

Кинетика и катализ, 2020, T. 61, № 1, стр. 85-98
Бифункциональный катализ на мезоуровне
a Laboratory of Industrial Chemistry and Reaction Engineering, Faculty of Science and Engineering, Johan Gadolin Process Chemistry Centre, Åbo Akademi University
FI-20500 Turku/Åbo, Finland
* E-mail: dmurzin@abo.fi
Поступила в редакцию 13.08.2019
После доработки 23.08.2019
Принята к публикации 25.08.2019
Аннотация
В обзоре рассмотрены требования к техническим бифункциональным катализаторам, которые кроме металлической фазы и кислотного носителя – цеолита, содержат различные компоненты, необходимые для обеспечения эффективного тепло- и массопереноса, а также химических или механических функций. Обсуждены некоторые каталитические реакции, где такие компоненты (например, связующее для экструзии) влияют на каталитические свойства. Описана стратегия введения металлов в формованный катализатор нанесением или на цеолит или на связующее, что влияет на расстояние между центрами различного типа и каталитические свойства полученных экструдатов.
1. ВВЕДЕНИЕ
Катализ как явление принято рассматривать на нескольких уровнях, условно подразделяя на нанометровый, миллиметровый и метровый, или микро-, мезо- и макроуровни [1–3]. При этом механизмы каталитических превращений на уровне активного центра, зерна катализатора или слоя, а также на уровне реактора характеризуются определенными особенностями взаимодействия отдельных составляющих. Естественно, что изучение каталитического процесса на уровне активного центра, исследование механизма и кинетики реакции, разработка метода приготовления катализаторов для лабораторных исследований составляют содержание большинства работ, публикуемых в научных журналах и посвященных катализу.
В то же время рассмотрение катализа на уровне зерна катализатора (т.е. на мезоуровне) представляет особый интерес для технического катализа (рис. 1).
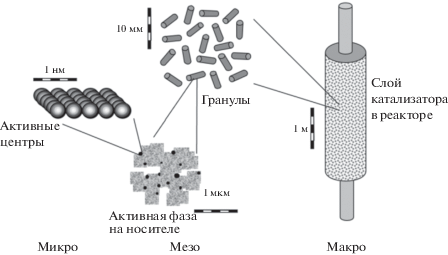
Катализаторы, применяемые в лабораторных исследованиях, как правило, представляют собой массивные или нанесенные катализаторы, используемые в виде порошков. Технические, или промышленные, катализаторы, используемые в виде гранул различных форм, наряду с активной фазой и носителем, как правило, содержат еще многочисленные компоненты, требующиеся для обеспечения эффективного тепло- и массопереноса, химических или механических (например, стабильность) функций (рис. 2).
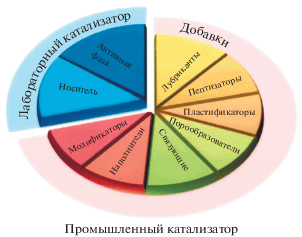
Так, для применения катализатора в стационарном слое промышленного реактора требуется увеличение размера зерна катализатора, чтобы не допустить слишком большого гидравлического сопротивления. Для этого на мезоуровне (т.е. миллиметровом) в состав промышленных катализаторов могут вводиться дополнительные компоненты. В результате состав промышленного катализатора может существенно отличаться от лабораторных аналогов, включая в себя для улучшения формуемости каталитических масс связующие вещества [4–7], модифицирующие добавки в виде дисперсантов [8], поверхностно-активные вещества (ПАВ) [9], водорастворимые полимеры, порогены и т.д. (рис. 2). Кроме того, формование суспензий с высоким содержанием твердой фазы может потребовать использования пептизаторов (например, азотной кислоты). Таким образом кислотность сформованных катализаторов может определяться свойствами пептизатора, а не активного компонента исследовательского катализатора как такового.
ФОРМОВАНИЕ КАТАЛИЗАТОРОВ
Фундаментальные работы в области катализа, выполняемые в университетах и научных организациях, в основном посвящены изучению связи состава исследовательских катализаторов с их физико-химическими и каталическими свойствами. Как отмечено выше, использование дополнительных компонентов в составе промышленных катализаторов может существенно изменить химический состав, кислотность, пористость, а также пространственное распределение активной фазы в зерне. Все это влияет на каталитические свойства полученных материалов. Фундаментальные работы в области приготовления формованных катализаторов, к сожалению, встречаются довольно редко, требуя знаний не только в области катализа, но также в области реологии, кинетики, массопереноса и инжиниринга химических реакций.
Методы экструзии и таблетирования широко используются для приготовления формованных гетерогенных катализаторов. Высокая производительность в сочетании с экономичностью метода и возможностью получения экструдатов различной формы являются явными достоинствами метода экструзии. Кроме того, этот довольно простой в смысле аппаратурного оформления метод позволяет получать пористые гранулы. При этом экструзия по сравнению с таблетированием дает бόльшие отклонения от средних размеров и приводит к гранулам меньшей механической прочности. Кроме того, экструзия также весьма чувствительна к влажности суспензий для формования, а в ряде случаев формование промышленного катализатора методом экструзии невозможно вообще (рис. 3).

Структура пор и механическая стабильность гранул зависят от свойств супензии и условий экструзии. Для использования экструдатов в катализе нужны транспортные поры размером 300–600 нм в дополнение к мезопорам (10–25 нм), что отличает такие гранулы от таблетированных катализаторов, имеющих в основном мономодальное распределение мезопор. Поскольку для экструзии используют суспензии, влияние давления при экструзии существенно менее значимо, чем при таблетировании, а, следовательно, влажность суспензии можно рассматривать как основной параметр, влияющий на вязкость пасты и на пористость экструдатов. Слишком большое количество жидкости препятствует формированию экструдатов, тогда как перемещение слишком сухих паст тоже невозможно. Несмотря на то, что добавки пластификаторов влияют положительно на пластичность катализаторной массы, перемещение суспензии в ходе экструзии, тем не менее, характеризуется сильным неньютоновским течением. Следует отметить, что сам по себе анализ течения суспензий не дает возможности прогнозировать ход экструзии и свойства образующихся гранул, тем не менее изучение реологии катализаторных суспензий важно для понимания и оптимизации метода экструзии.
Формовочная масса должна обладать определенными реологическими свойствами – с одной стороны, пластичностью для течения в экструдере и придания необходимой формы без разрушения, а, с другой стороны, также достаточной прочностью коагуляционной структуры, обеспечивающей механическую прочность сформованных материалов [7]. Однако излишне прочная коагуляционная структура приведет к более высоким энергозатратам при экструзии.
Экструзия высококонцентрированных суспензий отличается от экструзии полимеров, происходящей при плавлении формуемой массы. Реологические параметры, например, зависимость напряжения сдвига от вязкости [10, 11], а также дзета-потенциал частиц, отражающий электростатические взаимодействия и характеризующий стабильность дисперсных систем, определяют поведение суспензий при экструзии. При этом следует отметить, что обычно как реологические свойства, так и дзета-потенциал изучают в области низкоконцентрированных суспензий [12–14] и поэтому не соответствующих условиям экструзии реальных суспензий.
В качестве связующих используют неорганические вещества типа оксида алюминия, оксида кремния и глин, которые, в отличие от органических связующих, не разрушаются при прокаливании. Необходимость добавления связующего вещества при формовании катализаторов для получения прочных гранул может менять поверхностные и каталитические свойства формовочных смесей [15–17], как уже было отмечено выше.
Выбор связующего является в определенной степени копромиссом между требованиями реологии и механической стабильностью, с одной стороны, и каталитической активностью, с другой. Например, введение алюмосиликатов при экструзии монолитных блоков на основе оксида титана улучшает реологические свойства, однако негативно влияет при этом на катализ.
Получение катализатора на основе Ni/ZSM-5 со связующим оксидом алюминия обсуждалось в публикации [17]. Как было установлено, введение оксида алюминия повлияло на кислотные свойства катализатора. Кроме того, селективность в гидродеоксигенации циклогексанона зависела от количества связующего.
При формовании ZSM-5 с применением алюмофосфата AlPO4 и псевдобемита [18] образуется пространственная аморфная гель-фаза, в которую либо полностью, либо частично встраиваются кристаллиты цеолита.
Явное влияние связующего следует из работы [19], в которой было рассмотрено использование в качестве связующих компонентов бентонита и псевдобемита для превращения диметилового эфира в олефины. Применение псевдобемита повышает как стабильность катализатора, так и конверсию диметилового эфира и селективность по олефинам. Наличие пор большего размера при использовании γ-оксида алюминия приводит к уменьшению отложений кокса.
Связующее может обладать кислотными свойствами и влиять на распределение центров по их силе. Например, использование алюминофосфата как связующего меняет такое распределение (рис. 4), что видно из сравнения температурно-программируемой десорбции (ТПД) аммиака на исходном цеолите H-ZSM-5 и после добавления связующего [20]. Результаты ТПД аммиака, представленные на рис. 4, определенно указывают на изменения в кислотности при введении связующего, что было объяснено миграцией фосфора в структуру цеолита [20].
Рис. 4.
ТПД аммиака порошка H-ZSM-5 после прокаливания (1) и сформованной гранулы цеолита с AlPO4 в качестве связующего (2) [20].
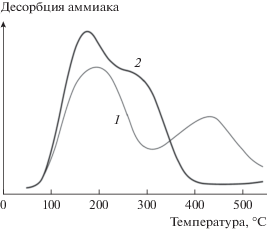
Применение связующего приводит также к неравномерному распределению активного компонента в грануле. В результате возможно образование зон с большей концентрацией активной фазы и, следовательно, с более высокой скоростью реакции, локальным перегревом и переходом реакции в диффузионную область. При таком сценарии реакция начинает определяться уже процессами массопереноса.
Влияние дисперсионной среды и полимерных добавок на реологические характеристики суспензий на основе цеолита MFI было изучено в работе [13]. Адсорбция на поверхности цеолита изменяет объемную долю твердой фракции суспензии и ее вязкость, что приводит к отклонениям от уравнения Кригера–Догерти для вязкости монодисперсных сферических упругих частиц
(1)
$\eta = {{\left( {1 - \frac{\varphi }{{{{\varphi }_{{\max }}}}}} \right)}^{{ - K{{\varphi }_{{\max }}}}}},$Реологическое поведение суспензий цеолитов осложнено не только тем, что частицы имеют разную форму, отличную от сферической, но и распределением частиц по размерам. Таким образом понимание реологии связано не только с приготовлением суспензий из имеющихся цеолитов и разных связующих, но и с синтезом цеолитов разного размера и морфологии. Существенное изменение вязкости обнаруживается при смешении частиц различного размера, когда параметр φ (для твердого вещества) превышает 0.5. Этот эффект проявляется более заметно при увеличении соотношения больших и маленьких частиц.
Для стационарного течения суспензий используют модели вида
связывающие напряжение сдвига τ с эффективной вязкостью η и скоростью сдвига γ.Напряжение сдвига суспензии формовочных масс обычно описывают уравнением типа Хершеля–Балкли
где τ0 – предельное напряжение сдвига, κ – коэффициент густоты потока, а n – показатель поведения жидкости. Для псевдопластичных жидкостей n < 1. Именно такое течение образуется в случае катализаторных паст со значениями n, близкими к 0.7. Малое значение показателя поведения жидкости соответствует менее значительным градиентам скорости в радиальном направлении и, соответственно, меньшим дефектам гранул после экструзии. Для сотовых катализаторов показатель поведения жидкости равен ~0.3.С точки зрения коллоидной химии, введение пептизаторов (например, растворов азотной, уксусной или муравьиной кислот) влияет на отклонения pH от изоэлектрической точки, когда поверхность не несет электрического заряда. В этом случае возможна аггломерация частиц в жидкой фазе. Следует отметить, что кислотные свойства пептизаторов влияют не только на каталитические свойства, но и на технологию производства катализаторов. Так, следует учитывать коррозионный эффект пептизаторов, а также необходимость очистки отходящих газов от оксидов азота, образующихся при прокаливании обработанных азотной кислотой материалов. Последнее приводит к удорожанию синтеза катализаторов, поэтому применение в качестве пептизатора азотной кислоты стоит рассматривать лишь тогда, когда другие методы регулирования реологических свойств не работают.
В качестве пластификаторов могут использоваться, например, производные целлюлозы, крахмал или поливиниловый спирт в количестве 3–5 мас. %, что соответствует полному покрытию доступной поверхности твердого вещества и вполне достаточно для достижения необходимых реологических свойств. При выборе пластификатора следует учитывать возможную адгезию суспензии к материалу экструдера, которая должна быть сведена к минимуму.
Добавление пластификаторов и порогенов обычно не влияет на каталитические свойства, так как они удаляются при термической обработке в атмосфере воздуха.
3. БИФУНКЦИОНАЛЬНЫЕ МЕТАЛЛ-КИСЛОТНЫЕ КАТАЛИЗАТОРЫ
Совмещение различных функций в одном катализаторе позволяет уменьшить общее время процесса, увеличивая при этом общий выход целевого продукта. Сложности осуществления каскадных или тандемных реакций, протекающих на одном катализаторе, связаны с тем, что различные реакции должны идти при одинаковых условиях. Но оптимальные условия для каждой реакции могут отличатьcя. Возможно также взаимное влияние интермедиатов каталитических реакций, протекающих на разных центрах. В некоторых случаях, как, например, в гидрокрекинге или изомеризации, такое влияние положительно, в других случаях оно может быть и негативным.
Скорости разных процессов, протекающих на одном катализаторе, должны быть по возможности близкими, иначе, например, на каком-то катализаторе может оказаться недостаточно субстрата. То есть промышленный катализатор должен содержать два или больше типов активных центров на таком расстоянии, чтобы скорость диффузии между центрами удовлетворяла кинетике реакций на этих центрах.
Наиболее распространенными в промышленности бифункциональными катализаторами подобного типа являются твердые кислоты – цеолиты, модифицированные металлами. В качестве примеров можно привести катализаторы гидроизомеризации и гидрокрекинга с двумя типами центров. Гидроизомеризация алканов увеличивает октановое число и протекает последовательно [21] через дегидрирование на металлических центрах, миграцию олефинов к кислотным центрам, скелетную изомеризацию (через карбениевые ионы) в разветвленные олефины, диффузию обратно к металлическим центрам и гидрирование до изоалканов (схема 1 ).
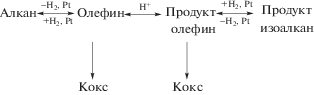
Схема 1 . Процесс скелетной изомеризации.
Эта реакция осложнена коксообразованием, поэтому концентрацию олефина стараются свести к минимуму, обеспечивая близкое расстояние между металлическими и кислотными центрами.
Скорость скелетной изомеризации олефинов выше, чем алканов, поэтому введение металла в цеолит ускоряет общий процесс, при этом присутствие металла повышает стабильность кислотных катализаторов, подавляя коксообразование. Для биметаллических катализаторов среднее расстояние между металлическими и кислотными центрами должно быть выбрано с учетом минимизации влияния массопереноса при слишком большом расстоянии, с одной стороны, и предотвращения нежелательного взаимодействия между центрами, если они расположены слишком близко, с другой. Критерий определения критического расстояния между разными центрами был предложен еще в 1962 г. [22].
Гидроизомеризация алканов довольно хорошо изучена как для легких [21], так и для С5–С6-алканов [23]. Последняя реакция широко используется в нефтепереработке, где также важна скелетная изомеризация и более тяжелых С10–С13- [24], а также С16–С18-алканов [25–27].
В работе [24] гидроизомеризация смеси C10–C13-алканов была исследована на нескольких платиновых катализаторах, нанесенных на цеолиты, содержащие разное количество тетраэдрических атомов (Т-атомы, 10 и 12). Последние были более активны, причем более высокий выход целевых моноразветвленных изомеров был получен в присутствии среднепористых цеолитов (10 Т-атомов), что связывают с молекулярной селективностью цеолитов с такими порами, подавляющими образование многоразветвленных изомеров.
Еще один интересный пример бифункционального катализа приведен на схеме 2 , которую можно рассматривать как метод улучшения цетанового числа в средних дистиллятах.
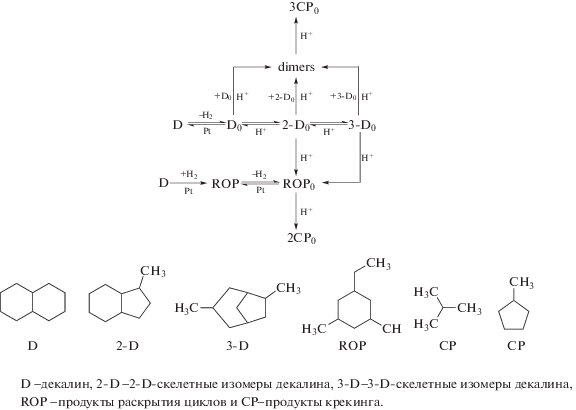
Схема 2 . Превращения цеолитов на металл-модифицированных катализаторах [28].
Такая реакция должна быть селективной к раскрытию цикла и при этом не приводить к крекингу и, соответственно, к снижению молекулярной массы. Так как крекинг происходит на тех же кислотных центрах катализатора, что и раскрытие цикла, то получить селективно продукты раскрытия циклов типа С10-алкилциклогексанов и С10-алкилциклопентанов довольно сложно.
При модифицировании цеолитов платиной превращения декалина соответствуют почти такой же схеме 2 . При этом начальная стадия та же, как и на схеме 1 , и представляет собой дегидрирование декалина, протекающее на металлических центрах. Протонирование олефина происходит уже на кислотных центрах Бренстеда, как и раскрытие цикла. При введении платины в цеолит общее число кислотных центров не меняется, а вот количество сильных кислотных центров уменьшается [29], что и подавляет нежелательный крекинг.
Однореакторный синтез ментола из цитронеллаля (схема 3 ) также возможен с использованием бифункциональных катализаторов [30–35].
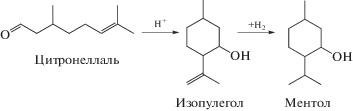
Схема 3 . Синтез ментола из цитронеллаля [30].
В работе [30] были исследованы платиновые катализаторы, нанесенные на цеолит TUD-1, промотированные вольфрамом с целью регулирования кислотности и дисперсности платины, и было установлено, что наиболее эффективным катализатором внутримолекулярной циклизации по Принсу является 1 мас. % Pt/W-TUD-1, обеспечивающий 96%-ный выход ментола. Для сравнения рутений (1%), нанесенный на цеолит Beta-25, исследованный в работах [31, 32], обеспечивал выход 93%. В литературе предложены и другие бифункциональные катализаторы, в которых различные металлы такие, как Pt [34] и Ir [35], были нанесены на цеолит Beta. В случае никеля [33] требовалась модификация цеолита также и цирконием для формирования центров Льюиса.
Особенностью реакции циклизации по Принсу на кислотных катализаторах является необходимость регулирования кислотности, так как при ее избыточности возможна нежелательная реакция дегидроксилирования.
В реакции гидроалкилирования бензола и этилбензола [36] для получения этилциклогексилбензола (схема 4 ) были предложены рутениевые катализаторы на цеолите Beta [36].
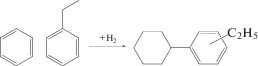
Схема 4 . Гидроалкилирование бензола и этилбензола на модифицированных металлами катализаторах.
Наибольшая селективность по целевому продукту составляла 42% при конверсии 78% на Ru–Ni/Beta при 170–190°C, 1 МПа и соотношении H2 : бензол : этилбензол, равном 2 : 1 : 1. Относительно невысокая селективность и наличие побочных продуктов полного гидрирования (циклогексан, этилциклогексан), а также частичного гидрирования с последующим алкилированием (циклогексилбензол, этилциклогексилбензол, диэтилциклогексилбензол), позволяет надеяться на дальнейшее улучшение эффективности катализаторов за счет оптимизации гидрирующих (металлических) и кислотных центров.
Селективное гидроалкилирование бензола (схема 5 ) на бифункциональном палладиевом катализаторе, нанесенном на цеолит Y с соотношением SiO2 : Al2O3, равным 5, 12, 30 и 60, описано в работе [37]. Было показано, что условия реакции и состав катализатора влияют на активность и селективность по циклогексилбензолу. Селективность по циклогексилбензолу определяется превращением циклогексена, который получается при селективном гидрировании бензола на металлических центрах, после чего циклогексен алкилируется на кислотных центрах. Таким образом, высокая селективность образования циклогексилбензола определяется оптимальным балансом кислотных и металлических центров. При конверсии 42% селективность по циклогексилбензолу была 75% на 0.2 мас. % Pd, нанесенном на H-Y-5 с соотношением оксида кремния к оксиду алюминия, равным 5.
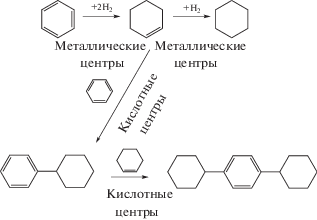
Схема 5 . Гидроалкилирование бензола на бифункциональном катализаторе.
Однореакторный синтез молочной кислоты на бифункциональном катализаторе типа Pt/Sn-MFI (схема 6 ) без использования оснований предложен в публикации [38].
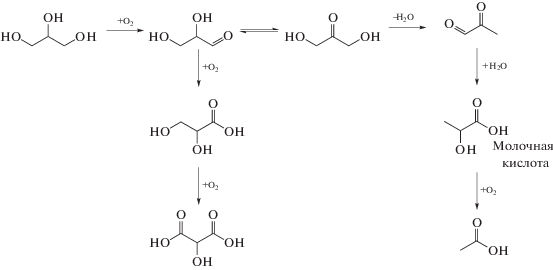
Схема 6 . Селективное окисление глицерина в молочную кислоту на катализаторе Pt–Sn/MFI.
В рамках предложенного механизма, селективное окисление до альдегида протекает на платине, тогда как на центрах Sn–MFI, обладающих льюисовской кислотностью, происходит образование дигидроксиацетона с последующим превращением в молочную кислоту. Селективность такого катализатора объясняется микропористой структурой, доступностью центров, содержащих олово, и присутствием центров с льюисовской кислотностью. В работе [38] на бифункциональном катализаторе Pt–Sn/MFI была достигнута селективность по молочной кислоте 80.5% при конверсии глицерина 90%.
Еще один пример бифункционального катализа связан с превращениями γ-валеролактона (ГВЛ) в пентановую (валерьяновую) кислоту [39]. Как было показано [39], для раскрытия цикла в молекуле ГВЛ необходимо присутствие кислотных центров (схема 7 ).
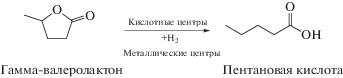
Схема 7 . Гидрирование γ-валеролактона на бифункциональном катализаторе.
Среди изученных в работе [39] бифункциональных катализаторов, платина, нанесенная на цеолит Beta, как в исходном состоянии, так и после его десиликации, проявила наибольшую активность в гидрировании ГВЛ до валерьяновой кислоты с селективностью, превышающей 98%. При этом не происходило вымывания из катализатора ни платины, ни алюминия. Активностью обладал и катализатор с золотом, нанесенным на цеолит Beta, подвергшийся десиликации.
Интересный пример, связанный с пространственной структурой биметаллического катализатора, был описан в работе [40] при тандемной альдольной конденсации фурфурола и ацетона с последующим гидрированием (схема 8 ).
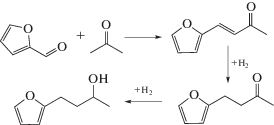
Схема 8 . Тандемная альдольная конденсация фурфурола и ацетона с последующим гидрированием.
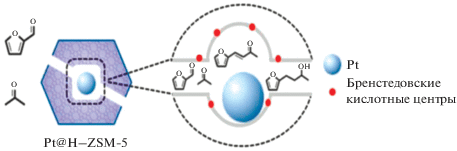
Авторы [40] разработали метод инкапсулирования наночастиц платины в цеолит типа MFI (т.е. ZSM-5) с высокой эффективностью (рис. 5), позволяющий довести выход продуктов гидрирования C8 до 87%, тогда как нанесенный Pt/HZSM-5 катализирует лишь гидрирование до фурфурилового спирта и декарбонилирование до фурана с их последующими превращениями.
4. РЕГУЛИРОВАНИЕ РАССТОЯНИЯ МЕЖДУ ЦЕНТРАМИ РАЗНОГО ТИПА В БИФУНКЦИОНАЛЬНЫХ КАТАЛИЗАТОРАХ
Примеры, приведенные в разделе 3, относились к введению металла в порошки цеолитов. Введение нецеолитной фазы в катализатор при формовании (см. раздел 2) может привести к изменению расстояния между центрами различного типа, а также дисперсности металла и кислотности. В этой связи интересно рассмотреть конкретные примеры такого влияния. Так, в гидроизомеризации этилбензола было обнаружено увеличение активности при формовании морденита с нанесенным Pt/Al2O3, а дисперсность металла была в 8 раз выше, чем при нанесении платины непосредственно на морденит [41].
Формование платины, нанесенной на экструдаты цеолита K-LTL, сформованного с основным связующим – оксидом алюминия, допированным MgO, привело к более высокой активности в реакции ароматизации по сравнению с активностью недопированного оксида алюминия или оксида кремния в качестве связующего [42].
Концепция близости активных центров была подвергнута детальному анализу в работах, посвященных катализаторам гидрокрекинга [43, 44]. Так, было показано, что селективность промышленного катализатора гидрокрекинга можно регулировать при неизбежном использовании связующего (оксида алюминия) за счет контроля на наноуровне расстояния между металлическими и кислотными центрами (рис. 6). В ходе гидрокрекинга сначала алканы превращаются в олефины на металлических центрах, затем мигрируют к кислотным центрам и подвергаются изомеризации и крекингу в алкены с более короткой цепью. Последующая миграция к металлическим центрам и гидрирование приводят к образованию алкана с меньшей длиной цепи, чем исходные алканы.
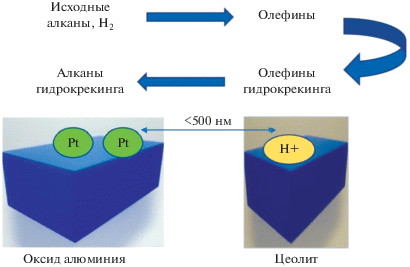
Согласно [43, 44], влияние расстояния между центрами следует рассматривать не только в диапазоне ~1 нм, но из-за присутствия связующего также в интервале 100–1000 нм. Если кластеры платины располагаются рядом с кислотными центрами, то образующиеся с более высокой концентрацией олефины более подвержены побочным реакциям с образованием кокса, способствуя более интенсивной дезактивации катализатора. Как было установлено, оптимальное расстояние между центрами составляет 500 нм [43, 44]. В этих работах исследовались промышленно доступные экструдаты, содержащие связующее и экструдат, а для нанесения платины только на связующее или только на цеолит применялась специальная методика. Промышленные катализаторы, исследованные в [44], имели определенное соотношение оксидов кремния и алюминия, а также один тип связующего.
В работе [46] были рассмотрены композитные материалы, состоящие из трех коммерческих цеолитов Beta с разной кислотностью (соотношение SiO2 : Al2O3 варьировалось от 25 до 300) и разным количеством бентонита (от 10 до 65 мас. %). Изменение свойств композитов с точки зрения кислотности и размера пор было обнаружено уже на стадии приготовления суспензий для последующей экструзии. Механическое воздействие в шаровой мельнице приводило к тому, что свойства композитов не могли быть описаны как линейные комбинации свойств цеолита и глины. Причина этого заключалась в химическом взаимодействии между цеолитом Beta и бентонитом при таком воздействии. В последующей работе авторов [47] была изучена циклизация цикронеллаля (схема 9 ) в проточном реакторе с орошением при 35–70°C. Использованные катализаторы были приготовлены экструзией цеолитов Beta и Y с различным соотношением бентонита и метилцеллюлозы в качестве неорганического и органического связующих соответственно. Полученные результаты каталитической активности и селективности сравнивали с результататами для той же реакции в реакторе с суспендированным катализатором.
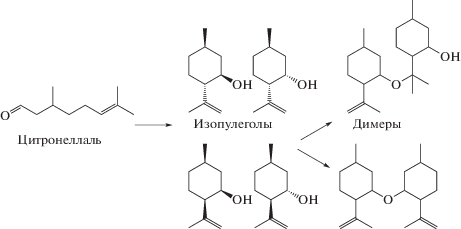
Схема 9 . Схема реакций при циклизации цитронеллаля.
В работе [47] было показано, что введение метилцеллюлозы и бентонита приводит к изменению текстурных и кислотных свойств экструдатов. Для формованных материалов с разным количеством бентонита, конверсия цитронеллаля варьировалась в интервале 53–71% через 2.5 ч в потоке (рис. 7), что авторы связали с изменением кислотности материалов.
Рис. 7.
Конверсия цитронеллаля (⚫) и сходимость массового баланса (⚪) в зависимости от времени для экструдатов H-Beta-25 с различным содержанием бентонита, мас. %: 0 (а), 10 (б), 30 (в), 100 (г). Условия реакции: 35°C, скорость потока раствора цитронеллаля 0.4 мл/мин, начальная концентрация субстрата 0.086 моль/л, масса катализатора 1 г.
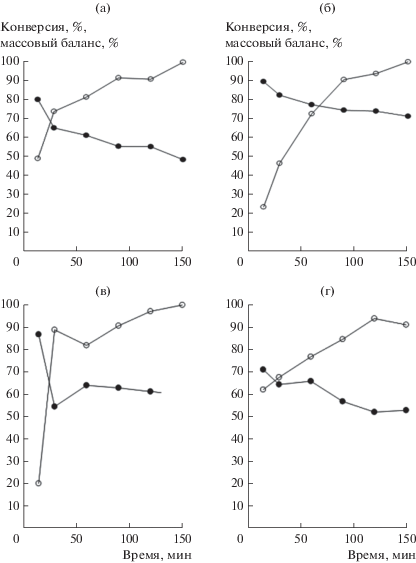
Результаты, представленные на рис. 7, указывают на дезактивацию катализатора даже при такой невысокой температуре реакции и на значительную активность самого связующего. При этом использование бентонита как связующего и наличие в нем примесей (MgO, K2O и Fe2O3) приводит к существенному образованию димеров (рис. 7) и снижению выхода изопулеголов. Наибольший выход пулеголов 63% был получен в отсутствие бентонита [47]. Энергия активации внутримолекулярной реакции циклизации цитронеллаля по Принсу была невысокой (21 кДж/моль для цеолита Beta с модулем 25 и 12 кДж/моль для бентонита), что однозначно указывает на наличие процесса массопереноса. Сравнение экспериментов в проточном реакторе на экструдированном катализаторе и в автоклаве на суспендированном катализаторе показали, что в реакции циклизации массоперенос не влиял на распределение изопулеголов. Однако поскольку реакция образования эфиров имеет более высокий кинетический порядок, селективность в области внутренней диффузии такой реакции уменьшается в соответствии с теоретическими представлениями. Таким образом, проведение реакции в диффузионной области более благоприятно с точки зрения уменьшения количества тяжелых продуктов конденсации.
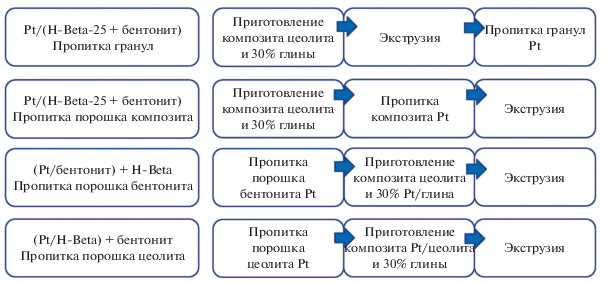
В продолжение этих работ [46, 47] был изучен синтез формованных бифункциональных катализаторов [48], когда до экструзии с 1% метилцеллюлозы платина наносилась либо на цеолит Beta-25, либо на связующее бентонит. Методология синтеза катализаторов показана на рис. 8.
Полученные катализаторы были охарактеризованы различными физико-химическими методами, включающими в том числе адсорбцию азота, сканирующую и просвечивающую электронную микроскопию и ИК-спектроскопию с пиридином в качестве молекулы-зонда. Катализаторы тестировали в реакции изомеризации н-гексана (схема 10 ), содержащего незначительные количества (1%) бензола. Опыты проводили в проточном реакторе при 200–350°C. При этом бензол подвергался гидрированию, скелетной изомеризации до метилпентана, раскрытию цикла с получением гексана и последующим привращениям согласно схеме 10 .
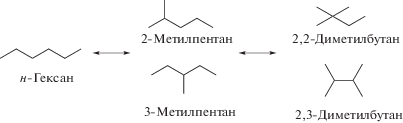
Схема 10 . Изомеризация н-гексана.
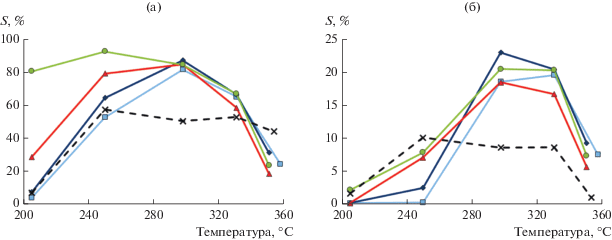
Наибольшая селективность по C6-изомерам (рис. 9а), включающим все разветвленные изомеры, такие как метилпентан и диметилбутан (рис. 9б), наблюдалась при 250°C для случая, когда Pt/H-Beta-25 подвергался экструзии вместе со связующим бентонитом. Этот же катализатор проявил наивысшую конверсию (23.4%) среди других изученных образцов.
Рис. 9.
Селективность по C6-изомерам (а) и дизамещенным бутанам (б) в зависимости от температуры, %: × – 100% H-Beta-25 (CP814E, порошок);  – 2 мас. % Pt/(H-Beta-25 + бентонит), платина нанесена после экструзии;
– 2 мас. % Pt/(H-Beta-25 + бентонит), платина нанесена после экструзии;  – 2 мас. % Pt/(H-Beta-25 + бентонит), платина нанесена до экструзии;
– 2 мас. % Pt/(H-Beta-25 + бентонит), платина нанесена до экструзии;  – 2 мас. % Pt/(бентонит) + H-Beta-25, платина нанесена до экструзии;
– 2 мас. % Pt/(бентонит) + H-Beta-25, платина нанесена до экструзии;  – 2 мас. % Pt/(H-Beta-25) + бентонит, платина нанесена до экструзии.
– 2 мас. % Pt/(H-Beta-25) + бентонит, платина нанесена до экструзии.
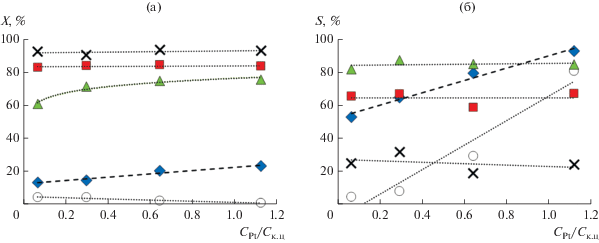
По сравнению с другими катализаторами этот образец обладал наименьшей брендстедовской и льюисовской кислотностью. Интересно, что все катализаторы существенно отличались отношением содержания платины к содержанию кислотных центров (CPt/Cк.ц = 0.07–1.12).
В рамках концепции идеального бифункционального катализатора для оптимальных результатов необходимо высокое значение отношения CPt/Cк.ц [25, 49–51]. При небольших значениях CPt/Cк.ц (<0.03) наблюдается сильная дезактивация катализатора и быстрое превращение неразветвленных алканов в продукты изомеризации и крекинга. При значениях, превышающих ≥0.17, активность в расчете на кислотный центр максимальна, при этом наблюдается медленная дезактивация с превращениями углеводородов в моно-, ди- и тризамещенные продукты, а также в продукты крекинга [49–51]. В статье [48] отмечается, что даже катализатор с наименьшим значением CPt/Cк.ц стабилен при наибольшей температуре реакции, а результаты соответствуют ожидаемой зависимости от CPt/Cк.ц (рис. 10), хотя при 200°С плато активности и селективности катализатора и не было достигнуто.



5. ЗАКЛЮЧЕНИЕ
В заключение хотелось бы отметить, что, количество фундаментальных работ, посвященных влиянию большого многообразия компонентов, вводимых в технические катализаторы, на их каталитические свойства незаслуженно мало. Одним из исключений является работа [52], где было показано, что экструзия приводит в том числе к деалюминированию из-за влияния пептизаторов и изменению структуры мезо- и макропор при удалении органических лубрикантов, изменяя каталитические свойства в реакции каталитического крекинга.
Результаты, представленные в научно-исследовательских работах по бифункциональным катализаторам, носят порой достаточно противоречивый характер. В течение долгого времени ключевым параметром для идеального бифункционального катализатора считалась близость между металлическими и кислотными центрами согласно критерию, предложенному в публикации [22]. Недавние работы показали однако, что такая близость совсем не обязательна для оптимальной селективности и стабильности. Так, согласно [44], близкое расстояние между двумя типами центров негативно влияет на селективность в реакции изомеризации. При этом, например, в работе [49] сделан вывод, что расстояние между центрами в микрометровом диапазоне не влияет на каталитические свойства. Более того, для реакции изомеризации гексана [48] было установлено, что ключевым параметром, влияющим на активность и селективность бифункциональных платиновых катализаторов в форме гранул, полученных экструзией, является соотношение металлических центров и кислотных центров цеолита Beta, причем метод приготовления катализаторов, включая способ введения платины, существенно влияет на такое соотношение.
Понимание фундаментальных причин влияния разнообразных компонентов в промышленных катализаторах и оптимального пространственного расположения металлических и кислотных центров с большой вероятностью приведет к получению более активных, селективных и стабильных катализаторов для ряда важных промышленно значимых процессов.
Список литературы
Слинько М.Г. // Кинетика и катализ. 1969. Т. 10. С. 12.
Vlachos D.G. //AIChE J. 2012. V. 58. P. 1314.
Maestri M. // Chem. Commun. 2017. V. 53. P. 10 244.
Mitchell S., Michels N.-L., Perez-Ramirez J. // Chem. Soc. Rev. 2013. V. 42. P. 6094.
Murzin D.Y. Engineering Catalysis. Berlin: Walter de Gruyter, 2013. 364 p.
Schüth F., Hesse M. Catalyst Forming. Handbook of Heterogeneous Catalysis / Eds Ertl G., Knözinger H., Schüth F., Weitkamp J. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 2008. 676 p.
Prokof’ev V. // Kinet. Catal. 2012. V. 53. P. 616.
Palmqvist L., Lyckfeld O., Carlström E., Davouts P., Kauppi A., Holmberg K. // Colloid Surface A. Physicochem. Eng. Aspect. 2006. V. 274. P. 100.
Yalçın T., Alemdar A., Ece Ö.I., Güngör N. // Mater. Lett. 2002. V. 57. P. 420.
Gordina N.E., Prokof’ev V.Y., Il’in A.P. // Glass. Ceram. 2005. V. 62. P. 282.
Zhou Z., Scales P.J., Boger D.V. // Chem. Eng. Sci. 2001. V. 56. P. 2901.
Zacahua-Tlacuatl G., Perez-Gonzalez J., Castro-Arelanno J., Balmori-Ramirez H. // Appl. Rheol. 2010. V. 20. P. 1.
Olanrewaju K.O., Bae T.-H., Nair S., Breedveld V. // Rheol. Acta. 2013. V. 53. P. 133.
Liu X., Mäki-Arvela P., Aho A., Vajglova Z., Gun’ko V., Heinmaa I., Kumar N., Eränen K., Salmi T., Murzin D.Yu. // Molecules. 2018. V. 23. 946.
Müller P., Russell A., Tomas J. // Chem. Eng. Sci. 2015. V. 126. P. 204.
Draper O., Blackburn S., Dolman G., Smalley K., Griffiths A. // J. Mater. Process. Technol. 1999. V. 92–93. P. 141.
Kong X., Liu J. // PLOS One. 2014. V. 9. P. e101744.
Lee Y.J., Kim Y.-W., Viswanadham N., Jun K.-W., Bae J.W. // Appl. Catal. A. Gen. 2010. V. 374. P. 18.
Pérez-Uriarte P., Gamero M., Ateka A., Diaz M., Aguayo A.T., Bilbao J. // Ind. Eng. Chem. Res. 2016. V. 55. P. 1513.
Freiding J., Patcas F.-C., Kraushaar-Czarnetzki B. // Appl. Catal. A. Gen. 2007. V. 328. P. 210.
Nieminen V., Kangas M., Salmi T., Murzin D.Yu. // Ind. Eng. Chem. Res. 2005. V. 44. P. 471.
Weisz P.B. // Adv. Catal. 1962. V. 13. P. 137.
Ахметов С.А., Сериков Т.П., Кузеев И.Р., Баязитов М.И. Технология и оборудование процессов переработки нефти и газа / Под ред. Ахметова С.А. С-Пб.: Недра, 2006. 868 с.
Zschiesche C., Himsl D., Rakoczy R., Reitzmann A., Freiding J., Wilde N., Gläser R. // Chem. Eng. Technol. 2018. V. 41. P. 199.
Batalha N., Pinard L., Bouchy C., Guillon E., Guisnet M. // J. Catal. 2013. V. 307. P. 122.
Huybrechts W., Vanbutsele G., Houthoofd K.J., Bertinchamps F., Laxmi Narasimhan C.S., Gaigneaux E.M., Thybaut J.W., Marin G.B., Denayer J.F.M., Baron G.V., Jacobs P.A., Martens J.A. // Catal. Lett. 2005. V. 100. P. 235.
Zhang M., Chen Y., Wang L., Zhang Q., Tsang C.-W., Liang C. // Ind. Eng. Chem. Res. 2016. V. 55. P. 6069.
Kangas M., Kubicka D., Salmi T., Murzin D.Yu. // Top. Catal. 2010. V. 53. P. 1172.
Kubicka D., Kumar N., Venäläinen T., Karhu H., Kubickova I., Österholm H., Murzin D.Yu. // J. Phys. Chem. B. 2006. V. 110. P. 4937.
ten Dam J., Ramanathan A., Djanashvili K., Kapteijn F., Hanefeld U. // RSC Adv. 2017. V. 7. P. 12 041.
Plösser J., Lucas M., Wärnå J., Salmi T., Murzin D.Yu., Claus P. // Org. Process Res. Dev. 2016. V. 20. P. 1647.
Plößer J., Lucas M., Claus P. // J. Catal. 2014. V. 320. P. 189.
Nie Y., Niah W., Jaenicke S., Chuah G.-K. // J. Catal. 2007. V. 248. P. 1.
Mertens P., Verpoort F., Parvulescu A.-N., De Vos D. // J. Catal. 2006. V. 243. P. 7.
Iosif F., Coman S., Parvulescu V., Grange P., Delsarte S., De Vos D., Jacobs P. // Chem. Comm. 2004. P. 1292.
Borodina I.B., Ponomareva O.A., Fajula F., Bousquet J., Ivanova I.I. // Micropor. Mesopor. Mater. 2007. V. 105. P. 181.
Kumar S.A., John M., Pai S.M., Ghosh S., Newalkar B.L. // Mol. Catal. 2017. V. 4422. P. 27.
Cho H.J., Chang C.-C., Fan W. // Green Chem. 2014. V. 16. P. 3428.
Derle S.N., Parikh P.A. // Biomass Convers. Biorefin. 2014. V. 4. P. 293.
Cho H.J., Kim D., Li J., Su D., Xu B. // J. Am. Chem. Soc. 2018. V. 140–411. P. 3514.
Fernandes L.D., Monteiro J.L.F., Sousa-Aguiar E.F., Martinez A., Corma A. // J. Catal. 1998. V. 177. P. 363.
Kumar M., Saxena A.K., Negi B.S., Viswanadham N. // Catal. Today. 2008. V. 130. P. 501.
Proeto G., Zecevic J., Friedrich H., de Jong K.P., de Jongh P.E. // Nature Mater. 2013. V. 12. P. 34.
Zecevic J., Vanbutsele G., de Jong K.P., Martents J.A. // Nature. 2015. V. 528. P. 245.
Gläser R. // Nature. 2015. V. 528. P. 197.
Vajglová Z., Kumar N., Peurla M., Peltonen J., Murzin D.Yu. // Catal. Sci. Technol. 2018. V. 8. P. 6150.
Vajglova Z., Kumar N., Mäki-Arvela P., Eränen K., Peurla M., Hupa L., Nurmi M., Toivakka M., Murzin D.Yu. // Ind. Eng. Chem. Res. 2019. V. 58. P. 16875. https://pubs.acs.org/doi/abs/10.1021/acs.iecr.9b02829
Vajglova Z., Kumar N., Peurla M., Hupa L., Semikin K., Sladkovskiy D., Murzin D.Yu. // Ind. Eng. Chem. Res. 2019. V. 58. P. 10875.
Gutierrez-Acebo E., Leroux C., Chizallet C., Schuurman Y., Bouchy C. // ACS Catal. 2018. V. 8. P. 6035.
Batalha N., Pinard L., Pouilloux Y., Guisnet M. // Catal. Lett. 2013. V. 143. P. 58.
Guisnet M. // Catal. Today. 2013. V. 218. P. 123.
Whiting G.T., Chung S.-H., Stosic D., Chowdhury A.D., van der Wal L.I., Fu D., Zecevic J., Travert A., Houben K., Baldus M., Weckhuysen B.M. // ACS Catal. 2019. V. 96. P. 4792.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ