. // Org. Lett. 2000. V. 2. P. 737.
Schmidbaur H., Schier A. // Organometallics. 2010. V. 20. P. 2.
Якобсон В.В., Гескин В.М., Клименко Н.М., Боженко К.В., Tемкин О.Н. // Теорет. и эксперим. химия. 1985. № 3. С. 303.
Klimenko N.M., Bozhenko K.V., Strumina E.V., Rykova E.A., Temkin O.N. // Mol. Structure (THEOCHEM) 1999. V. 490. P. 233.
Темкин О.Н. // Кинетика и катализ. 2014. Т. 55. № 2. С. 181.
McDade Ch., Bercaw J.E. // J. Organomet. Chem. 1985. V. 279. P. 281.
Frohnapfel D.S., Templeton J.L. // Coord. Chem. Rev. 2000. V. 206–207. P. 199.
Antonova A.B., Kolobova N.E., Petrovsky P.V., Lokshin B.V., Obezyuk N.S. // J. Organomet. Chem. 1977. V. 137. P. 55.
Kolobova N.E., Antonova A.B., Khitrova O.M., Antipin M.Yu., Struchkov Yu.T. // J. Organomet. Chem. 1977. V. 137. P. 69.
Антонова А.Б., Иогансон А.А. // Успехи химии. 1989. Т. 58. С. 1197.
De Angelis F., Sgamellotti A. // Organometallics. 2002. V. 21. № 13. P. 2715.
De Angelis F., Sgamellotti A., Re N. // Organometallics. 2002. V. 21. № 26. P. 5944.
Grotjahn D.B., Zeng Xi., CooksyA.L., Scott Kassel W., DiPasquale A.G., Zakharov L.N., Rheingold A.L. // Organometallics. 2007. V. 26. № 14. P. 3385.
De Angelis F., Sgamellotti A., Re N. // Organometallics. 2007. V. 26. № 14. P. 5285.
Vastine B.A., Hall M. // Organometallics. 2008. V. 27. P. 4325.
Tenorio M.J., Puerta M.C., Valegra P., Ortuno M.A., Ujaque G., Llendos A. // Inorg. Chem. 2013. V. 52. P. 8919.
Olivan M., Clot E., Eisenstein O., Caulton K.G. // Organometallics. 1998. V. 17. № 14. P. 3091.
Buil M.L., Eisenstein O., Esteruelas M.A., Garsia-Yebra C., Gutierrez-Puebla E., Olivan M., Onate E., Ruiz N., Tajada M. // Organometallics. 1999. V. 18. P. 4949.
Bassetti M., Cadierno V., Gimeno J., Pasquini C. // Organometallics. 2008. V. 27. № 19. P. 5009.
Muthoh Y., Imai K., Kimura Y., Ikeda Y., Ishii Y. // Organometallics. 2011. V. 30. P. 204.
Salvio R., Julia-Hernandez F., Pisciottni L., Mendoza-Merono R., Garcia-Granda S., Bassetti M. // Organometallics. 2017. V. 36. № 19. P. 3830.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ