

Кинетика и катализ, 2020, T. 61, № 1, стр. 63-84
Механизмы газофазного гидрохлорирования ацетилена: активные центры катализаторов, изотопные эффекты и стереоселективность реакции
Т. В. Краснякова a, b, Д. В. Никитенко a, С. А. Митченко a, c, *
a Институт физико-органической химии и углехимии им. Л.М. Литвиненко
Донецк, ул. Р. Люксембург, 70, Украина
b Луганский национальный университет имени Тараса Шевченко
Луганск, ул. Оборонная, 2, Украина
c ФГБОУ ВО Южно-Российский государственный политехнический университет им. М.И. Платова
346400 Новочеркасск, ул. Просвещения, 132, Россия
* E-mail: samit_RPt@mail.ru
Поступила в редакцию 08.05.2019
После доработки 17.07.2019
Принята к публикации 19.07.2019
Аннотация
Представлены результаты исследования механизмов газофазного гидрохлорирования ацетилена на хлоридных комплексах благородных металлов. Рассмотрены природа активных центров гетерогенных катализаторов, изотопные эффекты HCl/DCl, стереохимия продуктов реакции, возможные стадийные механизмы и механизмы отдельных стадий.
ВВЕДЕНИЕ
Винилхлорид (ВХМ) является мономером для производства одного из наиболее востребованных пластиков – поливинилхлорида (ПВХ). Благодаря химической устойчивости к разложению, прочности и отсутствию токсичности ПВХ находит широкое применение в медицине, электротехнике, автомобильной промышленности, строительстве и других областях [1–4]. Поливинилхлорид по объему мирового производства занимает третье место среди полимерных материалов после полиэтилена и полипропилена. По данным Ceresana [5] в 2016 году было потреблено более 42 млн тонн ПВХ – это около 16% от общего спроса на пластмассы. Согласно прогнозу [1] мировой спрос будет увеличиваться и далее – примерно на 2.3% в год вплоть до 2024 г., что обусловливает рост производства ВХМ. По состоянию на 2015 г. [6] лидером производства является Юго-Восточная Азия (60%), второе место занимает Северная Америка (14%), на долю стран Западной Европы приходится 10%, России и Балтии – 2%.
Начиная со второй половины прошлого века, в западных странах коммерческое получение ВХМ практически полностью ориентировано на его производство из этилена – продукта процессов переработки нефти и природного газа. Так называемый сбалансированный процесс получения винилхлорида состоит из трех стадий: оксихлорирования и прямого хлорирования этилена с образованием дихлорэтана (ДХЭ), протекающих в присутствии CuCl2 и FeCl3 соответственно, и термического разложения ДХЭ с выделением ВХМ [7]:
В отличие от других стран в Китае, крупнейшем мировом производителе ВХМ, благодаря дешевой электроэнергии и большим природным запасам каменного угля при ограниченных ресурсах ископаемых углеводородов основным способом промышленного получения ВХМ (до 80%) является прямое каталитическое гидрохлорирование ацетилена [8–11]:
Реакция экзотермическая и протекает с высокой селективностью при температуре ~180°С [12]. В качестве катализатора традиционно используется нанесенная на активированный уголь сулема (обычно 10–15 мас. %) – более 60% мировой добычи ртути в последние годы идет на производство катализаторов. Однако активность такого промышленного катализатора довольно быстро снижается за счет сублимации хлорида ртути. Это приводит к потере в совокупности 500–1000 т ртути в год за счет выбросов в окружающую среду даже при использовании современных систем улавливания ртути из абгазов [2, 11, 13]. Высокотоксичные ртутные выбросы негативно воздействуют на здоровье человека и наносят значительный ущерб окружающей среде.
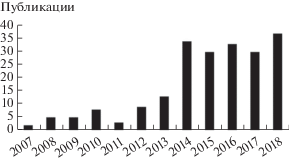
В октябре 2013 г. экологические проблемы, связанные с применением ртути при производстве ВХМ, были включены в контекст Минаматской конвенции по ртути [14]. Эта конвенция была ратифицирована в мае 2017 г. и является глобальным договором по защите здоровья человека и окружающей среды от неблагоприятных воздействий ртути, подписанным почти 140 странами. Она охватывает все аспекты, связанные с производством, использованием и хранением ртути. Ее пятая статья касается промышленных процессов, в которых применяется ртуть или ртутьсодержащие соединения: запрещено их использование на заводах по производству ВХМ, запускаемых позже 2017 г., в то время как уже существующие производственные мощности должны быть переведены на безртутные катализаторы к 2022 г. Сложившаяся ситуация в сочетании с ограниченностью природных запасов ртути мотивирует интенсивные поиски альтернативных экологически чистых гетерогенно-каталитических систем гидрохлорирования ацетилена – после 2012 г. наблюдается всплеск публикационной активности в этой области (рис. 1).
Рис. 1.
Динамика публикационной активности в области гетерогенно-каталитического гидрохлорирования ацетилена: данные Scopus, поисковые запросы “acetylene hydrochlorination”.
Подавляющее большинство работ (например, обзоры [2, 4, 15–18]) по гетерогенно-каталитическому гидрохлорированию ацетилена направлено на поиск новых и модифицирование известных катализаторов. В них мало уделяется внимания установлению механизмов протекания реакции, тогда как понимание последних позволило бы целенаправленно конструировать новые каталитические системы. Вместе с тем механизм каталитического гидрохлорирования ацетилена в гомогенных условиях достаточно хорошо изучен [12] (схема 1 ). Реакция начинается с π-координации тройной С≡С-связи к металлическому центру, что существенно облегчает [19] последующую атаку нуклеофилом. В случае атаки координированного ацетилена внешним нуклеофилом имеет место транс(анти)-присоединение последнего и металлокомплекса к тройной связи с образованием σ-винильного металлоорганического производного, в противном случае образуется продукт цис(син)-присоединения. Протолиз указанного комплекса дает конечный продукт с сохранением стереохимии интермедиата.
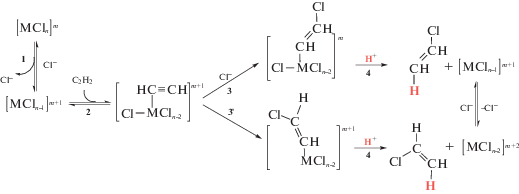
Схема 1 . Стадийный механизм гомогенно-каталитического гидрохлорирования ацетилена [12].
Исходя из взаимосвязи между гомогенным и гетерогенным катализом (например, [20–22]), можно ожидать реализацию в гетерогенных системах подобного стадийного механизма. Однако в этом случае некоторые детали остаются неясными. Во-первых, в гетерогенных условиях свободные хлорид-ионы (схема 1 , стадия 3) отсутствуют. Во-вторых, для осуществления реакции необходима предварительная π-координация ацетилена к комплексам металла с координационной вакансией, которые в растворах возникают в результате диссоциации связи М–Cl. В гетерогенных условиях нет сольватации, поэтому диссоциация термодинамически невыгодна и координационно ненасыщенные комплексы едва ли могут возникать таким путем. Указанные каталитически активные состояния на поверхности твердых тел можно генерировать механохимической обработкой [23]: предварительная механоактивация металлокомплексных солей K2PtCl6 [24], K2PtCl4 [25] и K2PdCl4 [26] в атмосфере ацетилена, этилена или пропилена приводит к формированию гетерогенных катализаторов газофазного гидрохлорирования ацетилена в пост-эффекте, т.е. после прекращения механической обработки.
Целью настоящей работы является обобщение представлений о механизмах газофазного каталитического гидрохлорирования ацетилена на гетерогенных катализаторах. Особое внимание уделено установлению природы активных состояний катализаторов. В качестве важного инструмента использован метод изотопных меток HCl/DCl, позволяющий определить стереохимию продуктов реакции, участие молекулы хлористого водорода в лимитирующей стадии и структуру ее переходного состояния [27].
МЕХАНОАКТИВИРОВАННЫЕ КАТАЛИЗАТОРЫ
Согласно представлениям об активных центрах в гетерогенно-каталитическом процессе участвует не вся поверхность катализатора, а только небольшая ее часть, которую обычно ассоциируют с дефектами на поверхности кристаллов. При традиционных способах получения катализаторов поверхностная концентрация активных центров зачастую слишком мала для их надежной идентификации. Механохимические методы приготовления катализаторов позволяют генерировать на поверхности дефекты, которые после прекращения механического воздействия сохраняются в сверхравновесных количествах благодаря заторможенной подвижности в твердых телах. Получаемые таким путем массивные катализаторы вряд ли могут составить конкуренцию нанесенным катализаторам в промышленном синтезе винилхлорида. Однако эти каталитические системы оказались весьма удобными для идентификации активных центров и установления механизма каталитического гидрохлорирования ацетилена.
Соль K2PtCl6 (система 1)
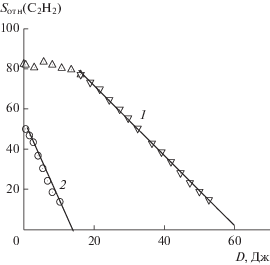
Кинетика гидрохлорирования ацетилена. Трибохимическая реакция. Соль K2PtCl6 в условиях непрерывной механической обработки катализирует [28] гидрохлорирование ацетилена газообразным HCl в отсутствии растворителя11. На кинетической кривой поглощения ацетилена при обработке порошка K2PtCl6 наблюдаются два участка [24] (рис. 2). В начале реакции имеет место индукционный период, по истечении которого ацетилен расходуется по нулевому порядку. Небольшие добавки K2PtCl4 к K2PtCl6 существенно уменьшают длительность индукционного периода (рис. 2), что свидетельствует о накоплении за это время PtII путем восстановления K2PtCl6 ацетиленом.
Рис. 2.
Типичная зависимость площади хроматографического пика ацетилена относительно инертного внутреннего стандарта (метана) от дозы поглощенной катализатором механической энергии в условиях непрерывной механообработки K2PtCl6 (0.25 г): 1 – реакция без добавок K2PtCl4; 2 – с добавкой 8 мол. % K2PtCl4.
Реакция в пост-эффекте. На предварительно измельченной в атмосфере воздуха соли K2PtCl6 поглощения ацетилена и накопления винилхлорида не наблюдается, а на обработанной аналогичным образом в атмосфере воздуха смеси K2PtCl6–K2PtCl4 (10 мол. %) винилхлорид образуется крайне медленно. Для получения активного катализатора необходима предактивация K2PtCl6 или, лучше, смеси K2PtCl6–K2PtCl4 в атмосфере ацетилена, этилена или пропилена [30]. В атмосфере HCl кинетика расходования ацетилена из газовой фазы замкнутого реактора в пост-эффекте, т.е. после прекращения механообработки, отвечает кинетическому уравнению первого порядка [31]. Реакция каталитическая, выход винилхлорида в расчете на поглощенный ацетилен близок к количественному. В присутствии DCl по данным 1Н-ЯМР-спектроскопии образуется исключительно продукт транс(анти)-присоединения:
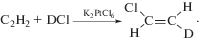
В реакции на бромидном катализаторе, полученном обработкой в атмосфере ацетилена солей K2PtBr6–K2PtBr4, в присутствии газообразного HCl образуется преимущественно винилхлорид.
Изотопные эффекты. Значения наблюдаемой константы скорости расходования ацетилена из газовой фазы замкнутого реактора в пределах экспериментальных ошибок совпадают в условиях примерно двадцатикратного и трехкратного избытка HCl по отношению к ацетилену [31]. Это может быть следствием неучастия молекулы HCl в лимитирующей стадии реакции или того обстоятельства, что изменением концентрации HCl на поверхности катализатора в этих условиях можно пренебречь. Последнее возможно, например, в случае предельного лэнгмюровского или полислойного заполнения поверхности молекулами хлористого водорода.
Участие молекулы хлористого водорода в лимитирующей стадии реакции должно приводить к появлению кинетического изотопного эффекта (КИЭ) при замене HCl на DCl. Действительно, оценка константы скорости протекания реакции в атмосфере чистого DCl дает значение [27], заметно меньшее величины $k_{{\text{н}}}^{{{\text{HCl}}}}$ – наблюдаемой константы в присутствии HCl (табл. 1), и КИЭ = 1.6.
Таблица 1.
Значения наблюдаемых констант скорости гидрохлорирования ацетилена и величины изотопных эффектов при катализе предварительно механоактивированными солями K2PtCl6 (система 1), K2PtCl4 (система 2) и K2PdCl4 (система 3)
Отметим, что значение изотопного эффекта γ, определенного из соотношения выходов не содержащего атома дейтерия и монодейтерированного изотопомеров винилхлорида, полученных в атмосфере DCl/HCl (табл. 1), превышает величину КИЭ. Различие значений изотопных эффектов указывает на участие молекул хлористого водорода с разрывом связи H–Cl в двух стадиях каталитической реакции – лимитирующей (КИЭ) и более быстрой (γ).
Состояния комплексов платины, генерированных механоактивацией соли. Для установления природы активных центров, вызывающих реакцию в пост-эффекте, важны следующие экспериментальные факты:
1. Механообработка соли K2PtCl6 приводит к генерированию [36] парамагнитных комплексов Pt3+ – [PtCl5]2– – и координационно ненасыщенных комплексов Pt4+ – [${\text{PtCl}}_{5}^{*}$]– – за счет, соответственно, гомолитического и гетеролитического разрыва связи Pt–Cl. Частицы Pt3+, образующиеся при механообработке соли K2PtCl6, локализуются на поверхности межзеренных границ, недоступной для адсорбции из газовой фазы [37], и поэтому не могут быть ответственны за протекание гетерогенной каталитической реакции. Существование в приповерхностных слоях комплексов [${\text{PtCl}}_{5}^{*}$]– показано в [36] методом парамагнитного зонда: в ЭПР-спектре обнаружены два сигнала адсорбированного поверхностью механоактивированной соли стабильного нитроксильного радикала ТЕМПО ((2,2,6,6-тетраметилпиперидин-1-ил)оксил). Один из сигналов отвечает нитроксильным радикалам с заторможенной вращательной подвижностью (адсорбция ТЕМПО на сильных акцепторных центрах [${\text{PtCl}}_{5}^{*}$]–), второй – радикалам с более высокой вращательной подвижностью, т.е. физически адсорбированным радикалам.
2. На предварительно измельченной в атмосфере воздуха соли K2PtCl6 гидрохлорирование ацетилена с заметной скоростью не идет. Для получения активного катализатора необходима предварительная обработка K2PtCl6 в атмосфере активных газов – ацетилена, этилена или пропилена. Такая обработка приводит, помимо отмеченного выше генерирования на поверхности катализатора координационно ненасыщенных комплексов [${\text{PtCl}}_{5}^{*}$]–, к частичному восстановлению Pt4+ до Pt2+, зафиксированному [30] методом РФЭС. Если порошок K2PtCl6 подвергался механической обработке на воздухе, то такого восстановления платины не наблюдалось. Таким образом, для осуществления реакции необходимо одновременное присутствие на поверхности катализатора комплексов PtII и PtIV.
Соль K2PtCl4 (система 2)
Образование комплексов с вакансией в координационной сфере при механоактивации соли. Обработка твердой соли K2PtCl4, в отличие от K2PtCl6, не приводит к гомолитическому разрыву связи Pt–Cl: образования каких-либо парамагнитных частиц в регистрируемых количествах не зафиксировано. В этом случае имеет место гетеролиз связи Pt–Cl с формированием координационно ненасыщенных комплексов [${\text{PtCl}}_{3}^{*}$]–:

Образование частиц с координационными вакансиями [${\text{PtCl}}_{3}^{*}$]– вытекает из следующих экспериментальных фактов:
1. Адсорбция ТЕМРО [37] на поверхности механоактивированной соли PtII указывает на наличие льюисовских кислотных центров. При нанесении 1.7 мкмоль/г ТЕМПО в ЭПР-спектре наблюдается единственный сигнал, отвечающий радикалам с заторможенной вращательной подвижностью. Увеличение количества вводимого радикала приводит к уширению линий ЭПР, вызванного, вероятно, обменным взаимодействием.
2. Свежеобработанная соль поглощает [37] из газовой фазы окись углерода с образованием карбонильного комплекса платины [PtCl3(CO)]–. ИК-спектр последнего в таблетке KBr содержит полосу νСО= 2115 см–1 валентного колебания координированной карбонильной группы22.
3. Предварительно механоактивированная соль поглощает из газовой фазы этилен [37], пропилен [39] или ацетилен [40] с образованием соответствующих π-комплексов платины(II) за счет π-координации непредельных соединений к генерированным механообработкой комплексам с координационной вакансией [${\text{PtCl}}_{3}^{*}$]–. Образование соли Цейзе – π-этиленового комплекса [PtCl3(C2Н4)]– – зафиксировано в [37] методом 1Н-ЯМР, а π-ацетиленового – методом ИК-спектроскопии диффузного рассеяния света (DRIFT) путем наблюдения [40] полосы 2096 см–1, отвечающей33 валентным колебаниям тройной С≡С-связи. Заметим, что в случае симметричных алкинов, в том числе ацетилена, валентные колебания тройной связи не приводят к изменению дипольного момента молекулы и поэтому неактивны в ИК-спектроскопии. Однако π-координация тройной связи к металлокомплексу нарушает симметрию ацетилена, что снимает запрет на проявление этих колебаний в ИК-спектре [38].
4. Деконволюция рентгенофотоэлектронных спектров Pt4f-уровня механоактивированной в ацетилене соли K2PtCl4 показывает, что поверхность образца является многофазной. Основная компонента с энергией связывания Eb(Pt4f7/2) = = 72.8 эВ отвечает Pt2+ в исходном комплексе K2PtCl4, а компоненту c большей на 0.6 эВ энергией связывания, Eb(Pt4f7/2) = 73.4 эВ, можно отнести к частицам с координационной вакансией ${{{\text{K}}}_{2}}{\text{PtCl}}_{3}^{ + }$ [40]. Отметим, что для обработанной в атмосфере ацетилена соли K2PtCl4 вклад комплексов платины с дефицитом хлорид-лигандов в координационной сфере ${{{\text{K}}}_{2}}{\text{PtCl}}_{3}^{ + }$ выше, чем для измельченной на воздухе. Это подтверждается примерно четырехкратным увеличением в спектре РФЭС интегральной интенсивности компоненты, отвечающей комплексам ${{{\text{K}}}_{2}}{\text{PtCl}}_{3}^{ + }{\text{.}}$ Такая ситуация объясняется, очевидно, стабилизацией комплексов ${{{\text{K}}}_{2}}{\text{PtCl}}_{3}^{ + }$ за счет π-координации к ним ацетилена. Рост величины отношения интегральных интенсивностей I(Pt4f)/I(Cl2p) для активированной в ацетилене соли также указывает на повышение поверхностной концентрации комплексов ${{{\text{K}}}_{2}}{\text{PtCl}}_{3}^{ + }{\text{.}}$ К аналогичным эффектам приводит предварительная механообработка соли платины в атмосфере пропилена.
Кинетика гидрохлорирования ацетилена. На поверхности механоактивированной в атмосфере воздуха соли K2PtCl4 реакция протекает очень медленно. Для получения активного катализатора необходима предварительная обработка K2PtCl4 в атмосфере ацетилена, этилена или пропилена. В атмосфере HCl в системе 2 расходование ацетилена из газовой фазы замкнутого реактора отвечает кинетическому уравнению нулевого порядка в отличие от системы 1, где убыль ацетилена следует уравнению первого порядка [31]. Реакция протекает в каталитическом режиме, выход винилхлорида в расчете на прореагировавший ацетилен близок к количественному. В реакции с дейтерохлоридом, также как и в системе 1, получается исключительно продукт транс(анти)-присоединения.
Аналогично системе 1, при проведении реакции в присутствии DCl наблюдаются два различных по величине изотопных эффекта (табл. 1) – определенный из кинетики реакции и из соотношения выходов недейтерованного и монодейтерированного винилхлорида, что подтверждает разрыв связи H–Cl в двух стадиях реакции.
Соль K2PdCl4 (система 3)
Генерирование комплексов с координационной вакансией. Так же как и в системе 2, механическая обработка соли палладия приводит к гетеролизу связи Pd–Cl и формирует комплексы с координационной вакансией [${\text{PdCl}}_{3}^{*}$]–

Об образовании в приповерхностных слоях механоактивированной соли комплексов [${\text{PdCl}}_{3}^{*}$]– и росте их поверхностной концентрации по мере увеличения времени обработки соли свидетельствуют следующие факты [42]:
1. Предварительная механообработка сухой соли K2PdCl4 в атмосфере ацетилена (пропилена) сопровождается выделением винилхлорида (изопропилхлорида). Источником атомов хлора в образующихся хлорорганических соединениях могут быть только комплексы K2PdCl4. Стехиометрическим следствием таких трибохимических реакций, помимо выделения указанных хлороорганических соединений, является также образование комплексов PdII с дефицитом хлорид-лигандов [${\text{PdCl}}_{3}^{*}$]–. Кинетика накопления винилхлорида в трибохимической реакции K2PdCl4 с ацетиленом имеет S-образный вид (рис. 3): на начальном участке кинетической кривой подводимая механическая энергия направляется преимущественно в канал развития поверхности, по окончании начального периода накопление C2H3Cl в процессе механоактивации идет эффективнее, что свидетельствует о трансформации поглощенной механической энергии главным образом в канал образования комплексов [${\text{PdCl}}_{3}^{*}$]–.
Рис. 3.
Зависимость количества ν, выделившегося в ходе механообработки винилхлорида, от поглощенной порошком K2PdCl4 удельной дозы механической энергии.
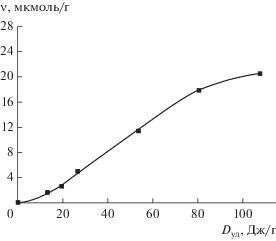
2. По данным микроэлементного анализа (рентгеновская энергодисперсионная спектроскопия EDS SEM) соотношение хлор/палладий монотонно уменьшается с ростом времени обработки соли в ацетилене, следовательно, доля приповерхностных комплексов палладия с дефицитом хлорид-лигандов возрастает по мере увеличения поглощенной порошком K2PdCl4 удельной дозы механической энергии.
3. В ИК-спектрах диффузного рассеяния света соли, экспонированной в атмосфере оксида углерода, зафиксированы две полосы: 1907 и 1972 см–1. ИК-спектры аналогично подготовленных образцов, но выдержанных 8 ч при температуре 120°С или находившихся при комнатной температуре в течение 6 мес, содержат только полосу 1907 см–1. Поэтому более коротковолновую полосу отнесли валентным колебаниям физически сорбированных молекул СО, а полосу 1907 см–1 – валентным колебаниям связи С–О в карбонильных лигандах [PdCl3(СО)]–. Интенсивность последней полосы растет с увеличением времени активации соли.
4. Молекулы пиридина также способны входить в координационную сферу комплексов PdII с вакансией. По данным 1Н-ЯМР-спектроскопии количество хемосорбированного пиридина в пределах экспериментальных ошибок одинаково для свежеприготовленных образцов и выдержанных 1.5 ч при 120°С, что свидетельствует о его прочном связывании. Указанная величина возрастает по мере увеличения времени предварительной механоактивации соли K2PdCl4.
Поверхностная концентрация комплексов с вакансией в координационной сфере выше для соли, активированной в атмосфере ацетилена по сравнению с обработанной на воздухе. Это подтверждают такие эксперименты [43]:
1. По данным энергодисперсионного анализа соотношение хлор/палладий для механоактивированной на воздухе соли выше, чем для образца, обработанного в атмосфере ацетилена.
2. Отношение интегральных интенсивностей фотоэлектронных пиков I(Pd3d)/I(Cl2p) больше для образца, активированного в ацетилене. Из этого следует, что атомное соотношение элементов Cl и Pd в приповерхностных слоях меньше для обработанного в ацетилене образца по сравнению с диспергированным на воздухе.
Кинетика гидрохлорирования ацетилена. Предварительная механоактивация в атмосфере ацетилена или пропилена сухой соли K2PdCl4 приводит к формированию катализатора гидрохлорирования ацетилена газообразным HCl [26]. Каталитическая реакция протекает при комнатной температуре в отсутствие непрерывной механической обработки. Аналогично системе 1, расходование ацетилена из газовой фазы замкнутого реактора в условиях избытка HCl описывается уравнением первого порядка. Выход винилхлорида в расчете на прореагировавший ацетилен близок к количественному. Подобно системам 1 и 2, в атмосфере HCl/DCl наряду с C2H3Cl образуется исключительно продукт транс(анти)-присоединения атомов хлора и дейтерия к тройной связи. Механоактивированная в атмосфере ацетилена (пропилена) соль K2PdCl4, в отличие от обработанной в аналогичных условиях соли K2PtCl4, способна активировать не только тройную С≡С-связь ацетилена, но и двойную С=С-связь в пропилене: последний количественно гидрохлорируется до изопропилхлорида.
Аналогично системам 1 и 2, проведение реакции при участии DCl сопровождается уменьшением скорости реакции. В системе 3 кинетический изотопный эффект HCl/DCl существенно отличается по величине от изотопного эффекта, определенного из соотношения изотопомеров винилхлорида (табл. 1), т.е. в системе 3 связь в молекуле HCl также претерпевает разрыв в двух стадиях каталитической реакции – лимитирующей и быстрой.
Зависимость активности катализатора от времени его предварительной механообработки. При возрастании времени предварительной активации соли происходит монотонное увеличение удельной поверхности и активности катализатора (рис. 4) [44]. Зависимость удельной поверхности катализатора от удельной дозы поглощенной порошком механической энергии имеет вид кривой с насыщением (рис. 4) и отвечает уравнению
(1)
${{S}_{{{\text{уд}}}}} = {{S}_{0}} + S_{{{\text{уд}}}}^{{\text{*}}}(1 - {{e}^{{ - {\eta }{{{\kern 1pt} }_{{{\text{S}}}}}{{D}_{{{\text{уд}}}}}}}}),$Рис. 4.
Зависимость удельной поверхности Sуд и приведенной к единице площади константы k* скорости расходования ацетилена от времени предварительной обработки соли K2PdCl4.
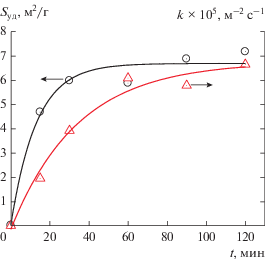
Увеличение активности катализатора по мере возрастания времени его предварительной механообработки может быть следствием двух причин: роста удельной поверхности соли и повышения поверхностной концентрации активных центров. Для исключения влияние первого фактора на величину наблюдаемой константы kн скорости расходования ацетилена ее отнесли к единице площади поверхности катализатора: k* = kн/(Sуд × m). Зависимость k* от удельной дозы поглощенной порошком механической энергии также имеет вид кривой с насыщением (рис. 4) и отвечает уравнению
(2)
$k* = k_{{{\text{макс}}}}^{{\text{*}}}(1 - {{e}^{{ - {\eta }{\kern 1pt} {{D}_{{{\text{уд}}}}}}}}),$Уравнения (1) и (2) удовлетворяют экспериментальным зависимостям при ηs = (8 ± 2) × 10–2 г/Дж и η = (3.2 ± 0.6) × 10–2 г/Дж соответственно. Значение ηs примерно в 3 раза превышает величину η, т.е. развитие поверхности и образование активных центров катализатора происходят в разных процессах. Выполнение уравнения (2) означает, что активность гетерогенного катализатора растет с увеличением времени предварительной механоактивации соли в атмосфере ацетилена вследствие повышения поверхностной концентрации активных центров.
Механизмы гидрохлорирования ацетилена в системах с механоактивированными катализаторами
Кинетическая модель гетерогенно-каталитической реакции. Ацетилен и HCl адсорбируются на поверхности катализатора и в адсорбированном виде принимают участие в бимолекулярной реакции гидрохлорирования ацетилена. Молекулы алкина и хлористого водорода не конкурируют за одни и те же участки на поверхности катализатора – в противном случае значения наблюдаемой константы скорости реакции отличались бы при различных парциальных давлениях HCl (системы 1 и 3) или ацетилена (система 2). Генерированные механообработкой приповерхностные комплексы металла с координационной вакансией несут локальный положительный заряд и электростатически взаимодействуют с полярными молекулами HCl. В отличие от ацетилена молекулы хлористого водорода не координируются при этом к металлическому центру и не занимают координационную вакансию. С другой стороны, образование π-ацетиленового комплекса не изменяет зарядового состояния активного центра катализатора и, следовательно, не уменьшает его электростатического взаимодействия с молекулами HCl. Таким образом, молекулы хлористого водорода и ацетилена адсорбируются на одних и тех же участках поверхности, не конкурируя однако из-за разных механизмов их сорбции: неполярные молекулы ацетилена связаны с металлическим центром посредством донорно–акцепторного взаимодействия, а полярные молекулы HCl – электростатическими силами.
Расходование ацетилена из газовой фазы замкнутого реактора в атмосфере HCl в системах 1 и 3 отвечает кинетическому уравнению первого порядка. В отличие от этого в системе 2 ацетилен убывает по нулевому порядку. Нулевой порядок по ацетилену в системе 2 объясняется быстрой его хемосорбцией на поверхности механоактивированной соли в результате π-координации ацетилена с приповерхностными комплексами платины(II), обладающими координационными вакансиями. Вследствие этого количество хемосорбированного субстрата практически не зависит от его парциального давления в газовой фазе, а определяется поверхностной концентрацией комплексов с вакансией [45]. Палладий(II), в отличие от PtII, не способен образовывать устойчивые π-комплексы [46] и в этом отношении подобен платине(IV). Видимо, это обстоятельство и является причиной одинаковых порядков реакции по ацетилену в системах 1 и 3.
Механоактивированные хлоридные комплексы PdII способны также активировать двойные связи С–С. В активации кратных С–С-связей комплексами металлов ключевую роль, по всей видимости, играют электрофильные свойства металлокомплексов. Способность соли K2PdCl4, в отличие от K2PtCl4, активировать также и двойную связь, очевидно, объясняется более высокой электрофильностью ацидокомплексов PdII по сравнению с PtII.
Оценка удельной каталитической активности (TOF) гидрохлорирования ацетилена на поверхности механоактивированных солей. В рассматриваемых системах при большом избытке одного из субстратов S1 по отношению к другому S2 (атмосфера S1) кинетика накопления винилхлорида отвечает уравнению первого порядка. Выполнение кинетического уравнения первого порядка означает, что в указанных условиях поверхностная концентрация [S2]адс адсорбированного S2 пропорциональна его парциальному давлению ${{P}_{{{{{\text{S}}}_{2}}}}}$, т.е. S2 адсорбируется по закону Генри:
где K – константа адсорбционного равновесия S2.В этом случае истинная (т.е. в отсутствие газовой фазы) константа k скорости псевдопервого порядка расходования S2 связана [31] с экспериментально определяемой величиной kн наблюдаемой константы скорости реакции выражением
где α = (RTK)–1, λ = ${{V}_{{\text{г}}}}S_{{{\text{уд}}}}^{{ - 1}}{{m}^{{ - 1}}},$ Vг – объем газовой фазы реактора, R – универсальная газовая постоянная, T – температура, Sуд – удельная поверхность катализатора, m – его масса.Уравнение (4) удовлетворяет экспериментальным зависимостям kн от массы катализатора (рис. 5) при значениях k и K, приведенных в табл. 2.
Рис. 5.
Зависимость наблюдаемой константы скорости реакции kн от массы катализатора для систем с механоактивированными солями: а – K2PtCl6, б – K2PtCl4, в – K2PdCl4.
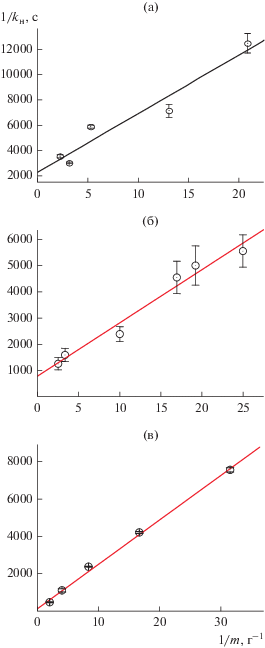
Таблица 2.
Удельная каталитическая активность механоактивированных катализаторов
| Система | k × 104, с–1 | K × 109, $\frac{{{\text{моль}}}}{{{{{\text{м}}}^{{\text{2}}}}\,{\text{Па}}}}$ | w × 107, $\frac{{{\text{моль}}}}{{{{{\text{м}}}^{{\text{2}}}}\,\,{\text{с}}}}$ | a × 1010, м | с × 1010, м | NМ × 106, $\frac{{{\text{моль}}}}{{{{{\text{м}}}^{{\text{2}}}}}}$ | TOF,$\frac{{{\text{мол}}{{{\text{ь}}}_{{{\text{ВХМ}}}}}}}{{{\text{мол}}{{{\text{ь}}}_{{{\text{кат}}}}}\,\,{\text{c}}}}$ |
|---|---|---|---|---|---|---|---|
| 1 (K2PtCl6) [47] | 4.4 ± 1.5 | 12 ± 6 | 5.3 | 9.78 [48] | 9.78 | 3.5* | ≥0.15 |
| 2 (K2PtCl4) [45] | 13 ± 4 | 10 ± 4 | 13 | 7.017 [49] | 4.131 | 5.7 | ≥0.23 |
| 3 (K2PdCl4) [44] | 5 ± 3 | 0.8 ± 0.4 | 0.4 | 7.054 [50] | 4.108 | 5.7 | ≥0.007 |
* Рассчитанное в соответствии с уравнением (5) значение удвоено с учетом нахождения в одной элементарной ячейке 2 атомов платины.
Полученные значения истинной константы скорости реакции k и константы K адсорбционного равновесия можно использовать для оценки нижнего предела удельной каталитической активности катализатора в рассматриваемых системах. Скорости w образования винилхлорида на 1 м2 катализатора при условии предельного заполнения поверхности молекулами S2 (см. выше) и парциальном давлении субстрата S1 1 атм приведены в табл. 2. Максимально возможное количество металла, расположенного на 1 м2 поверхности катализатора, NМ, можно оценить из параметров решетки a и c:
где Na – число Авогадро.Отсюда величина удельной каталитической активности катализатора при предельном заполнении поверхности молекулами S2 и парциальном давлении S1 1 атм
где w – скорость образования винилхлорида на 1 м2 катализатора.Одинаковый порядок реакции по ацетилену в системах 1 и 3 дает возможность сравнить удельную каталитическую активность соответствующих металлокомплексов в одинаковых условиях (табл. 2). Как видно, истинные константы скорости расходования ацетилена в системах 1 и 3 близки. Если считать, что в активации кратных С−С-связей комплексами металлов ключевую роль играют электрофильные свойства металлокомплексов, то примерное совпадение констант k в системах 1 и 3, очевидно, означает, что хлоридные комплексы PtIV и PdII близки по электрофильности. Нижний предел удельной каталитической активности в системе 3 примерно в 20 раз меньше, что обусловлено, главным образом, меньшей величиной константы адсорбционного равновесия ацетилена в этой системе. При равных парциальных давлениях ацетилена поверхностная концентрация субстрата в системе 1 на порядок больше, благодаря чему скорость его расходования выше.
Стадийный механизм каталитического гидрохлорирования ацетилена на механоактивированных комплексах платины и палладия. Во всех трех рассмотренных системах изотопные эффекты γ, измеренные в конкурентных условиях по соотношению выходов не содержащего дейтерий и дейтерированного винилхлорида, существенно отличаются от КИЭ (табл. 1). Наличие изотопного эффекта HCl/DCl указывает на разрыв связи в молекуле хлористого водорода. Поскольку атом водорода (дейтерия) входит в состав продукта в быстрой стадии (не лимитирующей), то лимитирующая стадия должна приводить к включению в состав продукта атома хлора из молекулы хлористого водорода.
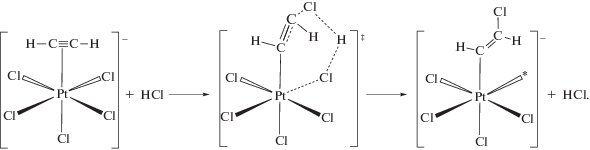
Реакции на механоактивированной соли K2PtCl6 (cистема 1). По аналогии с каталитическим гидроиодированием [51] ацетилена иодидными комплексами платины(IV) в водных растворах можно было полагать, что газофазное гетерогенное гидрохлорирование также протекает через промежуточное образование β-галогенвинильного производного платины(IV). Действительно, образование β-хлорвинильного комплекса платины(IV), PtIV–CH=CHCl – платинового аналога комплекса Бигинелли, постулированного в гидрохлорировании ацетилена на ртутном катализаторе HgCl2/C [12, 52], при механообработке соли K2PtCl6 в присутствии ацетилена и газообразного HCl в замкнутом реакторе удалось продемонстрировать [53] методом ЯМР. Для этого реакцию останавливали вакуумированием реактора и/или продуванием его сухим воздухом. Металлоорганическое производное платины экстрагировали метанолом, экстракт упаривали досуха без нагревания в токе сухого воздуха.
В 1Н-ЯМР-спектре экстракта в СD3OD наблюдаются два дублета с δ = 6.51 м. д. и δ = 5.69 м. д. и соотношением интенсивностей 1 : 1, 3JH–H = = 12.3 Гц, сопровождаемые платиновыми сателлитами 2JPt–H = 72.5 Гц и 3JPt–H = 28.2 Гц соответственно. 13C{1H}-ЯМР-спектр β-хлорвинильного комплекса платины(IV) содержит два синглета 114.5 и 96.6 м. д. с платиновыми сателлитами 2JPt–С = = 45.7 Гц и 1JPt–С = 749.6 Гц соответственно. Существенная разница в величинах констант спин–спинового взаимодействия 1J и 2J подтверждает σ-характер связи Pt–C, а достаточно высокое значение 3JH–H свидетельствует о транс(анти)-присоединении к тройной связи ацетилена атомов платины и хлора.
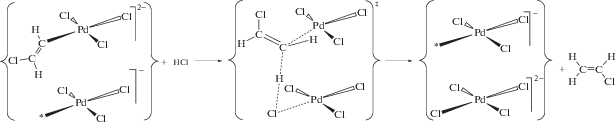
В системе твердая фаза K2PtCl6–газообразный HСl “внешние” нуклеофилы в количестве, сопоставимом с количеством образующегося винилхлорида, отсутствуют. Однако в реакции гидрохлорирования ацетилена получается продукт исключительно транс(анти)-присоединения. Кроме того, в реакции на бромидном катализаторе K2PtBr6–K2PtBr4 в присутствии небольшого количества газообразного HCl основным продуктом является винилхлорид. Следовательно, источником атомов хлора, входящих в состав продукта, служат молекулы HCl, принимающие участие в стадии хлорплатинирования ацетилена с промежуточным образованием PtIV–CH=CHCl. Это обеспечивает высокую стереоселективность реакции и объясняет наличие КИЭ.
Известно, что σ-органильные комплексы платины(IV) протолизу не подвергаются; для осуществления протодеплатинирования PtIV–CH=CHCl с выделением винилхлорида необходимо предварительное восстановление указанного интермедиата до соответствующего производного платины(II) (ср. с [51]). Как было отмечено выше, для получения активной формы катализатора необходима предварительная механообработка солей платины(IV) в атмосфере активных газов – ацетилена, этилена или пропилена. Такая обработка приводит к образованию на поверхности катализатора координационно ненасыщенных комплексов платины и, в отличие от обработки в инертной атмосфере, частичному восстановлению Pt4+ до Pt2+. Каталитическая реакция, очевидно, протекает на контактной паре PtII–комплекс PtIV с координационной вакансией (на схеме 2 контактная пара обведена фигурными скобками).
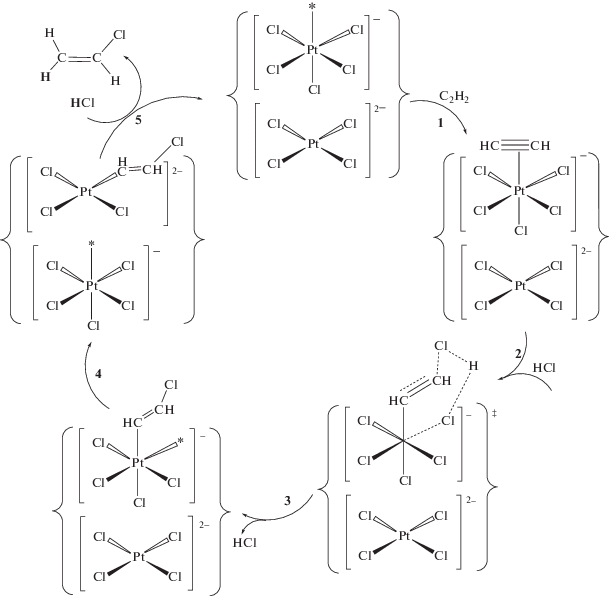
Схема 2 . Стадийный механизм каталитического гидрохлорирования ацетилена на механоактивированной соли K2PtCl6.
Реакция осуществляется через стадию хлорплатинирования ацетилена (схема 2 , стадии 1–3) координационно ненасыщенными комплексами [${\text{PtCl}}_{5}^{*}$]– с промежуточным образованием β-хлорвинильного производного PtIV. Роль PtII заключается в комплементарном восстановлении [54] указанного интермедиата до соответствующего производного PtII (стадия 4). Учитывая отсутствие в твердой фазе свободных хлорид-ионов, стехиометрическим следствием такой реакции должно быть образование, помимо органического производного PtII, комплекса PtIV с координационной вакансией. Протодеметаллирование β‑хлорвинильного производного PtII под действием HСl (стадия 5) приводит к выделению винилхлорида и регенерации активной формы катализатора – топологически связанной пары [PtCl4]2––[${\text{PtCl}}_{5}^{*}$]–.
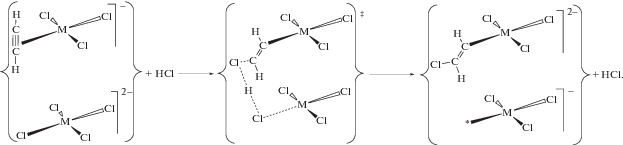
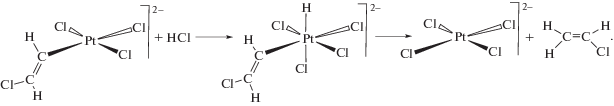
Механоактивированные соли K2PtCl4 (система 2) и K2PdCl4 (система 3). Стадийный механизм гидрохлорирования ацетилена газообразным HCl на поверхности механообработанных солей K2MCl4 (M = Pt, Pd) (схема 3 ) подобен стадийному механизму в системе 1 (схема 2 ) и включает π-координацию ацетилена к генерированным предварительной механообработкой комплексам с координационной вакансией [${\text{MCl}}_{3}^{*}$]–. Для случая M = Pt образование π-ацетиленовых комплексов прямо продемонстрировано методом DRIFT [40]. Последующее хлорметаллирование π-координированного ацетилена под действием молекулы HCl (лимитирующая стадия) дает ключевой интермедиат – хлорвинильное производное соответствующего металла. В реакции принимает участие соседний поверхностный комплекс металла, который служит донором хлорид-иона, стабилизирующим выделяющийся протон путем образования новой связи H–Cl. Такой способ осуществления реакции хлорметаллирования приводит к регенерации активных центров на поверхности катализатора – комплексов с координационной вакансией (здесь и далее в фигурные скобки заключены два соседних на кристаллографической плоскости (100) комплекса металла, которые участвуют в реакции). Быстрое протодеметаллирование хлорвинильного производного MII–CH=СHCl дает конечный продукт и ответственно за возникновение изотопного эффекта γ. Отметим, что для систем 1 и 2 получены одинаковые значения изотопного эффекта γ. Такое совпадение является следствием того обстоятельства, что в обеих системах в стадии протодеплатинирования принимают участие одинаковые частицы – хлорвинильные производные PtII. Вместе с тем значения КИЭ в платиновых системах 1 и 2 существенно различаются (табл. 1), что указывает на разные механизмы лимитирующей стадии хлорплатинирования ацетилена.
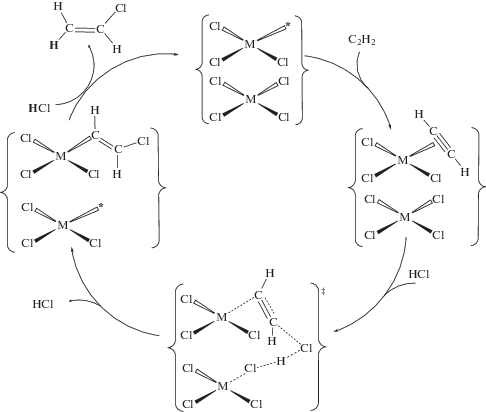
Схема 3 . Стадийный механизм каталитического гидрохлорирования ацетилена на предварительно механоактивированных солях K2MCl4 (M = Pt, Pd).
Таким образом, стадийный механизм реакций в системах 2 и 3 отличается от механизма реакции в системе 1 отсутствием дополнительной стадии комплементарного восстановления. Во всех рассмотренных системах лимитирующей стадией реакции является хлорметаллирование ацетилена c участием молекулы HCl. Поскольку гетеролиз связи H–Cl в отсутствие сольватации термодинамически крайне невыгоден, предположено, что разрыв ее компенсируется образованием новой связи H–Cl в результате гетеролиза менее прочных связей M–Cl (M = Pd, Pt), что приводит к регенерации на поверхности комплексов с координационной вакансией, служащих каталитически активными центрами.
В рамках стадийных механизмов (схемы 2 и 3 ) находит естественное объяснение наличие разных по величине изотопных эффектов, связанных с разрывом связи H–Сl в двух стадиях реакции: в скоростьопределяющем хлорметаллировании π-координированного ацетилена (КИЭ) и в быстрой стадии протодеметаллирования β-хлорвинильных производных PtII или PdII (γ). Приведенные механизмы позволяют также объяснить высокую стереоселективность реакции – образование продукта исключительно транс(анти)-присоединения молекулы хлористого водорода к тройной связи.
КИЭ и структура переходного состояния лимитирующей стадии. Согласно полуклассической теории [27] максимальная величина изотопного эффекта H/D при отсутствии вклада туннелирования достигается в случае линейного и симметричного переходного состояния и составляет 6.9 при 25°С. Симметричность переходного состояния предполагает нулевое изменение свободной энергии в ходе реакции, любое отклонение от нулевого изменения сопровождается уменьшением изотопного эффекта. К такому же результату приводит отклонение Н-атома от прямой, соединяющей Н-донор и Н-акцептор в переходном состоянии реакции.
В системах 2 и 3 переходное состояние осуществляется при участии двух соседних комплексов металла. Минимальное расстояние между комплексами на кристаллографической плоскости (100), составляющее ~4.1 Å [49, 50] и длины связей Н–Сl (1.274 Å) и С–Сl (1.719 Å) [55] геометрически позволяют реализоваться переходному состоянию, близкому к линейному (схема 4 ).
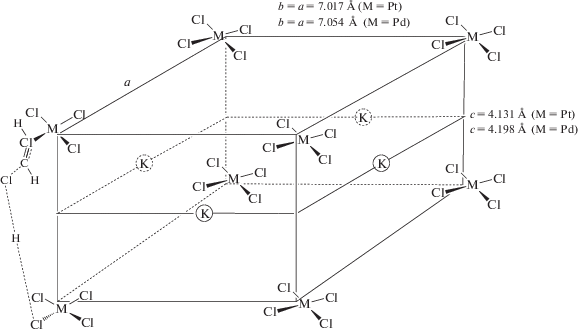
Схема 4 . Модель переходного состояния лимитирующей стадии в системах с предварительно механоактивированными солями K2MCl4 (M = Pt, Pd).
Поскольку кристаллические решетки K2PtCl4 и K2PdCl4 изоморфны и имеют близкие параметры [49, 50], различие КИЭ в системах 2 и 3 обусловлено скорее термодинамическим, а не геометрическим фактором. Число частиц, вступающих в реакцию (I) и образующихся в ней, одинаково, поэтому в первом приближении изменением энтропии можно пренебречь, тогда изменение свободной энергии определяется в основном энтальпийным вкладом.
Для обеспечения термодинамической движущей силы реакция (I) должна быть экзотермичной, т.е. теплота реакции отрицательна. Здесь связи H–Cl, M–Cl и M– (η2‑C2H2) разрываются, образуются новые связи H–Cl, C(sp2)–M и C(sp2)–Cl, а тройная связь C≡C трансформируется в двойную. Для оценки энтальпии реакции можно считать, что ΔH определяется главным образом энергиями диссоциации связей:
Разность теплот реакции (I) для M = Pt (ΔH2) и M = Pd (ΔH3) составляет
Учитывая, что ΔD(M–Cl) ≈ 29 кДж/моль [56], ΔD(C(sp2)–M) ≈ 34 кДж/моль [57] и хлоридные комплексы PtII образуют более стабильные π-комплексы, чем хлориды PdII [46], ΔH2 – ΔH3 ≈ ≈ ΔD(M–(η2-C2H2)) > 0, т.е. ΔH2 > ΔH3. Поскольку энтальпии реакции отрицательны, это означает, что ΔH2 ближе к нулевому значению, чем ΔH3. Соответственно степень симметрии переходного состояния в реакции (I) в системе 2 выше, чем в системе 3, что проявляется в более низком значении КИЭ для последней.
Существенно меньшая величина КИЭ в системе 1 по сравнению с системами 2 и 3 является, очевидно, следствием нелинейности ее переходного состояния. Действительно, в решетке K2PtCl6 минимальное расстояние между комплексами платины существенно больше (~9.7 Å [48]), и поэтому переходное состояние с участием двух соседних комплексов металла, подобное предложенному для систем 2 и 3, не может быть реализовано. Реакция (II) протекает через циклическое переходное состояние, нелинейностью которого определяется низкое значение КИЭ.
Стадия протодеметаллирования. Рассмотрим возможные механизмы стадии протодеметаллирования. Совпадение в пределах экспериментальных ошибок величины γ в системах 1 и 2 (табл. 1) является следствием участия в стадии протодеплатинирования одного и того же интермедиата – β‑хлорвинильного производного PtII. Существенное отличие величины γ в системе 3 от системы 2 свидетельствует о разных механизмах стадии протодеметаллирования в палладиевой и платиновых системах.
В системе 3 реакция протодепалладирования (III) может осуществляться как электрофильное замещение путем атаки α-атома углерода σ-винильного производного PdII атомом водорода молекулы HCl при электрофильном содействии соседнего44 комплекса с координационной вакансией. Локальный положительный заряд последнего оказывает ориентирующее действие на полярную молекулу HCl и стабилизирует образующийся хлорид-ион.
Число реагирующих и образующихся в реакции (III) частиц одинаково, поэтому можно пренебречь изменением энтропии и считать, что изменение свободной энергии определяется преимущественно энтальпийным членом. Будем также полагать, что энтальпия реакции (III) определяется, главным образом, энергиями диссоциации связей. Здесь связи Pd–C(sp2) и H–Cl разрываются, а H–C(sp2) и Pd–Cl образуются. Энергия диссоциации связи H–Cl (427 кДж/моль [58]) практически совпадает с энергией связи H–C(sp2) (431 кДж/моль [12]), т.е. разрыв первой связи компенсируется образованием второй. Энергии диссоциации связей Pd–C(sp2) и Pd–Cl также сопоставимы: расчеты методом DFT дают величины энергии диссоциации связей 164 кДж/моль для Pd–C(sp2) [57] и 141 кДж/моль для Pd–Cl [56]. Таким образом, реакция (III) практически термонейтральна, изменение свободной энергии в ней близко к нулю и, соответственно, переходное состояние почти симметрично, что обеспечивает высокое значение изотопного эффекта γ в стадии протодепалладирования.
Замена Pd на Pt кардинально не меняет ситуацию: в системах 1 и 2 энергии диссоциации связей Pt–Cl (169 кДж/моль [56]) и Pt–C(sp2) (195 кДж/моль [57]) тоже близки. Следовательно, переходное состояние стадии протодеплатинирования также должно быть почти симметричным. Существенно меньшая величина γ в этих системах является, очевидно, следствием нелинейности переходного состояния. Протодеплатинирование здесь может протекать через последовательность стадий окислительного присоединения молекулы HCl к σ-винильному комплексу PtII с промежуточным образованием винилгидридного производного PtIV [59] и последующего восстановительного элиминирования из указанного интермедиата винилхлорида:
Трудно сказать, какая из стадий реакции протодеплатинирования по механизму (IV) – окислительное присоединение HCl или восстановительное элиминирование винилхлорида – является лимитирующей. Однако каждая из них протекает через нелинейное переходное состояние, что может быть причиной существенно меньшей величины изотопного эффекта.
НАНЕСЕННЫЕ КАТАЛИЗАТОРЫ
В промышленности экономически оправданным является использование нанесенных катализаторов, применение которых позволяет существенно снизить расход дорогостоящих металлов и повысить скорость реакции за счет большей дисперсности активного компонента. Среди представленных в литературе каталитических систем газофазного гидрохлорирования ацетилена механизмы реакции наиболее детально изучены для нанесенных на активированный уголь хлоридных комплексов золота и палладия. Поэтому мы ограничились рассмотрением только этих систем.
Хлоридные комплексы палладия(II) на активированном угле
Состояние комплексов на поверхности угля. Состояние нанесенных на активированный уголь хлоридных комплексов PdII было детально изучено в работах [60–70]. Установлено, что адсорбция поверхностью активированного угля палладийхлористоводородной кислоты Н2PdCl4 приводит к образованию комплексов дихлорида палладия с активными центрами А матрицы угля и выделению хлороводорода [65]:
Кинетика, изотопные эффекты HCl/DCl и стереоселективность реакции гидрохлорирования ацетилена. Нанесенные палладиевые катализаторы активны в гидрохлорировании ацетилена газообразным НCl [например, 71–76]. Для нанесенных на активированный уголь K2PdCl4 или Н2PdCl4 установлено [76], что подобно системе с механоактивированной солью K2PdCl4 кинетика расходования ацетилена из газовой фазы замкнутого реактора в атмосфере НCl отвечает уравнению первого порядка. Аналогично рассмотренным выше системам с механоактивированными солями платины и палладия, в системе с нанесенным палладием проведение реакции в присутствии DCl сопровождается заметным снижением наблюдаемых констант скорости расходования ацетилена по сравнению с гидрохлорированием в атмосфере HCl [76]. Подобно механоактивированным катализаторам в системах с нанесенными палладиевыми катализаторами наблюдаются два разных по величине изотопных эффекта – кинетический и полученный из соотношения H2C=CHCl и DHС=CHCl, что указывает на разрыв связи H–Cl в двух стадиях реакции.
В отличие от рассмотренных систем с механоактивированными солями платины и палладия при протекании реакции в изотопной смеси HCl/DCl в присутствии K2PdCl4/C или Н2PdCl4/C наряду с недейтерированным винилхлоридом образуются продукты как транс(анти)-, так и цис(син)-присоединения атомов хлора и дейтерия к тройной С≡С-связи ацетилена [76]. При образовании продукта транс(анти)-присоединения молекула хлористого водорода участвует в двух стадиях – лимитирующем хлорметаллировании и протодепалладировании. В случае продукта цис(син)-присоединения HCl принимает участие только в быстрой стадии протолиза: атом хлора входит в состав винилхлорида из координационной сферы металлокомплекса. Наличие двух маршрутов реакции гидрохлорирования ацетилена – с выделением продуктов цис(син)- и транс(анти)-присоединения – обусловливает меньшее наблюдаемое значение КИЭ в системах с нанесенным палладием по сравнению с системой 3.
Механизм реакции. Совпадающие в пределах экспериментальных ошибок величины эффективных энергий активации и кинетических изотопных эффектов (табл. 3) позволяют предполагать сходные механизмы реакции в системах с K2PdCl4/C и Н2PdCl4/C.
Таблица 3.
Значения эффективных энергий активации и кинетических изотопных эффектов КИЭ для систем с нанесенными палладиевыми катализаторами
| Система | Е, кДж/моль | КИЭ |
|---|---|---|
| K2PdCl4/C | 30 ± 1 | 1.25 ± 0.06 |
| Н2PdCl4/C | 32 ± 3 | 1.29 ± 0.20 |
Наличие двух маршрутов реакции с образованием продуктов цис(син)- и транс(анти)-присоединения можно связать с существованием на поверхности активированного угля двух форм PdCl2. Подобно системе с механоактивированной солью палладия, продукт транс(анти)-присоединения получается при участии протяженных кластеров α-PdCl2. На краях кластеров олигомерные цепочки, содержащие четырехкоординированные атомы палладия, терпят разрыв, формируя комплексы с координационными вакансиями. Расстояние между атомами хлора в соседних олигомерных цепочках в структуре α-PdCl2 [76, 77] (схема 5 ) близко к таковому между атомами хлора в соседних комплексах палладия в системе 3 (схема 4 ). Такое совпадение межатомных расстояний позволяет реализовать в системах с нанесенным палладием подобный системе 3 механизм реакции (схема 3 ) с участием двух соседних комплексов, один из которых – комплекс с координационной вакансией.
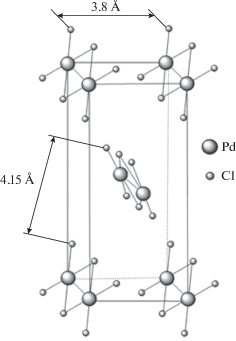
Схема 5 . Структура элементарной ячейки α-PdCl2.
Продукт цис(син)-присоединения образуется на малых кластерах дихлорида палладия. Реакция протекает (схема 6 ) путем внутрисферной атаки хлорид-лиганда на π-ацетиленовый комплекс PdII – в состав винилхлорида входит Cl-атом из координационной сферы металлокомплекса. На этой стадии получаются хлорвинильное производное палладия и вакансия в координационной сфере металла. Последующее быстрое протодеметаллирование при атаке молекулой HCl атома углерода винильного производного PdII приводит к выделению конечного продукта и регенерации исходного металлокомплекса.
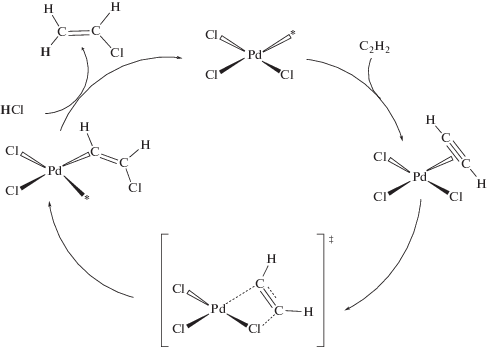
Схема 6 . Механизм образования продукта цис(син)-присоединения в системах с нанесенными катализаторами K2PdCl4/C и Н2PdCl4/C.
Комплексы золота(III) на активированном угле
В настоящее время наиболее изученной и перспективной в плане промышленного использования является система с нанесенными на активированный уголь комплексами золота(III), AuIII/C [2, 13, 15, 78–91]. В 2010–2011 гг. в Китае были проведены успешные ее испытания на пилотной установке, точно воспроизводящей полномасштабный промышленный трубчатый реактор гидрохлорирования ацетилена (длина слоя катализатора 3 м, внутренний диаметр 50 мм, загрузка катализатора Au ((0.1 мас. %)/C ~2 кг). На протяжении продолжительной (более 12000 ч) непрерывной работы пилотной установки селективность по винилхлориду была выше 99%, а конверсия ацетилена при скорости его подачи 0.6 кг/ч превышала 85% (рис. 6).
Рис. 6.
Сравнение производительности газофазного гидрохлорирования ацетилена на ртутном катализаторе HgCl2 (10 мас. %)/C (1) и золотом катализаторе Au (0.1 мас. %)/C в пилотном реакторе (2) и в полномасштабном коммерческом кожухотрубчатом реакторе (3) по данным [2].
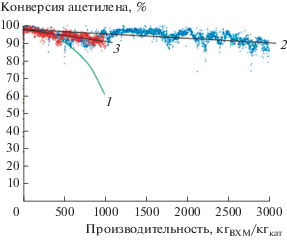
В 2012 г. вслед за успешными пилотными испытаниями этого катализатора было проведено его полномасштабное тестирование в кожухотрубчатом реакторе, содержащем 790 труб [2]. В реактор было загружено около 1.6 т катализатора, он был введен в эксплуатацию и проработал в таких же условиях более 4500 ч в непрерывном режиме. Конверсия ацетилена составляла >90%, а селективность превышала 99%. При достижении производительности (1 т ВХМ)/(кг катализатора) конверсия ацетилена на золотом катализаторе в кожухотрубчатом реакторе практически совпадает с величиной, полученной в пилотном реакторе, и существенно превышает степень конверсии ацетилена на ртутном катализаторе (рис. 6). Результаты этих демонстрационных испытаний послужили источником эксплуатационных справочных данных для полномасштабного промышленного применения катализаторов Au (0.1 мас. %)/C в трубчатых реакторах с неподвижным слоем.
Каталитически активные состояния комплексов золота на поверхности угля. Методами просвечивающей электронной микроскопии HAADF-STEM высокого разрешения и in situ спектроскопии тонкой структуры рентгеновского поглощения XAFS показано [2, 13, 89–91], что при нанесении на активированный уголь относительно небольшого (до 2 мас. %) количества хлорида золота(III) из раствора в царской водке получается катализатор с нанесенными моноядерными хлоридными комплексами AuI и AuIII. Дискретные наночастицы металлического золота, а также димерные комплексы золота обнаружены не были [13], наблюдалось лишь атомарно-диспергированное золото, распределенное по всему углеродному носителю.
При повышении температуры до 180–200°C (температура, при которой обычно ведут каталитическую реакцию) соотношение AuIII/AuI уменьшается, т.е. нанесенные комплексы золота(III) в атмосфере инертного газа почти полностью восстанавливаются до хлорида золота(I) с сохранением высокой дисперсности металла на поверхности носителя [90, 91]. В атмосфере HCl окислительное состояние комплексов Au практически не изменяется, а при подаче реакционной смеси газов (HCl/C2H2 = 1.1) гидрохлорирование идет с индукционным периодом, в течение которого одновременно с ростом скорости реакции увеличивается содержание хлоридов золота(III), достигая в стационарном режиме соотношения AuI/AuIII ~ ~ 1.5 [90]. Наночастицы металлического золота заметной каталитической активности не проявляют. Эти наблюдения позволили авторам [13, 90, 91] сделать вывод, что активными центрами нанесенных на активированный уголь хлоридов золота являются его окисленные состояния, причем для протекания реакции важно одновременное присутствие как AuI, так и AuIII. Отметим, что активные состояния нанесенных золотосодержащих катализаторов в виде окислительно-восстановительных пар AuI–AuIII подобны рассмотренным выше для механоактивированной соли PtIV, где активными центрами катализатора служат топологически связанные пары PtII–PtIV.
Активность AuIII, нанесенного на активированный уголь, уменьшается в процессе гидрохлорирования. Основной причиной дезактивации катализатора в ходе реакции является восстановление AuIII и AuI до металлического золота [78], которое обнаружено методами 197Au Мессбауэроской спектроскопии и рентгенофотоэлектронной спектроскопии [83]. Еще одной причиной снижения активности считают [13, 80, 83, 84, 86, 91] зауглероживание поверхности катализатора продуктами олигомеризации ацетилена и/или винилхлорида. Последний процесс вносит заметный вклад в дезактивацию катализатора, экспонированного в атмосфере чистого ацетилена, без HCl. В этом случае методом неупругого рассеяния нейтронов показано [91] образование на поверхности катализатора олигомеров ацетилена, которые могут служить предшественниками углеродных нановолокон, наблюдавшихся в качестве побочного продукта гидрохлорирования ацетилена в промышленных условиях [89]. Хлористый водород, наоборот, играет стабилизирующую роль: скорость дезактивации катализатора в условиях реакции определяется молярным соотношением HCl/C2H2 – чем оно выше, тем медленнее теряется активность. Поскольку хлориды золота не сублимируют и разлагаются при довольно высоких температурах (269°С для AuI, ~160°С для AuIII), никаких потерь металла не наблюдалось [78–81, 83].
Кинетика и стереоселективность реакции гидрохлорирования ацетилена. Кинетика реакции гидрохлорирования отвечает общему кинетическому уравнению второго порядка, первого по каждому из реагентов – по ацетилену и хлористому водороду [80, 91].
Результатом гидрохлорирования алкинов могут быть четыре продукта: образованные путем цис(син)- и транс(анти)-присоединения молекулы НCl к тройной С–С-связи алкина, а также по правилу Марковникова и против него. Региоселективность определяли в реакции гидрохлорирования замещеных алкинов [83, 84, 89]. На скорость реакции существенное влияние оказывают стерические факторы: конверсия алкина уменьшается в ряду ацетилен (около 40%) > гексин-1 (10%) > фенилацетилен (7%) > гексин-2 (2%) [83]. В случае реакции гексина-1 с хлороводородом из четырех указанных выше возможностей реализуется только две: основным является продукт транс(анти)-присоединения HCl по правилу Марковникова, в следовых количествах образуется продукт транс(анти)-присоединения HCl против правила Марковникова. Такая же ситуация имеет место в гидрохлорировании фенилацетилена.
Таким образом, подобно каталитическому гидрохлорированию ацетилена на рассмотренных выше механоактивированных катализаторах, винилхлорид при катализе нанесенными на активированный уголь хлоридами золота образуется исключительно путем транс(анти)-присоединения молекулы HCl к тройной С–С-связи.
Механизм реакции гидрохлорирования ацетилена на нанесенных комплексах золота. Основным и единственным инструментом, который использовался для установления механизма реакции гидрохлорирования ацетилена при катализе нанесенными комплексами золота, было квантовохимическое моделирование методами DFT [83, 87, 92–100]. Эти расчеты должны отвечать следующим экспериментально установленным фактам: во-первых, активными состояниями катализатора являются изолированные моноядерные катионные комплексы золота (single atom sites) [13, 90, 91]; во-вторых, реакция приводит к продукту транс(анти)-присоединения молекулы HCl к тройной связи ацетилена [83, 84, 89]. Указанным условиям удовлетворяют только работы проф. Хатчингса (Hutchings) c соавт. [83, 87]. Согласно расчетам других авторов [92–100] стереоселективность реакции отвечает цис(син)-присоединению и, кроме того, в качестве активных состояний катализатора рассматриваются частицы инертного металлического золота [96–98] или биядерные комплексы AuI [93] и AuIII [93, 94, 99]. Поэтому эти исследования здесь не обсуждаются.
DFT-расчеты [83, 87] показали, что оба реагента – хлористый водород и ацетилен – способны координироваться к Т-образным (т.е. имеющим координационную вакансию) комплексам AuCl3 (схема 7 ). Первоначальная координация HCl (комплекс 2) согласно расчетам понижает энергию системы на 105 кДж/моль, тогда как координация C2H2 (комплекс 3) – на 168 кДж/моль, т.е., как и следовало ожидать, алкин является лучшим лигандом, чем хлористый водород. Это, по мнению авторов, объясняет необходимость небольшого избытка HCl в реакционной смеси (обычно используется смесь с молярным соотношением HCl/C2H2 = 1.1). С другой стороны, расчеты показали, что координация ацетилена в отсутствие HCl приводит к блокированию активных центров катализатора в результате образования стабильного металлоцикла 5 с энергией –213 кДж/моль. Указанное блокирование координационных вакансий в AuCl3 может служить причиной наблюдаемой в отсутствие гидрохлорида дезактивации катализатора ацетиленом [83, 89, 90].
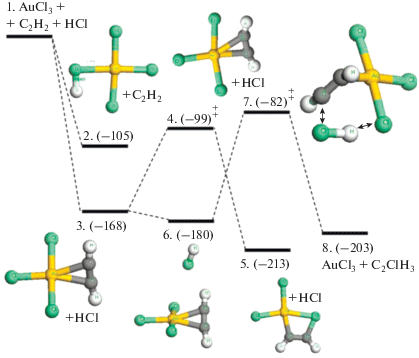
Схема 7 . Энергетический профиль реакции гидрохлорирования ацетилена газообразным HCl на AuCl3/C катализаторе согласно данным [2, 84, 85].
Согласно расчетам реакция начинается с π-координации ацетилена к металлокомплексу AuIII. Скоростьопределяющей стадией является хлораурирование ацетилена: в переходном состоянии происходит разрыв связи в молекуле HCl с присоединением уходящего атома хлора к атому углерода π-координированного ацетилена, а атома водорода – к одному из хлорид-лигандов. Другой атом углерода образует σ-связь с металлоцентром, которая расположена в транс(анти)-положении к образующейся связи C(sp2)–Cl. Стереохимия получающегося таким путем интермедиата задается этим переходным состоянием, поскольку последующее протодеаурирование протекает с сохранением стереохимической конфигурации металлоорганического субстрата.
По мнению авторов [83] рассмотренный механизм каталитического гидрохлорирования ацетилена на AuIII подобен механизму реакции при катализе механоактивированными комплексами K2PtCl6 [31]: в обеих системах каталитически активными состояниями служат электрофильные комплексы с координационными вакансиями и молекулы хлористого водорода участвуют в стадии хлорметаллирования ацетилена, обусловливая высокую стереоселективность процесса. Эту аналогию можно распространить также на причину, по которой необходимо одновременное присутствие в каталитических системах комплексов в меньшей степени окисления – PtII и AuI. Известно [101], что протодеаурирование металлорганических производных AuI протекает намного легче, чем органических производных золота(III). Для осуществления протолиза R–AuIII требуется наличие лигандов с сильным транс-влиянием в транс-положении к органильному лиганду [102]. Поэтому роль AuI в системе AuCl3/C, подобно PtII в случае катализа механоактивированной солью K2PtCl6, может заключаться в комплементарном восстановлении хлорвинильного производного AuIII до соответствующего комплекса золота(I). Перенос электронов в этом случае может осуществляться, например, через электропроводящий носитель – активированный уголь.
ЗАКЛЮЧЕНИЕ
Использование изотопно меченых реагентов позволяет получать крайне важную информацию о механизмах гетерогенно-каталитического гидрохлорирования ацетилена: устанавливать стереоселективность реакции, выявлять ее лимитирующую стадию, моделировать структуру переходного состояния. Так, в системах с механоактивированными хлоридными комплексами платины и палладия, а также нанесенными на активированный уголь золотосодержащими катализаторами стереохимия продукта реакции отвечает исключительно транс(анти)-присоединению атомов водорода и хлора к тройной связи ацетилена. На нанесенных палладиевых катализаторах образуются продукты как транс(анти)-, так и цис(син)-присоединения.
Наличие разных по величине изотопных эффектов HCl/DCl в системах с комплексами Pt и Pd – определенного из кинетики реакции (КИЭ) и из соотношения выходов изотопомеров продукта (γ) – указывает на участие молекулы хлористого водорода в двух стадиях реакции. КИЭ отвечает лимитирующей стадии хлорметаллирования, в которой в состав продукта входит атом хлора, а γ – более быстрому протодеметаллированию с включением в продукт атома водорода (дейтерия).
Численные значения величин изотопных эффектов позволяют моделировать структуру переходного состояния отдельных стадий реакции. В системах с механоактивированными солями K2PtCl4 и K2PdCl4 близость величин КИЭ и их высокие значения являются следствием одинаковой геометрии переходного состояния лимитирующей стадии реакции: в изоморфных кристаллических решетках K2PtCl4 и K2PdCl4 с близкими параметрами хлорметаллирование π‑координированного ацетилена под действием HCl протекает через линейное переходное состояние. Совпадение величин γ в системах с комплексами K2PtCl4 и K2PtCl6 указывает на участие в стадии протодеметаллирования в этих двух системах одного и того же интермедиата – σ-винильного производного PtII. Значительное отличие в значениях изотопных эффектов стадии протодеметаллирования винильных производных PtII и PdII под действием газообразного хлористого водорода свидетельствует о разных механизмах указанной реакции. Высокое значение γ отвечает линейному симметричному переходному состоянию стадии протодепалладирования, осуществляющегося путем электрофильного замещения SE2. Существенно меньшая величина γ для протодеплатинирования может быть следствием протекания этой реакции через нелинейное переходное состояние в последовательности стадий окислительное присоединение HCl – восстановительное элиминирование винилхлорида.
Для гетерогенно-каталитического гидрохлорирования ацетилена необходимо наличие на поверхности катализатора активных центров – комплексов металла с координационными вакансиями. В модельных системах с K2МCli (М = PtIV, PtII, PdII) координационно ненасыщенные комплексы [МCli – 1]– генерируются при механоактивации указанных солей в атмосфере непредельных соединений.
Рассмотренные металокомплексные катализаторы содержат различные металлы в разных степенях окисления. Тем не менее, во всех системах при образовании продукта транс(анти)-присоединения механизм реакции можно представить единой схемой (схема 8 ).
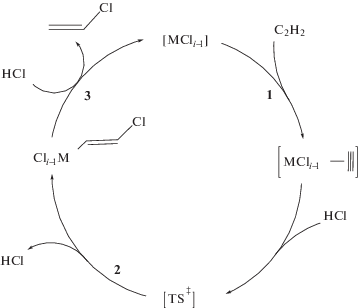
Схема 8 . Обобщенный механизм каталитического гидрохлорирования ацетилена с образованием продукта транс(анти)-присоединения С2Н2.
Как отражено на схеме 8 , реакция начинается с π-координации ацетилена к комплексу металла с координационной вакансией (стадия 1). Последующее скоростьопределяющее хлорплатинирование π‑координированного ацетилена под действием молекулы HCl (стадия 2) приводит к образованию β-хлорвинильного производного соответствующего металла, быстрый распад которого в системах K2МCl4 (М = Pt, Pd) и PdCl2/С в результате протодеметаллирования (стадия 3) замыкает каталитический цикл. При этом выделяется винилхлорид и регенерируется активный центр катализатора. В системе с механоактивированной солью K2PtCl6 (возможно, и в случае нанесенных катализаторов AuCl3/C) протодеметаллированию предшествует комплементарное восстановление хлорвинильных производных PtIV (AuIII) до соответствующих соединений PtII (AuI).
Список литературы
Ciacci L., Passarini F., Vassura I. // Resour. Conserv. Recycl. 2017. V. 123. P. 108.
Johnston P., Carthey N., Hutchings G.J. // J. Am. Chem. Soc. 2015. V. 137. P. 14 548.
Flid M.R. // Catalysis in Industry. 2009. V. 1. P. 285.
Meng X., Ding Q., Wang Q., Duan L. // Energy Env. Focus. 2014. V. 3. № 1. P. 37.
Polyvinyl Chloride (PVC) – Market Study Ceresana, URL: http://www.ceresana.com/en/market-studies/plastics/polyvinyl-chloride/ (дата обращения: 06.05.2019).
Schellerer K.-M., Kufner Th., Mieden O., Vogel E. // Kunstoffe Intern. 2016. V. 10. P. 28.
Magistro A.J., Cowfer J.A. // J. Chem. Educ. 1986. V. 63. P. 1056.
Zhang J., Liu N., Li W., Dai B. // Front. Chem. Sci. Eng. 2011. V. 5. P. 514.
Dai B., Wang Q., Yu F., Zhu M. // Scientific Reports. 2015. V. 5. Article number: 10553.
Zhou K., Jia J., Li C., Xu H., Zhou J., Luo G., Wei F. // Green Chem. 2015. V. 17. P. 356.
Ren W., Duan L., Zhu Z., Du W., An Z., Xu L., Zhang C., Zhuo Y., Chen C. // Env. Sci. Technol. 2014. V. 48. P. 2321.
Темкин O.Н., Шестаков Г.К., Трегер Ю.A. Ацетилен: химия, механизмы реакций и технология. Москва: Химия, 1991. 429 с.
Liu X., Conte M., Elias D., Lu L., Morgan D.J., Freakley S.J., Johnston P., Kiely C.J., Hutchings G.J. // Catal. Sci. Technol. 2016. V. 6. P. 5144.
Minamata Convention on Mercury, http://www.mercuryconvention.org/ (дата обращения: 06.05.2019).
Davies C.J., Miedziak P.J., Brett G.L., Hutchings G.J. // Chin. J. Catal. 2016. V. 37. P. 1600.
Zhong J., Xu Y., Liu Z. // Green Chem. 2018. V. 20. P. 2412.
Trotuş I.-T., Zimmermann T., Schüth F. // Chem. Rev. 2014. V. 114. P 1761.
Xu H., Luo G. // J. Ind. Eng. Chem. 2018. V. 65. P. 13.
Eisenstein O., Hoffmann R. // J. Am. Chem. Soc. 1980. V. 102. № 19. P. 6148.
Жермен Дж. Каталитические превращения углеводородов. Москва: Мир, 1982. 310 с.
Казанский Б.В. // Успехи химии. 1988. Т. 57. № 12. С. 1937.
Лунин В.В., Романовский Б.В. // Вестн. Моск. ун-та. 1999. Сер. 2. Химия. Т. 40. № 5. С. 293.
Митченко С.А. // Теорет. и эксперим. химия. 2007. Т. 43. № 4. С. 199.
Митченко С.А. // Кинетика и катализ. 1998. Т. 39. № 6. С. 936.
Шубин А.А., Митченко Р.С., Вдовиченко А.Н. // Журн. орг. химии. 2006. Т. 42. № 5. С. 783.
Митченко С.А., Краснякова Т.В., Жихарев И.В. // Теорет. и эксперим. химия. 2008. Т. 44. № 5. С. 306.
Меландер Л., Сондерс У. Скорости реакций изотопных молекул. Москва: Мир, 1983. 344 с.
Митченко С.А, Анаников В.П., Белецкая И.П. // Журн. орг. химии. 1998. Т. 34. № 12. С. 1859.
Сильченко Л.А., Панова С.А., Шестаков Г.К., Темкин О.Н. // Кинетика и катализ. 1997. Т. 38. № 6. С. 861.
Митченко С.А., Хомутов Е.В., Шубин А.А., Шульга Ю.М. // Теорет. и эксперим. химия. 2003. Т. 39. № 4. С. 248.
Mitchenko S.A., Khomutov E.V., Shubin A.A., Shul’ga Yu.M. // J. Mol. Catal. A: Chem. 2004. V. 212. P. 345.
Barker C., James P.L., Yarwood J. // Faraday Discuss. Chem. Soc. 1977. № 64. P. 188.
Митченко С.А., Краснякова Т.В., Жихарев И.В. // Теорет. и эксперим. химия. 2012. Т. 48. № 3. С. 147.
Митченко С.А., Краснякова Т.В. // Кинетика и катализ. 2014. Т. 55. № 6. С. 741.
Митченко С.А., Краснякова Т.В., Жихарев И.В. // Кинетика и катализ. 2009. Т. 50. № 5. С. 764.
Митченко С.А., Хомутов Е.В., Коваленко В.В., Белецкая И.П. // Кинетика и катализ. 2002. Т. 43. № 4. С. 509.
Mitchenko S.A., Khomutov E.V., Kovalenko V.V. // Inorg. Chim. Acta. 2001. V. 320. № 1–2. P. 31.
Collman J.P., Hegedus L.S., Norton J.R., Finke R.G. Principles and Applications of Organotransition Metal Chemistry. University Science Books, Mill Valley, California, 1987. P. 859.
Митченко Р.С., Шубин А.А., Краснякова Т.В. // Теорет. и эксперим. химия. 2006. Т. 42. № 5. С. 306.
Mitchenko S.A., Krasnyakova T.V., Mitchenko R.S., Korduban A.M. // J. Mol. Catal. A: Chemical. 2007. V. 275. P. 101.
Наканиси К. Инфракрасные спектры и строение органических соединений. Москва: Мир, 1965. С. 216.
Митченко С.А., Краснякова Т.В., Жихарев И.В. // Теорет. и эксперим. химия. 2010. Т. 46. № 1. С. 32.
Krasnyakova T.V., Zhikharev I.V., Mitchenko R.S., Burkhovetski V.I., Korduban A.M, Kryshchuk T.V., Mitchenko S.A. // J. Catal. 2012. V. 288. P. 33.
Митченко С.А., Краснякова Т.В., Жихарев И.В. // Кинетика и катализ. 2009. Т. 50. № 5. С. 764.
Митченко Р.С., Шубин А.А., Вдовиченко А.Н. // Теорет. и эксперим. химия. 2006. Т. 42. № 3. С. 173.
Херберхольд М. π-Комплексы металлов. Комплексы с моноолефиновыми лигандами. Москва: Мир, 1975. С. 134.
Митченко С.А., Хомутов Е.В., Шубин А.А., Белецкая И.П. // Кинетика и катализ. 2004. Т. 45. № 3. С. 417.
JCPDS: International Centre for Diffraction Data, № 25-1203.
JCPDS: International Centre for Diffraction Data, №. 9-367.
JCPDS: International Centre for Diffraction Data, №. 12-412.
Митченко С.А., Замащиков В.В., Шубин А.А. // Кинетика и катализ. 1993. Т. 34. № 3. С. 479.
Флид М.Р., Трегер Ю.А. Винилхлорид: химия и технология. В 2-х книгах. Книга 1. Москва: Калвис, 2008. 584 с.
Митченко С.А., Хомутов Е.В., Шубин А.А. // Теорет. и эксперим. химия. 2003. Т. 39. № 2. С. 88.
Luinstra G.A., Wang L., Stahl S.S., Labinger J.A., Bercaw J.E. // J. Organomet. Chem. 1995. V. 504. P. 75.
Гордон А., Форд Р. Спутник химика. Москва: Мир, 1976. 541 с.
Paulusse J.M.J., Sijbesma R.P. // Chem. Commun. 2008. P. 4416.
Ananikov V.P., Musaev D.G., Morokuma K. // Organometallics. 2005. V. 24. P. 715.
Гурвич Л.В., Карачевцева Г.В., Кондратьева Н.В., Лебедев Ю.А. Энергии разрыва химических связей. Потенциалы ионизации и сродство к электрону. Москва: Наука, 1974. С. 351.
Hill G.S., Rendina L.M., Puddephatt R. // Organometallics. 1995. V. 14. P. 4966.
Simonov P.A., Filimonova S.V., Kryukova G.N., Moroz E.M., Likholobov V.A., Kuretzky T., Boehm H.P. // Carbon. 1999. V. 37. P. 591.
Simonov P.A., Moroz E.M., Chuvilin A.L., Kolomiichuk V.N., Boronin A.I., Likholobov V.A. // Stud. Surf. Sci. Catal. 1995. V. 91. P. 977.
Moroz E.M., Simonov P.A., Bogdanov S.V., Chuvilin A.L. // Mater. Sci. Forum. 2000. V. 321. P. 1074.
Ryndin Yu.A., Alekseev O.S., Simonov P.A., Likholobov V.A. // J. Mol. Catal. 1989. V. 55. P. 109.
Симонов П.А., Троицкий С.Ю., Лихолобов В.А. // Кинетика и катализ. 2000. Т. 41. № 2. С. 281.
Simonov P.A., Romanenko A.V., Prosvirin I.P., Moroz E.M., Boronin A.I., Chuvilin A.L., Likholobov V.A. // Carbon. 1997. V. 35. № 1. P. 73.
Симонов П.А., Семиколенов В.А., Лихолобов В.А., Воронин А.И., Ермаков Ю.И. // Изв. АН СССР. Сер. хим. 1988. № 12. С. 2719.
Симонов П.А., Чувилин А.Л., Лихолобов В.А. // Изв. АН СССР. Сер. хим. 1989. № 9. С. 1952.
Симонов П.А., Мороз Э.М., Лихолобов В.А., Плаксин Г.В. // Изв. АН СССР. Сер. хим. 1990. № 7. С. 1478.
Simonov P.A., Romanenko A.V., Prosvirin I.P., Kryukova G.N., Chuvilin A.L., Bogdanov S.V., Moroz E.M., Likholobov V.A. // Stud. Surf. Sci. Catal. 1998. V. 118. P. 15.
Simakova O.A., Simonov P.A., Romanenko A.V., Simakova I.L. // React. Kinet. Catal. L. 2008. V. 95. P. 3.
Song Q.L., Wang S.J., Shen B.X., Zhao J.G. // Pet. Sci. Technol. 2010. V. 28. P. 1825.
Wang L., Wang F., Wang J. // Catal. Commun. 2015. V. 65. P. 41.
Wang L., Wang F., Wang J., Tang X., Zhao Y., Yang D., Jia F., Hao T. // React. Kinet. Mech. Cat. 2013. V. 110. P. 187.
Wang L., Wang F., Wang J. // Catal. Commun. 2016. V. 83. P. 9.
Wang F., Wang L., Wang J., Zhao Y., Wang Y., Yang D. // React. Kinet. Mech. Cat. 2015. V. 114. P. 725.
Краснякова Т.В., Никитенко Д.В., Хомутова Е.В., Митченко С.А. // Кинетика и катализ. 2017. Т. 58. № 5. С. 551.
Evers J., Beck W., Gobel M., Jakob S., Mayer P., Oehlinger G., Rotter M., Klapotke T.M. // Angew. Chem. Int. Ed. 2010. V. 49. P. 5677.
Nkosi B., Coville N.J., Hutchings G.J. // J. Chem. Soc. Chem. Commun. 1988. P. 71.
Nkosi B., Coville N.J., Hutchings G.J. // Appl. Catal. 1988. V. 43. P. 33.
Nkosi B., Coville N.J., Hutchings G.J., Adams M.D., Friedl J., Wagner F.E. // J. Catal. 1991. V. 128. P. 366.
Nkosi B., Adams M.D., Coville N.J., Hutchings G.J. // J. Catal. 1991. V. 128. P. 378.
Hutchings G.J. // Gold Bull., 1996, V. 29. P. 123.
Conte M., Carley A.F., Heirene C., Willock D.J., Johnston P., Herzing A.A., Kiely C.J., Hutchings G.J. // J. Catal. 2007. V. 250. P. 231.
Hutchings G.J. // Top. Catal. 2008. V. 48. P. 55.
Hutchings G.J. // Gold Bull. 2009. V. 42. P. 260.
Conte M., Davies C.J., Morgana D.J., Davies T.E., Elias D.J., Carley A.F., Johnston P., Hutchings G.J. // J. Catal. 2013. V. 297. P. 128.
Conte M., Davies C.J., Morgan D.J., Carley A.F., Johnston P., Hutchings G.J. // Catal. Letters. 2014. V. 144. P. 1.
Hutchings G.J. // Top. Catal. 2014. V. 57. P. 1265.
Johnston P., Carthey N., Hutchings G.J. // J. Am. Chem. Soc. 2015. V. 137. P. 14 548.
Malta G., Freakley S.J., Kondrat S.A., Hutchings G.J. // Catal. Commun. 2017. V. 53. P. 11 733.
Malta G., Kondrat S.A., Freakley S.J., Davies C.J., Dawson S., Liu X., Lu L., Dymkowski K., Fernandez-Alonso F., Mukhopadhyay S., Gibson E.K., Wells P.P., Parker S.F., Kiely C.J., Hutchings G.J. // ACS Catal. 2018. V. 8. P. 8493.
Zhao Y., Zhao F., Kang L. // J. Mol. Model. 2018. V. 24. P. 61.
Gu J., Du Q., Han Y., He Z., Li W., Zhang J. // Phys. Chem. Chem. Phys. 2014. V. 116. P. 25498.
Zhang J., He Z., Lib W., Han Y. // RSC Adv. 2012. P. 4814.
Zhu M., Kang L., Su Y., Zhang S., Dai B. // Can. J. Chem. 2013. V. 91. P. 120.
Gong W., Zhao F., Kang L. // Comput. Theor. Chem. 2018. V. 1130. P. 83.
Zhao F., Wang Y., Kang L. // Can. J. Chem. 2016. V. 94. P. 842.
Wang Y., Zhu M., Kang L., Dai B. // RSC Adv. 2014. V. 4. P. 38 466.
Xu H., Meng S., Luo G. // Catal. Sci. Technol. 2018. V. 8. P. 1176.
Wan F., Chao S., Guan Q., Wang G.C., Li W. // Catal. Commun. 2017. V. 101. P. 120.
Wegner H.A., Auzias M. // Angew. Chem. Int. Ed. 2011. V. 50. P. 8236.
Реутов О.А., Курц А.Л., Бутин К.П. Органическая химия. Москва: МГУ, 1999. 2336 с.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ