

Кинетика и катализ, 2020, T. 61, № 1, стр. 5-44
Теоретическое изучение деоксигенации сложных эфиров на малых кластерах интерметаллидов Pt–Sn
Д. Е. Завелев a, *, Г. М. Жидомиров b, c, М. В. Цодиков a
a ФГБУН Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Ленинский просп., 29, Россия
b ФГБУН Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Академика Лаврентьева, 5, Россия
c Национальный исследовательский университет “Высшая школа экономики”, физический факультет
101000 Москва, Мясницкая ул., 20, Россия
* E-mail: metalian@ips.ac.ru
Поступила в редакцию 01.04.2019
После доработки 02.07.2019
Принята к публикации 19.07.2019
Аннотация
Методом теории функционала плотности изучены механизмы реакций, протекающих при деоксигенации сложных эфиров на Pt и интерметаллидных Pt–Sn-катализаторах: реакций гидродеоксигенации (как по последовательному, так и по синхронному механизмам), реакций, начинающихся с разрыва одной из связей C–C, C–O, C–H или с синхронного разрыва связей C–O и C–H, а также реакций пиролиза сложных эфиров. Расчеты показали, что на Pt, а также на интерметаллидах Pt–Sn с небольшим содержанием олова активационные барьеры реакций, не сопровождающихся потерями углеродной массы на образование оксидов углерода и метана, сопоставимы с активационными барьерами реакций крекинга, приводящих к такой потере. По мере увеличения содержания Sn в интерметаллидах активационные барьеры реакций крекинга существенно повышаются, тогда как барьеры реакций, не ведущих к потере углеродной массы, увеличиваются незначительно. Этим может объясняться наблюдаемая высокая селективность биметаллических катализаторов Pt–Sn (1 : 5) в реакции гидродеоксигенации сложных эфиров и триглицеридов жирных кислот.
Одной из главных тенденций развития промышленной органической химии в XXI в. является постепенное замещение нефтяной сырьевой базы возобновляемыми источниками сырья. Так, на 2013 г. биомасса составляла примерно 10% от первичных энергоресурсов [1]; в том же году возобновляемое сырье составило те же 10% от сырьевой базы промышленной органической химии Европейского союза [2]. Значительная часть продукции промышленной органической химии приходится на топливо и топливные компоненты. Несмотря на активное развитие в ряде стран как солнечной, ветровой и других видов альтернативной энергетики, так и использование аккумуляторов, суперконденсаторов и топливных элементов для различных видов транспорта, полный отказ от традиционного моторного топлива (бензина, авиационного и дизельного топлива) в ближайшие десятилетия вряд ли возможен. Поэтому не теряет своей актуальности проблема совершенствования технологий получения углеводородных компонентов моторных топлив из возобновляемого сырья.
Биодизель получил широкое распространение с 90-х гг. XX в. Биодизелем называют топливо на основе моноалкиловых (метиловых, реже этиловых) эфиров жирных кислот, полученных из растительных или животных жиров [3]. Однако применение такого топлива в чистом виде вместо традиционного дизельного топлива сильно затруднено по целому ряду причин. Например, биодизель не пригоден для использования при низких температурах, окисляется при хранении и транспортировке, обладает коррозионной активностью [4]. При его сгорании образуется большее количество оксидов азота, а получающиеся твердые частицы имеют меньшие размеры, что позволяет им проникать на слизистую оболочку легких, провоцируя возникновение раковых заболеваний [5].
Помимо метиловых эфиров жирных кислот существуют и другие виды дизельного топлива из возобновляемого сырья: гриндизель, биодизель по Фишеру–Тропшу, уайтдизель, гибридный биодизель [6]. Гриндизель, представляющий собой смесь алифатических (преимущественно парафиновых) углеводородов, получают каталитическим гидрированием жиров (как правило, растительных). Он не содержит ароматических соединений, оксигенатов и серы. По своим эксплуатационным и экологическим показателям гриндизель превосходит биодизель и близок к нефтяному дизельному топливу [6].
При обработке триглицеридов жирных кислот (ТГЖК) водородом на гетерогенных катализаторах протекают параллельно несколько реакций – декарбонилирование, декарбоксилирование и гидродеоксигенация [7]:
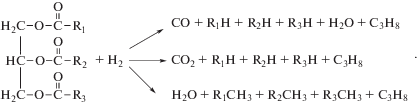
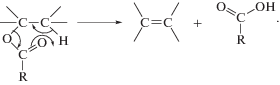
Также при повышенных температурах обычно идут реакции крекинга, протекающие по радикальному механизму и приводящие к алканам и алкенам как с четным, так и с нечетным количеством атомов углерода и ряду других продуктов [8, 9]. Вероятно, возможен и пиролиз по согласованному механизму, характерный для сложных эфиров [10]:
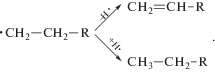
При термолизе сложноэфирной связи триглицеридов и последующих превращениях возможны следующие реакции [11]:
При этом из C–O-связей в ТГЖК и сложных эфирах наиболее лабильными являются связи со стороны глицерина, т.е. второй путь в реакции (III) является более вероятным, а из связей C–C наиболее неустойчивы связи, образуемые аллильными атомами углерода (при наличии остатков непредельных карбоновых кислот) [12].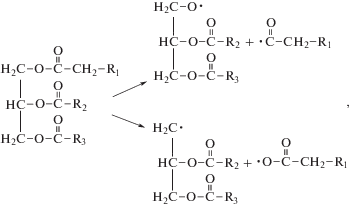
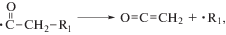
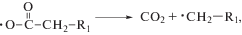
Очевидно, что при небольшом времени контакта селективности реакций будут определяться их активационными барьерами: чем выше барьер, тем меньше доля конкретной реакции. Также вполне очевидно, что активационные барьеры, а значит, и соотношение продуктов сильно зависят от катализатора.
Исторически первым катализатором гидрогенизации сложных эфиров был медно-хромовый катализатор CuO ⋅ CuCr2O4 (катализатор Адкинса), применявшийся для гидрогенизации сложных эфиров до насыщенных спиртов [13]. Роль оксида меди заключалась в ингибировании дальнейшего гидрирования спиртов до алканов, тогда как более активные частицы, такие как Ni и Co, способствовали такому гидрированию (через промежуточное образование алкена [14]). Оксид меди также способствует и насыщению C=C-связей, т.е. ненасыщенные сложные эфиры дают при гидрогенизации насыщенные спирты [14]. В настоящее время по причине большой токсичности в данном процессе используют медные катализаторы, промотированные другими оксидами металлов на различных носителях.
Гидродеоксигенация сложных эфиров и карбоновых кислот в алканы с тем же числом углеродных атомов на медно-никелевых катализаторах также известна с 30-х гг. прошлого века [15]. Вместе с тем Адкинс и др. [16] получали на никеле Ренея углеводороды из первичных спиртов с потерей одного атома углерода. Аналогично при использовании нанесенного никелевого катализатора, промотированного медью, деоксигенация сложных эфиров и ТГЖК проходит с потерей одного атома углерода [17].
При деоксигенации тристеарина из водорослей на никеле, нанесенном на Al2O3, ZrO2 или LaCeO2, а также на слоистом двойном гидроксиде Ni–Al существенно (особенно при увеличении температуры) влияние реакции крекинга на состав продуктов деоксигенации [18]. При деоксигенации стеарилстеарата на никеле, нанесенном на ZnO–Al2O3, по мере роста отношения Zn : Al в подложке падает селективность по н‑гептадекану и растет селективность по октадеканалю; при этом селективность по н‑октадекану также падает [19]. При гидрогенолизе пальмоядрового масла на Ni–MoS2/γ‑Al2O3 образуются углеводороды C10–C12 с селективностью 58%, а селективность гидродеоксигенации составляет ~63% [20]. В работе [21] было показано, что при обработке водородом отработанного пищевого масла на CoMoS основными реакционными путями являются декарбонилирование и декарбоксилирование. Гидродеоксигенация же является побочной реакцией и протекает преимущественно через промежуточное образование спирта, а не альдегида. Также показано, что льюисовские кислотные центры подложки могут играть важную роль в селективности процесса по реакции гидродеоксигенации. При гидроконверсии в жидкой фазе стеариновой и пальмитиновой кислот на β‑Mo2C, нанесенном на углеродные нанотрубки, удалось достичь выходов н-октадекана и н-гексадекана 91.2 и 92.2% соответственно [22, 23]. При гидроконверсии касторового масла на NiMo/Al2O3-катализаторе выход октадекана достигает 57%. При этом существенен также выход гептадекана, а доля продуктов крекинга заметно меньше [24]. Гидроконверсия пальмитиновой кислоты на Ni2P и MoP, взятых как в чистом виде, так и нанесенных на γ-Al2O3, при значительной степени конверсии приводит, в основном, к пентадекану. Существенная селективность по гексадекану наблюдалась только при низкой степени конверсии [25]. При гидроконверсии рапсового масла на никеле, промотированном MoO3 и WO3 и нанесенном на γ-Al2O3 или γ‑Al2O3–ZSM-5, образуются как продукты декарбонилирования и декарбоксилирования, так и продукты деоксигенации (с небольшим перевесом), а также продукты крекинга и изомеризации. При этом как в исходных, так и в промотированных образцах частицы интерметаллидов не обнаружены [26].
В работе [27], в которой изучали биметаллические катализаторы Cu–In, Cu–Ga, Cu–Sn, Cu–Tl, Cu–Cd, нанесенные на γ-Al2O3, было показано, что Cu–In обладает наибольшей активностью (среди упомянутых биметаллических систем) в реакции восстановления каприловой кислоты водородом в 1-октанол. Ni–In/γ-Al2O3 также обладает более высокой активностью, чем промышленный адкинсовский катализатор в той же реакции. Экспериментально было доказано присутствие интерметаллидов Cu2In и Ni2In в катализаторах, полученных добавлением 10% In2O3 к Cu/γ-Al2O3 и Ni/γ-Al2O3.
Гидрогенолиз пальмитиновой кислоты на промышленном 1% Pt/γ-Al2O3-катализаторе приводит, в основном, к пентадекану (выход ~70–75%) [28]. При использовании же Pt/SAPO-11 в качестве катализатора гидрогенолиза ТГЖК соотношение C17 : C18 составляет ~2.5 [29]. В зависимости от соотношения компонентов (Pt, Al, Si) в таком катализаторе меняется активность не только в реакциях деоксигенации, но и в реакциях крекинга и изомеризации [30], которые даже могут стать основными. При гидрогенолизе каприловой кислоты на Pd/SiO2 и Pd/С также преобладают реакции декарбонилирования, и основным продуктом оказывается н-гептан [31]. Аналогично при гидролизе рапсового масла с последующим гидрированием на Pd/C основным продуктом является гептадекан [32] как и при деоксигенации олеиновой кислоты на Pd/SBA‑16 [33]. При использовании Ni, нанесенного на полые шарики ZrO2, в продуктах гидрогенолиза также преобладает н-гептан [34].
Для повышения селективности гидродеоксигенации и подавления декарбоксилирования было предложено проводить реакцию в избытке CO2 [35], что позволило на Fe/SBA-15 получить октадекан из олеиновой кислоты с выходом 83.3%.
Хорошие результаты были получены при катализе биметаллическими системами на основе металлов из триады платины. При деоксигенации олеиновой кислоты на платине, промотированной WOx, нанесенной на ZrO2, была достигнута селективность по октадекану 89.4% при исчерпывающей конверсии [36]. При использованни же в качестве подложки γ-Al2O3 селективность по октадекану составила 67.1–80.8% [37]. Однако схожая каталитическая система WOx–Pt/TiO2 при обработке масла ятрофы проявляет существенно меньшую селективность в реакции гидродеоксигенации [38].
При обработке метилпальмитата водородом на Pt–Sn/SAPO-11 соотношение н- и изоалканов в продуктах зависит от соотношения Sn : Pt в катализаторе (при этом отдельной идентификации алканов C15 и C16 в продуктах не проводилось) [39]. При гидротермальном декарбоксилировании стеариновой, олеиновой и линолевой кислот на Pt/C образуются алканы C18 (преимущественно) и C17, при этом алкены в продуктах реакции не были обнаружены. При использовании в качестве катализаторов PtSnx/C, где х = 1/3, 1 и 3, выход алканов С17 увеличивается в 2–3 раза по сравнению с Pt/C, при этом активность катализатора падает; также в продуктах появляются алкены С18. При этом в ходе реакции источником водорода служит вода [40].
При использовании Ru–Sn/γ-Al2O3 удалось с селективностью ~75% получить олеиловый спирт из метиолеата, причем максимальный выход был достигнут при атомном соотношении Sn : Ru = 4 [41]. При этом была выдвинута следующая гипотеза о строении активных центров. При Sn : Ru < 4 частицы рутения и SnO2 перемешаны на поверхности носителя, и между ними нет взаимодействия. При 4 < Sn : Ru < 5.5 смешанные частицы рутения с оксидами олова образуются на поверхности частиц рутения, на поверхности носителя, а также возможно покрытие частиц рутения оксидами олова. При соотношении Sn : Ru > 5.5 происходит полное покрытие поверхности катализатора оксидами олова с уменьшением количества смешанных активных центров, ответственных за образование ненасыщенного спирта. Кроме частиц Ru0, на поверхности возможно формирование и частиц сплавов (интерметаллидов) Ru–Sn.
Таким образом, в настоящее время не существует промышленных каталитических процессов получения линейных алканов и α-олефинов из ТГЖК без потерь углеродной массы.
В лаборатории каталитических нанотехнологий ИНХС РАН были получены Pt–Sn-катализаторы, нанесенные на γ-Al2O3 методом пропитки, с содержанием Pt 0.4 мас. % и было показано, что катализатор с мольным соотношением Pt : Sn = 1 : 5 проявляет высокую селективность в реакции деоксигенации триглицеридов жирных кислот (ТГЖК) и сложных эфиров в углеводороды без потерь углеродной массы на образование С1, С2 и оксидов углерода [42–44]. Мольное соотношение Pt : Sn в исследованных катализаторах составляло 1 : 1, 1 : 3 и 1 : 5. В качестве предшественников для катализаторов с соотношениями 1 : 1 и 1 : 3 использовали (PPh3)2PtCl2 и SnCl2, а с соотношением 1 : 5 – гетерометаллический комплекс со связями Pt–Sn, полученный по оригинальной методике [45]. В работах [42–44] также было показано, что в частицах катализатора присутствуют нанокластеры интерметаллидов Pt–Sn, что дает основания предполагать, что именно биметаллические активные компоненты отвечают за отличные от промышленного катализатора Pt/γ-Al2O3 каталитические свойства полученной системы в исследуемой реакции. Однако природа активных центров и возможные механизмы протекания реакции экспериментальным путем установлены не были.
Ранее считалось [15], что гетерогенно-каталитическая гидродеоксигенация сложных эфиров протекает через промежуточное образование полуацеталя:
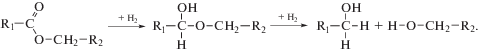
В работе [41] также предполагается, что восстановление сложных эфиров протекает через промежуточное образование полуацеталя:
Описаны и другие возможные механизмы: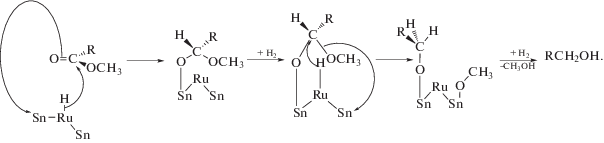
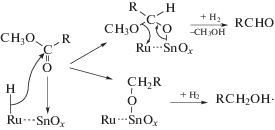
Еще одной возможной схемой протекания гидродеоксигенации ТГЖК, а также побочных процессов декарбонилирования и декарбоксилирования является промежуточное образование насыщенной карбоновой кислоты [46] (первая стадия – насыщение C=C-связей в кислотных фрагментах – опущена):
Более подробная схема параллельных реакционных каналов для декарбонилирования и декарбоксилирования (т.е. без гидродеоксигенации и крекинга) пропионовой кислоты на поверхности Ru(0001) представлена в работе [47]: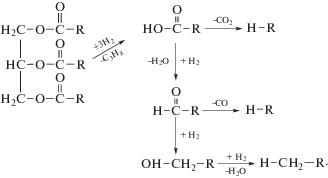
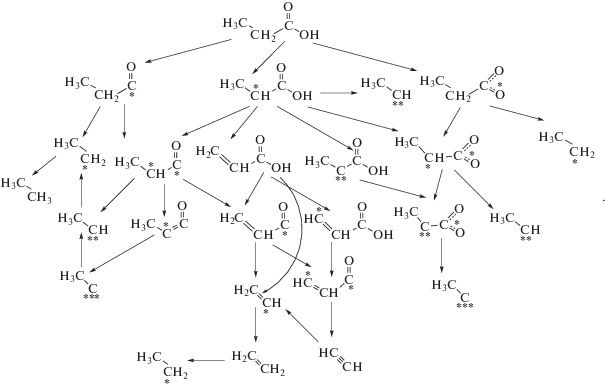
Таким образом, исследуемая реакция гидродеоксигенации ТГЖК и сложных эфиров на нанесенных Pt–Sn-катализаторах является весьма сложной для изучения из-за значительного количества реакционных каналов, относительный вклад каждого из которых, очевидно, зависит как от используемого катализатора, так и от условий проведения процесса. В таких случаях распространенным подходом является теоретическое моделирование, поэтому в данной работе были использованы методы вычислительной квантовой химии.
В предыдущей нашей работе [48] были изучены малые кластеры, моделирующие активные центры интерметаллидов Pt–Sn, рассмотрена молекулярная адсорбция водорода, диссоциация молекул водорода на платиновых активных центрах и переход адсорбированных атомов водорода с Pt на Sn. В ней было найдено, что введение Sn заметно уменьшает сродство Pt к водороду. При этом с ростом содержания Sn облегчается переход атомов H на атомы Sn, и был сделан вывод, что спилловер водорода в интерметаллидах Pt–Sn не должен приводить к существенному накоплению водорода на олове, т.е. атомы Sn, в отличие от атомов Pt, не могут, скорее всего, играть роль центров адсорбции водорода, активного в реакции деоксигенации.
В настоящей работе изучены возможные механизмы реакций с водородом и без него, в которые могут вступать ТГЖК при катализе металлической платиной и интерметаллидами Pt–Sn. Поскольку было установлено, что в ходе гидрогенолиза сложных эфиров на рассматриваемой каталитической системе образуются с высокой селективностью продукты гидродеоксигенации [43] (как и в случае с триглицеридами [44]), то логично предположить близкий характер протекания реакций в обоих случаях, и поэтому объектом изучения данной работы были выбраны механизмы реакций сложных эфиров.
ДЕТАЛИ РАСЧЕТОВ
Расчеты проводили в рамках теории функционала плотности (DFT) с использованием функционала электронной плотности PBE [49] и базисного набора SBK (с соответствующими остовными псевдопотенциалами для тяжелых атомов) [50, 51] по программе ПРИРОДА 17 [52, 53]. Для систем с открытыми электронными оболочками использовали неограниченный метод Кона–Шэма.
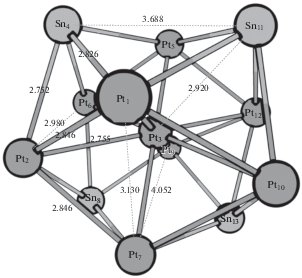
В предыдущей нашей работе [48] были предложены модельные кластеры для исследования взаимодействия активных центров поверхности интерметаллидов Pt–Sn с водородом. Эти же кластеры использовали и в данной работе, но для интерметаллида Pt3Sn был взят новый кластер – Pt9Sn4 (рис. 1), обладающий большей устойчивостью, вместо использованного ранее кластера Pt6Sn2. Расчеты показали, что основное спиновое состояние данного кластера триплетное.
Рис. 1.
Модельный кластер Pt9Sn4 интерметаллида PtSn3. Здесь и далее межатомные расстояния приведены в ангстремах.
В качестве модельного сложного эфира в расчетах использовали этилацетат.
Моделирование реакций проводили по нескольким методикам.
1. Строили первое приближение для предреакционного комплекса (адсорбция реагентов на поверхности катализатора), оптимизировали геометрию и результат тестировали на отсутствие мнимых частот в спектре гармонических колебаний. Далее проводили сканирование поверхности потенциальной энергии по координате образуемой или разрываемой связи. Если энергия системы не возрастала монотонно, получали возможные первые приближения для поиска переходного состояния (структура, соответствующая явно выраженному максимуму на кривой полной энергии в зависимости от заданной координаты, или же, при наличии срыва на кривой, несколько точек – последняя точка перед срывом и промежуточные структуры, получающиеся в начале оптимизации геометрии при следующем шаге после срыва). После расчета частот гармонических колебаний всех предложенных структур выбирали наиболее пригодные для поиска переходного состояния структуры (т.е. структуры, для которых наибольшая мнимая частота существенно превосходила остальные мнимые частоты, если они есть, и при этом соответствовала возможной реакции). Далее проводили поиск переходного состояния с последующим тестированием его по наличию единственной мнимой частоты в спектре гармонических колебаний и ее соответствию протекающей реакции. После этого проводили спуск по координате реакции в долины реагентов и продуктов (с последующей дооптимизацией структур). В случае нескольких стадий интермедиат, образующийся после одной стадии, служил отправной точкой для сканирования поверхности потенциальной энергии следующей стадии.
2. Найденное ранее переходное состояние “переносили” на другие активные центры или другой модельный кластер. Далее проводили анализ частот гармонических колебаний, после чего действовали аналогично описанному в п. 1.
3. Аналогично могли “переносить” и предреакционные комплексы и интермедиаты. При необходимости поверхности потенциальной энергии могли сканироваться в направлении от интермедиата к предреакционному комплексу, если прямое сканирование не позволяло получить хорошего начального приближения для поиска переходного состояния.
Использование методов, описанных в пп. 2 и 3, позволило найти существенное количество возможных переходных состояний (и соответствующих им реакционных каналов), которые было бы невозможно обнаружить, используя только “классический” метод, описанный в п. 1.
Значения энтальпии рассчитывали для температуры 400°С сложением полной энергии и энтальпийного вклада, полученного по модели гармонических колебаний. При такой температуре и общем давлении в системе не более нескольких атмосфер реагенты и продукты реакции находятся в газообразном состоянии (поэтому не требуется учитывать влияние растворителя).
Так как в нашей предыдущей работе [48] было показано, что диссоциация молекулярного водорода на атомах платины во всех изученных кластерах протекает с незначительными активационными барьерами, то при расчете энтальпий адсорбции в предреакционных комплексах с атомами водорода на атомах платины полагали, что эти комплексы образуются из кластера, молекулы этилацетата и молекулы водорода.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Расчеты показали, что реакция гидродеоксигенации начинается со стадии переноса атома водорода с атома платины на карбонильный атом углерода. При этом такой механизм справедлив для всех рассматриваемых модельных кластеров. Структуры предреакционного комплекса, переходного состояния и интермедиата для кластера Pt6Sn5 представлены на рис. 2а–2в. Дальнейшее протекание реакции возможно по следующим трем каналам. 1) Перенос второго атома водорода на атом –O– с разрывом связи C–O и образованием этанола и ацетальдегида (рис. 2г, 2д). 2) Перенос второго атома водорода на атом O= с образованием полуацеталя и последующим его разложением на этанол и ацетальдегид (рис. 2е–2и). 3) Разрыв связи C–O с образованием ацетальдегида и оксиэтильной группы на атоме Pt (рис. 2к–2п). В последнем случае для получения этанола, очевидно, необходима стадия гидрирования, для чего потребуется перенос водорода на соответствующий атом платины или хемосорбция еще одной молекулы водорода. Далее возможные последующие стадии (десорбция ацетальдегида, адсорбция дополнительной молекулы водорода, образование этанола и его десорбция, дегидратация этанола, гидрирование этилена, а также реакция восстановления ацетальдегида до этанола), вероятно, протекают с существенно меньшими активационными барьерами, и поэтому их не рассчитывали в данной работе. Энтальпии активации основных стадий данных реакционных каналов, а также стандартные изменения энтальпии для каждой из стадий суммированы в табл. 1. (Активационные барьеры промежуточных стадий, не приведенные в табл. 1, не рассчитывались, так как барьеры внутреннего вращения этилацетата вряд ли могут составлять более нескольких ккал/моль, а значит, заведомо не могут являться значимыми в исследуемых реакциях.)
Рис. 2.
Структуры, соответствующие локальным минимумам и переходным состояниям в реакции гидродеоксигенации этилацетата на кластере Pt6Sn5: предреакционный комплекс (а), переходное состояние I (б) и интермедиат I (в). Переходное состояние II (г) и постреакционный комплекс (д) двухстадийной реакции гидродеоксигенации этилацетата на кластере Pt6Sn5. Переходное состояние II (е), интермедиат II (ж), переходное состояние III (з) и постреакционный комплекс (и) реакции гидродеоксигенации этилацетата на кластере Pt6Sn5, протекающей через полуацеталь. Переходное состояние II (к), интермедиат II (л), переходное состояние III (м), интермедиат III (н), переходное состояние IV (о) и постреакционный комплекс (п) реакции гидродеоксигенации этилацетата на кластере Pt6Sn5, протекающей через разрыв связи C–O.
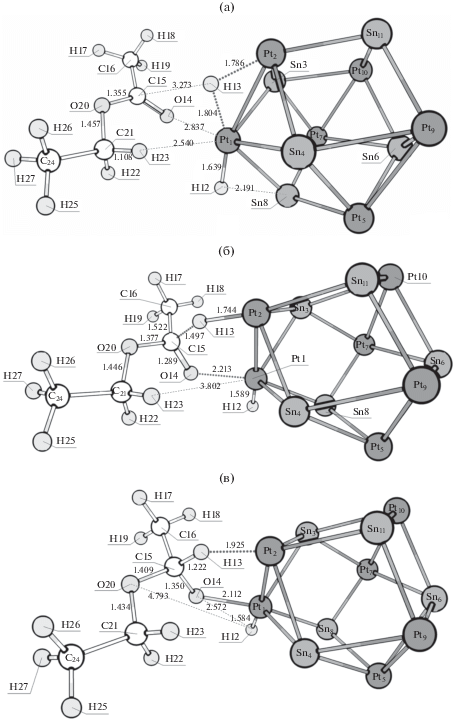
Рис. 2.
Продолжение.
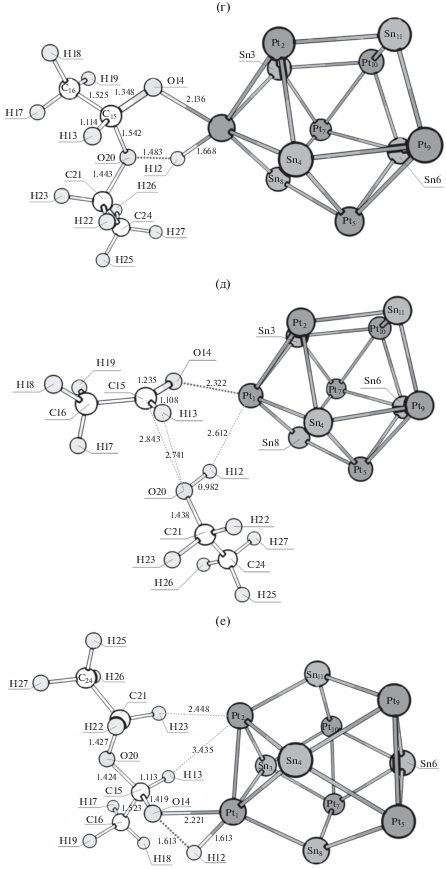
Рис. 2.
Продолжение.
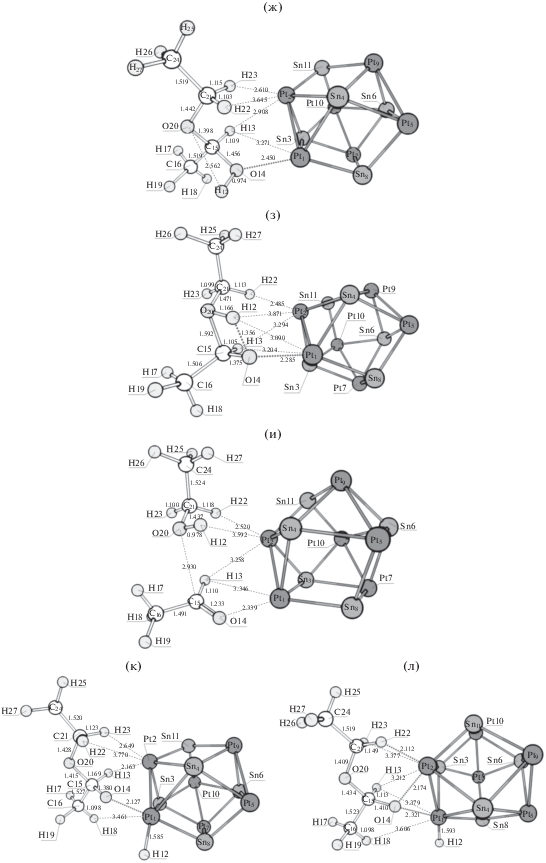
Рис. 2.
Окончание.
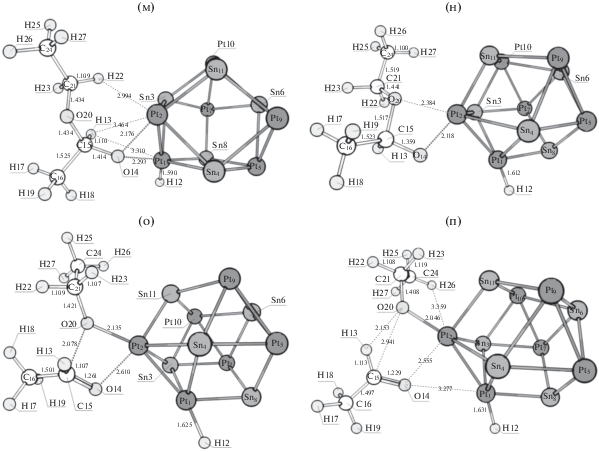
Таблица 1.
Гидродеоксигенация этилацетата на модельных кластерах, начинающаяся с переноса атома водорода на карбонильный углерод
| Модельный кластер, спиновая мультиплетность (M) и начальная координация этилацетата | Образование предреакционного комплекса из кластера, молекулы CH3COOC2H5 и молекулы H2 | Перенос первого атома водорода с Pt на C= | Промежуточные стадии* | Перенос второго атома водорода с Pt на –O– с образованием этанола и ацетальдегида | ||
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | ||||||
| Pt6 (Pt) M = 5 (для переноса второго атома водорода необходима хемосорбция второй молекулы H2) |
–46.5 | 20.6 | 14.6 | –13.5/1.0** | 2.1 | –8.6 |
| Pt9Sn4 (Pt3Sn) M = 3 CH3COOC2H5 на Pt CH3COOC2H5 на Pt CH3COOC2H5 на Pt |
–36.7 –40.9 –36.1 |
25.8 21.5 21.6 |
16.2 20.2 20.6 |
0.7 0.7 –7.2 |
14.2 9.8 7.8 |
5.4 –3.0 3.6 |
| Pt6Sn5 (PtSn) M = 1 CH3COOC2H5 на Pt CH3COOC2H5 на Sn CH3COOC2H5 на Sn |
–4.7 –4.2 0.8 |
13.9 16.2 10.7 |
14.5 –1.8 2.8 |
–4.6 0.2 0.1 |
9.3 9.2 9.5 |
–4.6 7.6 7.8 |
| Pt6Sn8 (Pt2Sn3, PtSn2) M = 3 CH3COOC2H5 на Sn | 0.9 | 12.0 | 7.8 | 0.4 | 8.2 | –4.7 |
| Pt2Sn6 (PtSn4) M = 1 CH3COOC2H5 на Sn |
–17.7 | 13.6 | 4.8 | – | 12.3 | 12.2 |
| * Прочерк в данном столбце означает, что промежуточных стадий между двумя основными нет. ** Для двух последовательных промежуточных стадий. | ||||||
| Модельный кластер и начальная координация этилацетата | Промежуточные стадии* | Перенос второго атома водорода (H на O=) с образованием полуацеталя | Промежуточные стадии* | Распад полуацеталя** | ||
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | ||||||
| Pt6 (Pt) M = 5 | –14.4 | 23.8 | 5.3 | – | – | – |
| Pt9Sn4 (Pt3Sn) M = 3 CH3COOC2H5 на Pt |
0.9 | 25.1 | 5.5 | –0.9 | 27.0 | –0.9 |
| CH3COOC2H5 на Pt | – | 20.5 | 0.5 | 5.5 | 27.3 | –5.0 |
| CH3COOC2H5 на Pt | –7.2 | 13.2 | –5.1 | 3.1 | 28.6 | –4.6 |
| Pt6Sn5 (PtSn) M = 1 | ||||||
| CH3COOC2H5 на Pt | –7.7 | 21.7 | –7.4 | – | 26.9 | 5.6 |
| CH3COOC2H5 на Sn CH3COOC2H5 на Sn |
–0.8 –2.2 |
15.1 18.5 |
5.3 10.0 |
–3.7 –16.7 |
26.8 36.7 |
5.6 6.4 |
| Pt6Sn8 (Pt2Sn3, PtSn2) M = 3 CH3COOC2H5 на Sn | – | 10.8 | –11.4 | – | 30.3 | 7.9 |
| Pt2Sn6 (PtSn4) M = 1 CH3COOC2H5 на Sn |
–2.1 | 23.2 | 12.1 | –1.2 | 30.7 | 5.4 |
| * Прочерк в данном столбце означает, что промежуточных стадий между двумя основными нет. ** Прочерк в данном столбце означает, что найти реакционный канал распада полуацеталя для соответствующего кластера не удалось. | ||||||
| Модельный кластер и начальная координация этилацетата | Промежуточные стадии | Разрыв связи C–O **** | ||||
| $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673*** | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |||
| ккал/моль | ||||||
| Pt6 (Pt) M = 5 | – | – | – | – | ||
| Pt9Sn4 (Pt3Sn) M = 3 CH3COOC2H5 на Pt |
* | –0.5 | 1.9 | –4.3 | ||
| CH3COOC2H5 на Pt | – | – | 9.8 | 1.1 | ||
| CH3COOC2H5 на Pt | * | –7.2 | 11.7 | 5.9 | ||
| Pt6Sn5 (PtSn) M = 1 CH3COOC2H5 на Pt |
1.1/0.6** | –7.6/–4.1** | 4.1 | –0.1 | ||
| CH3COOC2H5 на Sn | – | – | – | – | ||
| CH3COOC2H5 на Sn | – | – | – | – | ||
| Pt6Sn8 (Pt2Sn3, PtSn2) M = 3 | * | –0.2 | 9.9 | 5.6 | ||
| CH3COOC2H5 на Sn | * | –0.8 | 10.1 | 6.2 | ||
| Pt2Sn6 (PtSn4) M = 1 CH3COOC2H5 на Sn |
* | –1.1 | 13.2 | 6.5 | ||
| * Переходные состояния для ряда промежуточных стадий найти не удалось. ** Для двух последовательных промежуточных стадий. *** Прочерк в данном столбце означает (при заполненных двух следующих столбцах), что промежуточных стадий между двумя основными нет. **** Прочерки в данных столбцах означают, что соответствующие реакционные каналы обнаружить не удалось. | ||||||
Активационный барьер первой (общей) стадии этой группы реакционных каналов уменьшается по мере роста содержания олова в кластерах. При этом для канала через полуацеталь лимитирующей стадией будет распад полуацеталя (вероятно, в газовой фазе такому распаду соответствует существенно больший реакционный барьер, нежели в жидкой). Для обоих каналов, связанных с разрывом связи C–O для всех рассмотренных кластеров лимитирующей стадией является стадия переноса первого атома водорода. Также смоделирована газофазная реакция разложения полуацеталя этанола и ацетальдегида на этанол и ацетальдегид. Расчетное значение энтальпии активации составило 34.0 ккал/моль, а стандартного изменения энтальпии – 5.7 ккал/моль. Таким образом, влияние катализатора на эту стадию не столь существенно, понижение активационного барьера заметно только при малом содержании олова (однако смоделировать такую же реакцию на кластере Pt6 не удалось).
Еще одной возможной реакцией с участием водорода является разрыв связи C–O с одновременным переносом атома водорода с Pt на карбонильный углерод или на сложноэфирный кислород. Такие реакционные каналы действительно были найдены. Структуры предреакционного комплекса, переходного состояния и интермедиата для кластера Pt6Sn5 для каждого из вариантов представлены на рис. 3 и 4. Энтальпии активации первых стадий данных реакционных каналов, а также стандартные изменения энтальпии для первых стадий приведены в табл. 2.
Рис. 3.
Структуры, соответствующие локальным минимумам и переходному состоянию I в реакции гидродеоксигенации этилацетата (с одновременным разрывом связи C–O и переносом водорода с платины на карбонильный углерод) на кластере Pt6Sn5: предреакционный комплекс (а), переходное состояние I (б) и интермедиат I (в) канала с переносом водорода на атом C=.
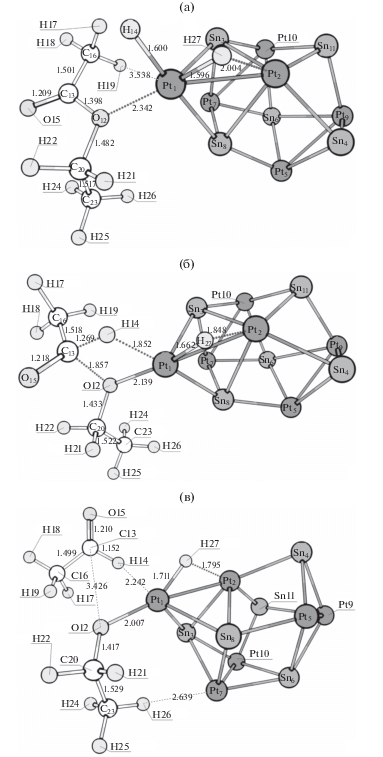
Рис. 4.
Структуры, соответствующие локальным минимумам и переходному состоянию I в реакции гидродеоксигенации этилацетата (с одновременным разрывом связи C–O и переносом водорода с платины на карбонильный углерод) на кластере Pt6Sn5: предреакционный комплекс (а), переходное состояние I (б) и интермедиат I (в) канала с переносом водорода на атом –O–.
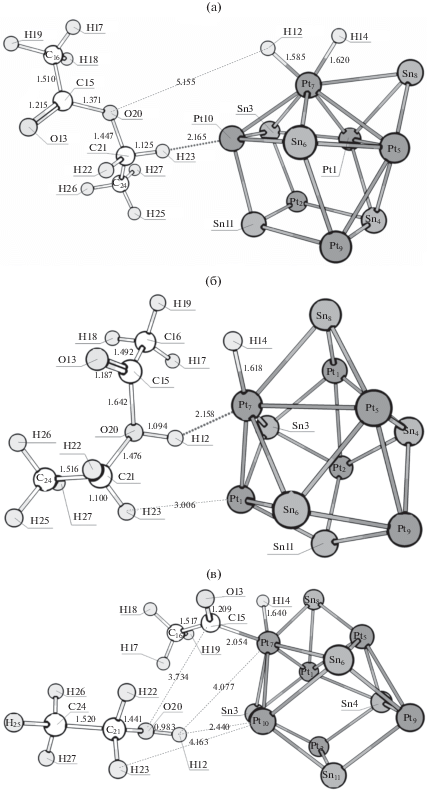
Таблица 2.
Гидродеоксигенация этилацетата на модельных кластерах через синхронный разрыв связи C–O и перенос атома водорода
| Модельный кластер | Образование предреакционного комплекса из кластера, молекулы CH3COOC2H5 и молекулы H2 | Перенос атома водорода на C= с синхронным разрывом связи C–O и образованием ацетальдегида и поверхностного алкоголята | Образование предреакционного комплекса из кластера, молекулы CH3COOC2H5 и молекулы H2 | Перенос атома водорода на –O– с синхронным разрывом связи C–O и образованием уксусной кислоты и поверхностного ацетила | ||
|---|---|---|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | ||||||
| Pt6 (Pt) | –35.4 | 19.9 | 13.9 | –52.8 | 28.9 | 14.6 |
| Pt9Sn4 (Pt3Sn) | –25.4 | 24.4 | 8.3 | –41.5/–46.9* | 21.2/37.6* | 6.8/5.9* |
| Pt6Sn5 (PtSn) | 1.9 | 18.2 | 11.1 | –15.7 | 40.4 | 7.2 |
| Pt6Sn8 (Pt2Sn3, PtSn2) | 12.6 | 25.2 | 14.5 | – | – | – |
| Pt2Sn6 (PtSn4) | – | – | – | – | – | – |
Как можно видеть, первый реакционный канал (с переносом водорода на C=) возможен для всех кластеров, кроме Pt2Sn6, а второй (с переносом водорода на –O–) – только для кластеров с малым содержанием олова. При этом для первого канала по мере роста содержания олова в кластере реакционные барьеры оказываются выше, чем для реакционного канала, в котором разрыв связи C–O происходит одновременно с переносом второго атома водорода. Второй канал становится маловероятным уже для кластера Pt6Sn5, а для кластеров Pt6Sn8 и Pt2Sn6 обнаружить его не удалось.
Также среди реакций с участием водорода стоит отметить разрыв связи C–O с одновременным переносом атома водорода с Pt на метиленовый углерод с образованием этана и поверхностного ацетата. Структуры, соответствующие предреакционному комплексу, переходному состоянию и интермедиату, представлены на рис. 5, а расчетные данные по энтальпиям – в табл. 3. Активационные барьеры для всех кластеров составляют не менее 20 ккал/моль.
Рис. 5.
Структуры, соответствующие локальным минимумам и переходному состоянию I в реакции гидродеоксигенации этилацетата (с одновременным разрывом связи C–O и переносом водорода с платины на метиленовый углерод) на кластере Pt2Sn6: предреакционный комплекс (а), переходное состояние I (б) и интермедиат I (в) канала с переносом водорода на атом –O–.
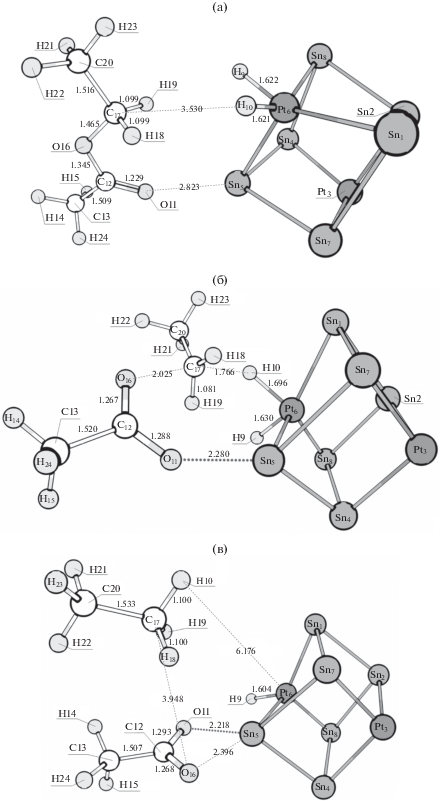
Таблица 3.
Гидродеоксигенация этилацетата на модельных кластерах через синхронный разрыв связи C–O и перенос атома водорода на метиленовый углерод
| Модельный кластер и особенности строения продукта или адсорбции реагентов | Образование предреакционного комплекса | Синхронный разрыв связи C–O и перенос атома H с образованием этана и поверхностного ацетата | |
|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | |||
| Pt6 (Pt) | –50.8 | 20.4 | –26.5 |
| Pt9Sn4 (Pt3Sn) поверхностный ацетат на Pt и Sn поверхностный ацетат на 2 атомах Pt поверхностный ацетат на Sn |
–44.0 –39.7 –32.7 |
26.4 28.3 33.5 |
–3.1 –19.4 –15.7 |
| Pt6Sn5 (PtSn) CH3COOC2H5 адсорбируется по O= на Pt CH3COOC2H5 адсорбируется по O= на Sn |
–5.6 4.5 |
21.4 20.4 |
–25.6 –32.9 |
| Pt6Sn8 (Pt2Sn3, PtSn2) (CH3COOC2H5 адсорбируется по O= на Sn) |
1.3 | 26.7 | –18.9 |
| Pt2Sn6 (PtSn4) (CH3COOC2H5 адсорбируется по O= на Sn) |
–16.8 | 26.4 | –23.0 |
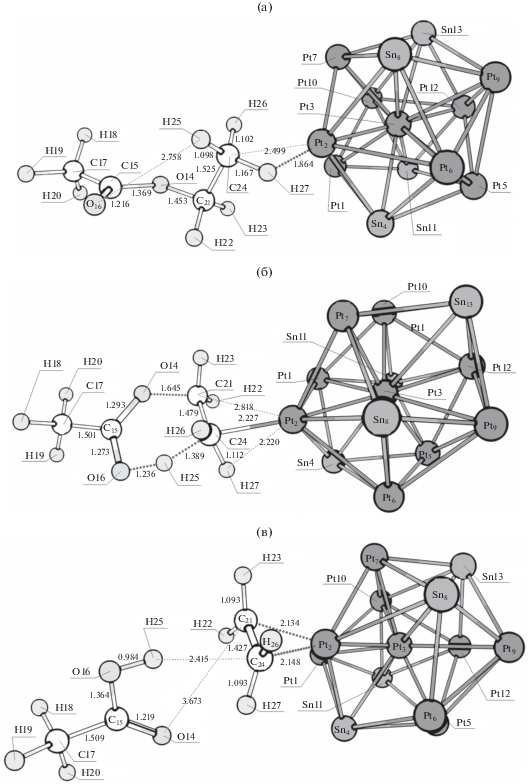
Другой возможной начальной стадией данного процесса, как ни парадоксально, может являться перенос одного из атомов водорода с этилацетата на атом Pt катализатора с последующим разрывом одной из связей. Расчеты показали, что дегидрогенизации может подвергаться любой из связанных с водородом атомов углерода в этилацетате. Структуры предреакционных комплексов, переходных состояний и интермедиатов для случаев дегидрогенизации метильного атома углерода в этильной группе, метиленового атома углерода в этильной группе и метильного атома углерода в ацильной группе представлены на рис. 6–8 соответственно. Энтальпии активации первых стадий данных реакционных каналов, а также стандартные изменения энтальпии для первых стадий приведены в табл. 4–6 соответственно.
Рис. 6.
Структуры, соответствующие предреакционному комплексу (а), переходному состоянию I (б), интермедиату I (в), переходному состоянию II (г) и постреакционному комплексу (д) в реакции дегидрирования этилацетата по метильному углероду этильной группы с последующим разрывом связи C–O на кластере Pt6Sn5.
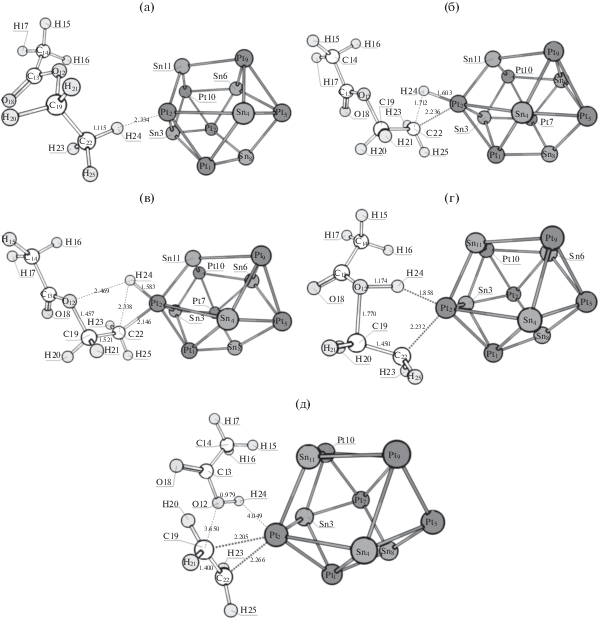
Рис. 7.
Структуры, соответствующие предреакционному комплексу (а), переходному состоянию I (б) и интермедиату I (в) в реакции дегидрирования этилацетата по метиленовому углероду этильной группы, а также интермедиату Iа (г), переходному состоянию II (д) и постреакционному комплексу (е) последующего разрыва связи C–O с образованием ацетальдегида и поверхностного ацетила на кластере Pt6Sn5. Структуры, соответствующие переходному состоянию II (ж) и постреакционному комплексу (з) последующего разрыва связи C–O с образованием уксусной кислоты и поверхностного этилидена на кластере Pt6Sn5.
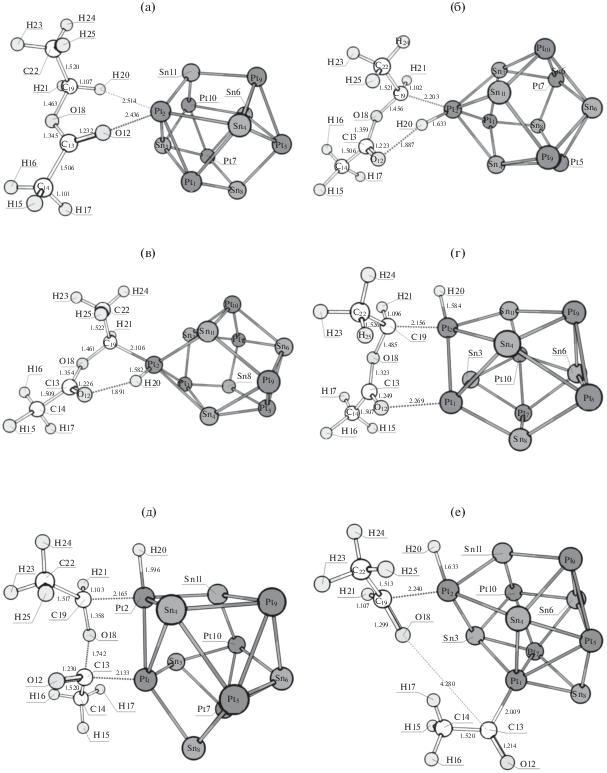
Рис. 7.
Окончание
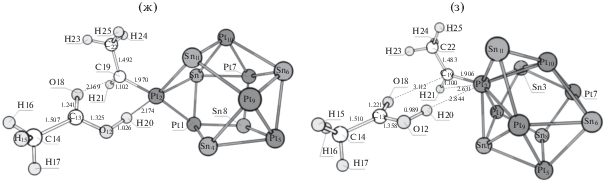
Рис. 8.
Структуры, соответствующие предреакционному комплексу (а), переходному состоянию I (б), интермедиату I (в), интермедиату Iа (г), переходному состоянию II (д) и постреакционному комплексу (е) в реакции дегидрирования этилацетата по метильному углероду ацетильной группы с последующим разрывом связи C–O с образованием кетена и поверхностной этоксильной группы на кластере Pt9Sn4.
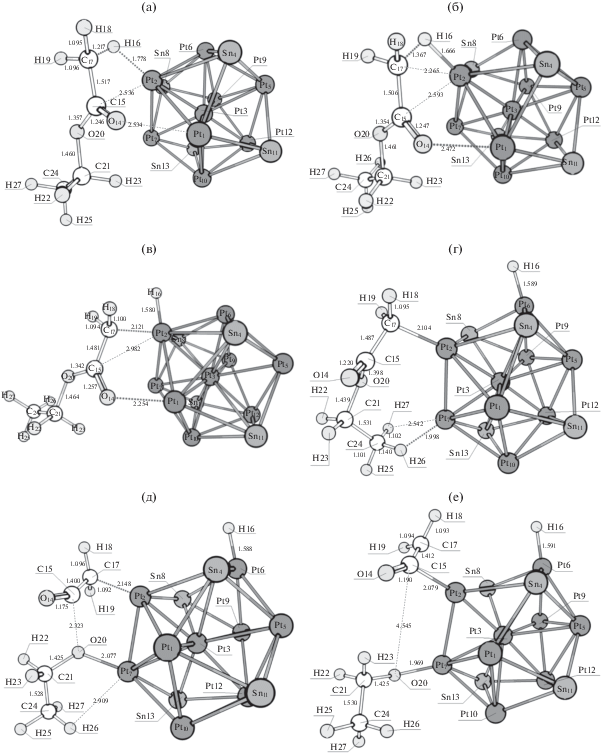
Таблица 4.
Дегидрирование этилацетата по метильному атому углерода этильной группы на модельных кластерах с последующим разрывом одной из связей
| Модельный кластер | Адсорбция молекулы CH3COOC2H5 на кластере | Перенос атома водорода с C на Pt | Промежуточная стадия* | Разрыв связи C–O с образованием уксусной кислоты и этилена** |
Промежуточная стадия* | Разрыв связи C–O с образованием этилена и поверхностного ацетата** |
|||
|---|---|---|---|---|---|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | |||||||||
| Pt6 (Pt) 2 разных варианта начальной координации |
–16.5 –17.2 |
–0.2 –0.0 |
–15.8 –13.1 |
– – |
– – |
– – |
1.7 1.7 |
14.3 16.7 |
–28.5 –43.5 |
| Pt9Sn4 (Pt3Sn) (CH3COOC2H5 адсорбируется по O= на Pt) |
–19.9 | 7.8 | 0.6 | – | – | – | 2.8 | 10.6 | –25.2 |
| Pt6Sn5 (PtSn) | 0.8 | 20.4 | 18.2 | – | 15.8 | –18.3 | – | – | – |
| Pt6Sn8 (Pt2Sn3, PtSn2) | –0.3 | 25.5 | 21.4 | 0.8 | 17.4 | –15.9 | – | – | – |
| Pt2Sn6 (PtSn4) (CH3COOC2H5 адсорбируется по O= на Sn) |
–2.3 | 19.2 | 9.6 | – | – | – | – | 19.9 | –18.6 |
Таблица 5.
Дегидрирование этилацетата по метиленовому атому углерода на модельных кластерах с последующим разрывом одной из связей
| Модельный кластер и начальная координация этилацетата | Адсорбция молекулы CH3COOC2H5 на кластере | Перенос атома водорода с C на Pt | Промежуточная стадия* | Разрыв связи C–O с образованием ацетальдегида и поверхностного ацетила** | Промежуточная стадия* |
Разрыв связи C–O
с образованием уксусной кислоты и поверхностного этилидена** |
|||
|---|---|---|---|---|---|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | |||||||||
| Pt6 (Pt) (координирует по O= на Pt и H CH2-группы на соседней Pt) |
–17.0 | 0.5 | –20.9 | 4.3 | 11.7 | –5.1 | – | – | – |
| Pt9Sn4 (Pt3Sn) | –6.0 | 3.6 | –0.1 | –5.1 | 11.3 | –10.5 | –8.9 | 7.4 | 4.6 |
| (координирует по H CH2-группы на Pt) | –5.4 | 1.5 | –6.8 | –5.1 | 16.0 | 6.5 | – | 5.9 | 0 |
| Pt6Sn5 (PtSn) | –3.1 | 24.8 | 23.3 | –8.0 | 18.5 | –4.3 | – | 5.1 | –14.7 |
| (координирует по O= на Pt) | –4.7 | 25.0 | 23.1 | –5.9 | 12.9 | –9.2 | –0.5 | 6.6 | 4.8 |
| Pt6Sn8 (Pt2Sn3, PtSn2) (координирует по O= на Pt) |
–2.1 | 25.5 | 24.3 | – | – | – | 2.8 | 2.6 | –0.9 |
| Pt2Sn6 (PtSn4) (координирует по H CH2-группы на Pt) |
7.8 | 13.8 | 6.6 | – | – | – | –2.1 | 6.0 | 4.7 |
Таблица 6.
Дегидрирование этилацетата по ацетильному атому углерода на модельных кластерах с последующим разрывом одной из связей C–O
| Модельный кластер и особенности протекания реакции | Адсорбция молекулы CH3COOC2H5 на кластере | Перенос атома водорода с C на Pt | Промежуточная стадия*** | Разрыв связи C–O с переносом H с Pt на O и образованием кетена и этанола**** | Промежуточная стадия*** | Разрыв связи C–O с образованием кетена и поверхностной этоксильной группы**** | |||
|---|---|---|---|---|---|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | |||||||||
| Pt6 (Pt)* (при дегидрировании O= идет на Pt) |
–20.9 –22.7 |
–1.1 0.4 |
–21.6 –16.9 |
– – |
– – |
– – |
0.4 7.9 |
39.0 20.3 |
20.8 17.0 |
| Pt9Sn4 (Pt3Sn) (при дегидрировании O= идет на Pt) |
–5.2 | –1.5 | –16.2 | – | – | – | –7.0 | 18.2 | 4.9 |
| Pt6Sn5 (PtSn)** | |||||||||
| (при дегидрировании O= идет на Sn) | –5.2 | 26.4 | 16.7 | –0.2 | 15.8 | 8.5 | – | – | – |
| (при дегидрировании O= идет на Pt) | –5.2 | 17.6 | 8.5 | – | – | – | – | 24.9 | 23.0 |
| Pt6Sn8 (Pt2Sn3, PtSn2)* | –0.2 | 22.7 | 12.8 | – | – | – | –0.2 | 20.8 | 22.1 |
| (при дегидрировании O= идет на Sn) | –0.4 | 24.0 | 12.8 | – | – | – | – | 20.8 | 22.1 |
| Pt2Sn6 (PtSn4) (при дегидрировании O= идет на Sn) |
–2.3 | 18.2 | 17.1 | – | – | – | –3.3 | 14.9 | 12.0 |
* Рассчитано два похожих варианта. ** Рассчитано два разных варианта протекания реакции, начинающихся с одинакового предреакционного комплекса. *** Прочерк в данном столбце означает (при заполненных двух следующих), что промежуточных стадий между двумя основными нет. **** Прочерки в данных столбцах означают, что соответствующие реакционные каналы не реализуются.
В реакции дегидрогенизации по метильному атому углерода этильной группы молекула этилацетата координируется карбонильным кислородом по платине или по олову или же непосредственно водородом по платине. Энергия активации переноса водорода на чистую платину близка к нулю, она немного возрастает при переходе к кластеру Pt9Sn4 и существенно возрастает при переходе к остальным кластерам. Второй стадией данного реакционного канала (не считая промежуточных стадий, связанных с внутренним вращением, реакционные барьеры которых мы не определяли) является разрыв связи C–O с образованием этилена и уксусной кислоты (или поверхностного ацетата). Лимитирующей в данном случае может являться как первая стадия (при существенном содержании олова), так и вторая (при небольшом содержании олова). Для кластера Pt2Sn6 барьеры стадий оказались близки.
В реакции дегидрогенизации по метиленовому атому углерода этильной группы молекула этилацетата может координироваться карбонильным кислородом по платине или же непосредственно переносимым водородом по платине, что, впрочем, не влияет на структуру переходного состояния первой стадии, но оказывает влияние на активационный барьер. На второй стадии происходит разрыв одной из связей C–O, в результате чего могут с незначительным барьером образовываться уксусная кислота и поверхностный этилиден или же с более существенным барьером – ацетальдегид и поверхностный ацетил (этот вариант второй стадии реализуется только для кластеров с контактами Pt–Pt).
В реакции дегидрогенизации по метильному атому углерода ацетильной группы молекула этилацетата координируется непосредственно переносимым водородом по платине. При этом в ходе дегидрогенизации молекула может координироваться карбонильным кислородом как по платине, так и по олову. На кластерах Pt6 и Pt9Sn4 барьер первой стадии оказывается незначительным, для остальных кластеров он составил более 17 ккал/моль. На второй стадии (являющейся лимитирующей для первых двух кластеров) происходит разрыв связи C–O с образованием кетена и этанола или поверхностной этоксильной группы. При этом для чистой Pt (в одном из вариантов) барьер второй стадии весьма существенен, а в остальных случаях он составляет не более 25 ккал/моль, что для кластеров Pt6Sn5, Pt6Sn8 и Pt2Sn6 сопоставимо с энтальпией активации первой стадии.
Для всех вариантов дегидрогенизации также были предприняты попытки найти канал разрыва связи C=O на второй стадии. Расчеты показали, что такой канал теоретически возможен для всех кластеров и всех видов первой стадии. При этом активационные барьеры составили не менее 48 ккал/моль, что делает такую реакцию маловероятной. Также были предприняты попытки найти канал разрыва одной из связей C–C на второй стадии, но таких каналов найти не удалось.
Следующей группой реакционных каналов являются реакционные каналы, начинающиеся с разрыва связи C–O в этилацетате. Структуры предреакционных комплексов, переходных состояний и интермедиатов для отщепления этильной и оксиэтильной групп представлены на рис. 9 и 10 соответственно. Энтальпии активации первых стадий данных реакционных каналов, а также стандартные изменения энтальпии для первых стадий приведены в табл. 7 и 8 соответственно.
Рис. 9.
Структуры, соответствующие предреакционному комплексу (а), переходному состоянию I (б) и интермедиату I (в) разрыва связи C–O с отщеплением этильной группы на кластере Pt6Sn5.
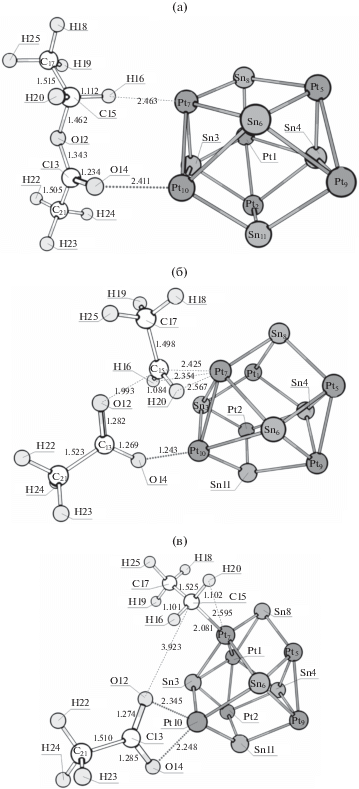
Рис. 10.
Структуры, соответствующие предреакционному комплексу (а), переходному состоянию I (б) и интермедиату I (в) разрыва связи C–O с отщеплением оксиэтильной группы на кластере Pt9Sn4.
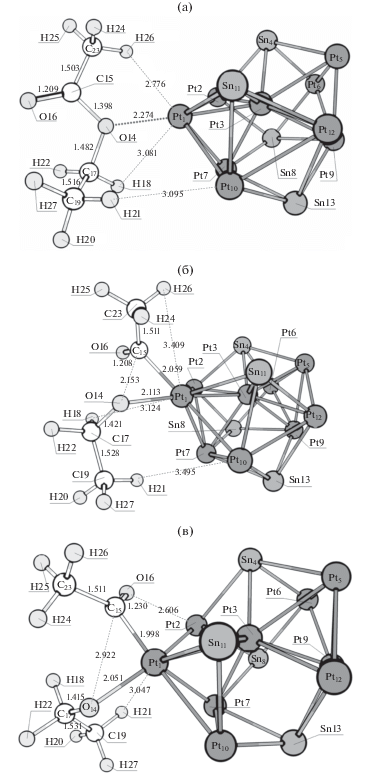
Таблица 7.
Разрыв связи C–O в этилацетате (отщепление этильной группы)
| Модельный кластер и начальная координация этилацетата | Адсорбция молекулы CH3COOC2H5 на кластере | Разрыв связи C–O | |
|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | |||
| Pt6 (Pt) | – | – | – |
| Pt9Sn4 (Pt3Sn)* (CH3COOC2H5 адсорбируется по H CH2-группы на Pt) |
–7.4/–5.9 | 20.6/23.5 | –23.6/–18.8 |
| Pt6Sn5 (PtSn) CH3COOC2H5 адсорбируется по O= на Pt CH3COOC2H5 адсорбируется по O= на Pt и по H CH2-группы на соседней Pt CH3COOC2H5 адсорбируется по O= на Sn |
–3.6 –4.3 4.4 |
30.7 25.0 24.0 |
–19.2 –10.2 14.1 |
| Pt6Sn8 (Pt2Sn3, PtSn2) CH3COOC2H5 адсорбируется по O= на Pt CH3COOC2H5 адсорбируется по O= на Sn и по H CH3-группы Et на соседней Pt CH3COOC2H5 адсорбируется по O= на Sn и по H CH2-группы на соседней Pt |
1.7 –0.3 –0.9 |
25.1 38.4 29.6 |
19.1 37.9 1.4 |
| Pt2Sn6 (PtSn4) (CH3COOC2H5 адсорбируется по O= на Sn и по H CH2-группы на соседней Pt) |
–5.0 | 24.7 | –18.7 |
Таблица 8.
Разрыв связи C–O в этилацетате (отщепление оксиэтильной группы)
| Модельный кластер и начальная координация этилацетата | Адсорбция молекулы CH3COOC2H5 на кластере | Разрыв связи C–O | |
|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | |||
| Pt6 (Pt) (CH3COOC2H5 координируется по C=O на соседних атомах Pt) |
–21.7 | 10.5 | –8.9 |
| Pt9Sn4 (Pt3Sn) (CH3COOC2H5 координируется по –O– на Pt) CH3CO координируется на Pt по C= CH3CO координируется на 2 атома Pt по C= и O= |
–9.0 –12.3 |
8.1 8.6 |
4.4 7.3 |
| Pt6Sn5 (PtSn) CH3COOC2H5 координируется электростатически CH3COOC2H5 координируется по –O– на Pt |
–1.1 –0.1 |
17.5 22.2 |
9.4 15.3 |
| Pt6Sn8 (Pt2Sn3, PtSn2) (CH3COOC2H5 координируется по O= на Pt) |
–3.5 | 30.3 | 24.8 |
| Pt2Sn6 (PtSn4) | – | – | – |
Отщепление этильной группы не идет на чистой Pt. Для кластеров Pt–Sn активационные барьеры получаются примерно одного порядка, а роль олова заключается лишь в возможности реализации данного канала.
Отщепление оксиэтильной группы, наоборот, протекает с небольшим барьером на Pt и кластере Pt9Sn4. При переходе к последующим кластерам активационный барьер возрастает, а на кластере Pt2Sn6 данная реакция не идет.
Также следует отметить, что разрыв связи C=O не может рассматриваться в качестве первой стадии процесса: если для чистой Pt было получено значение энтальпии активации такого канала, равное 23.6 ккал/моль, то для всех исследованных кластеров Pt–Sn оно составило не менее 40 ккал/моль.
Далее рассмотрим реакционные каналы, в которых синхронно разрываются связи C–O и C–H (что в результате приводит к тем же продуктам, что и в случае последовательного разрыва связей C–H и C–O). Структуры предреакционных комплексов, переходных состояний и интермедиатов представлены на рис. 11–13 соответственно. Энтальпии активации первых стадий данных реакционных каналов, а также стандартные изменения энтальпии для первых стадий приведены в табл. 9–11 соответственно.
Рис. 11.
Структуры, соответствующие предреакционному комплексу (а), переходному состоянию I (б) и интермедиату I (в) синхронного разрыва связей C–O и C–H с образованием этилена (π-комплекс на Pt) на кластере Pt9Sn4. Структуры, соответствующие предреакционному комплексу (г), переходному состоянию I (д) и интермедиату I (е) синхронного разрыва связей C–O и C–H с образованием этилена (электростатический комплекс) на кластере Pt9Sn4.
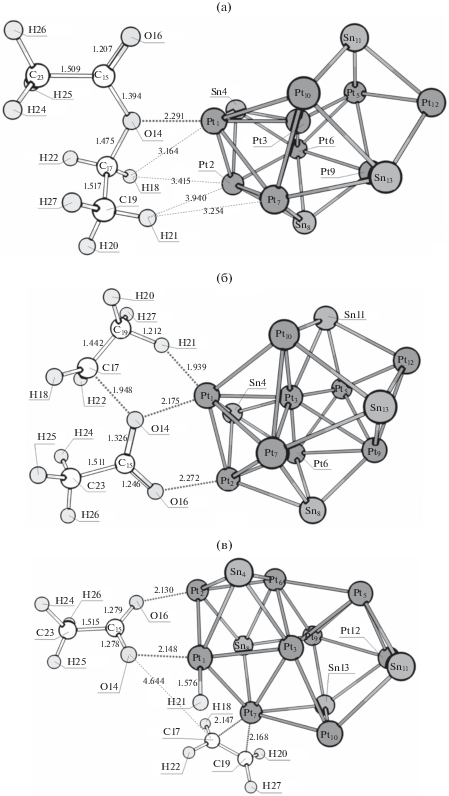
Рис. 11.
Окончание
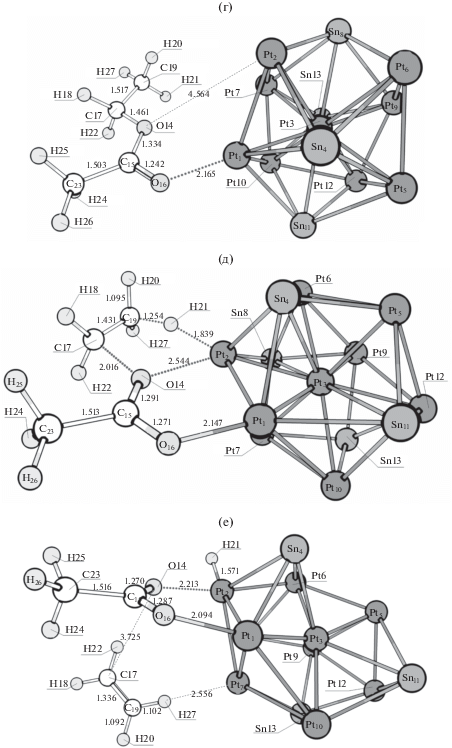
Рис. 12.
Структуры, соответствующие предреакционному комплексу (а), переходному состоянию I (б) и интермедиату I (в) синхронного разрыва связей C–O и C–H с образованием ацетальдегида, протекающего на кластере Pt9Sn4.
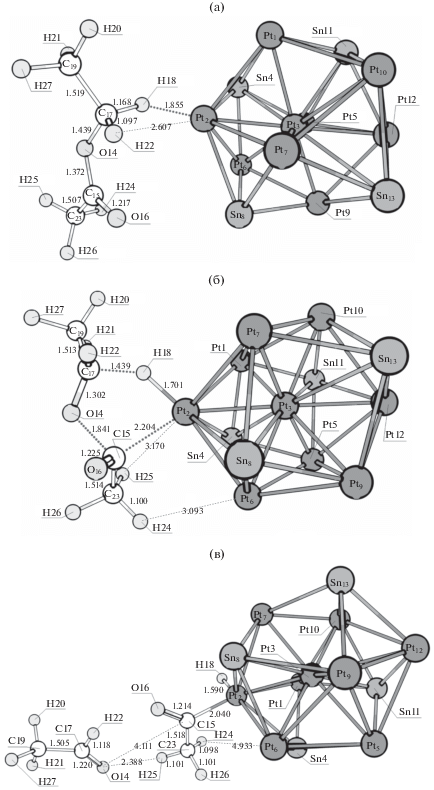
Рис. 13.
Структуры, соответствующие предреакционному комплексу (а), переходному состоянию I (б) и интермедиату I (в) синхронного разрыва связей C–O и C–H с образованием кетена, протекающего на кластере Pt9Sn4.
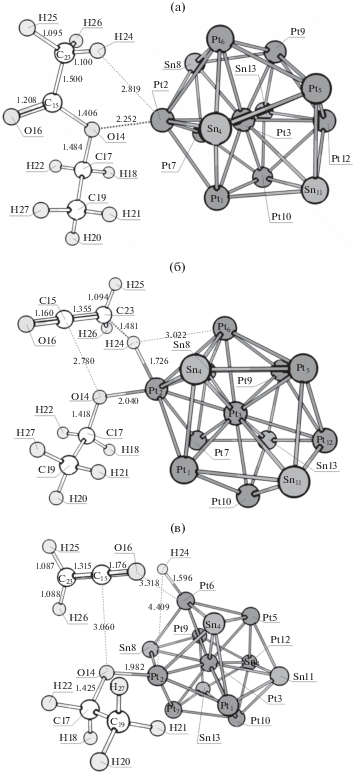
Таблица 9.
Синхронный разрыв связей C–O и C–H в этилацетате (с образованием этилена и поверхностного ацетата)
| Модельный кластер и начальная координация этилацетата | Адсорбция молекулы CH3COOC2H5 на кластере | Разрыв связей C–O и C–H | |
|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | |||
| Pt6 (Pt) (CH3COOC2H5 координируется по O= на Pt) |
–10.7 | 14.1 | –54.0 |
| Pt9Sn4 (Pt3Sn) CH3COOC2H5 координируется по –O– на Pt CH3COOC2H5 координируется по O= на Pt CH3COOC2H5 координируется по O= на Sn |
–6.4 –11.4 –6.5 |
10.7 18.4 23.7 |
–47.8 –6.2 –39.2 |
| Pt6Sn5 (PtSn) CH3COOC2H5 координируется по –O– на Pt CH3COOC2H5 координируется по O= на Pt CH3COOC2H5 координируется по O= на Sn |
–1.1 0.3 –1.6 |
28.9 16.7 27.1 |
20.2 5.6 7.6 |
| Pt6Sn8 (Pt2Sn3, PtSn2) | – | – | – |
| Pt2Sn6 (PtSn4) (CH3COOC2H5 координируется по –O– на Pt) |
8.4 | 13.4 | –6.3 |
Таблица 10.
Синхронный разрыв связей C–O и C–H в этилацетате (с образованием ацетальдегида и поверхностного ацетила)
| Модельный кластер и начальная координация этилацетата | Адсорбция молекулы CH3COOC2H5 на кластере |
Разрыв связей C–O и C–H | |
|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | |||
| Pt6 (Pt) (CH3COOC2H5 координируется по O= и H CH2-группы на одном атоме Pt) |
–15.4 | 51.7 | –5.4 |
| Pt9Sn4 (Pt3Sn) (CH3COOC2H5 координируется по H CH2-группы на Pt) |
–6.0 | 27.7 | 0.4 |
| Pt6Sn5 (PtSn) (CH3COOC2H5 координируется по O= на Pt) |
–5.2 | 49.5 | 27.5 |
| Pt6Sn8 (Pt2Sn3, PtSn2) (CH3COOC2H5 координируется по O= на Pt) |
–2.5 | 47.7 | 28.7 |
| Pt2Sn6 (PtSn4) (CH3COOC2H5 координируется по O= на Pt) |
1.4 | 36.6 | 6.4 |
Таблица 11.
Синхронный разрыв связей C–O и C–H в этилацетате (с образованием кетена)
| Модельный кластер и начальная координация этилацетата | Адсорбция молекулы CH3COOC2Н5 на кластере |
Разрыв связей C–О и C–H с образованием кетена и этанола |
Разрыв связей C–O
и C–H с образованием кетена и поверхностной этоксильной группы |
||
|---|---|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | |||||
| Pt6 (Pt) CH3COOC2H5 координируется по –O– и H из Ac-группы на соседних атомах Pt CH3COOC2H5 координируется по –O– и H из Ac-группы на том же атоме Pt |
–9.8 –12.6 |
– – |
– – |
22.1 26.0 |
20.6 –11.3 |
| Pt9Sn4 (Pt3Sn) (CH3COOC2H5 координируется по –O– на Pt) |
–9.1 | – | – | 37.4 | 20.3 |
| Pt6Sn5 (PtSn) | 0.3 | 59.3 | 18.6 | – | – |
| Pt6Sn8 (Pt2Sn3, PtSn2) (CH3COOC2H5 координируется по O= на Sn) |
–0.5 | 62.0 | 27.3 | – | – |
| Pt2Sn6 (PtSn4) | – | – | – | – | – |
Реакционный канал, приводящий к этилену и поверхностному ацетату, реализуется для всех кластеров, кроме Pt6Sn8. При этом активационные барьеры различались в некоторых пределах, однако установить какую-либо корреляцию не представлялось возможным. Впрочем, логично предположить, что в экспериментальных условиях этот канал может реализовываться на всех изучаемых кластерах.
Реакционный канал, приводящий к ацетальдегиду и поверхностному ацетилу, имеет достаточно существенные (по сравнению с другими реакционными каналами) активационные барьеры, поэтому вероятность протекания реакции через него невелика.
Реакционный канал, приводящий к кетену и поверхностной этоксильной группе, возможен на чистой Pt. На кластере Pt9Sn4 он маловероятен. На кластерах Pt6Sn5 и Pt6Sn8 реакционный барьер слишком велик, а на кластере Pt2Sn6 такой реакционный канал не реализуется вообще.
Следующим важным реакционным каналом является канал, начинающийся с разрыва связи C–C в ацетильной группе. Структуры предреакционных комплексов, переходных состояний и интермедиатов для данного канала приведены на рис. 14, энтальпии активации первых стадий, а также стандартные изменения энтальпии для первых стадий – в табл. 12. Как можно видеть, активационные барьеры данного канала невелики на чистой Pt и лишь при небольшом содержании Sn, а по мере его роста они резко возрастают.
Рис. 14.
Структуры, соответствующие предреакционному комплексу (а), переходному состоянию I (б) и интермедиату I (в) разрыва связи C–С в ацетильной группе на кластере Pt9Sn4.
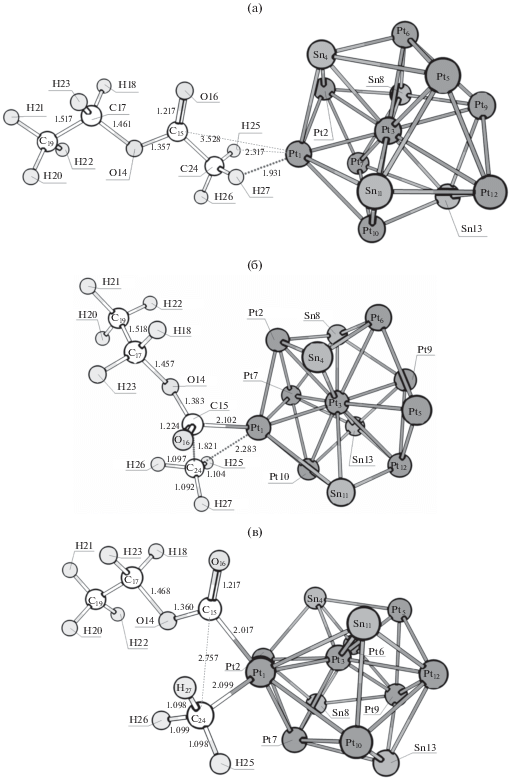
Таблица 12.
Разрыв связи C–C в ацетильной группе этилацетата (крекинг по α-углеродному атому)
| Модельный кластер и начальная координация этилацетата | Адсорбция молекулы CH3COOC2H5 на кластере |
Разрыв связи C–C с образованием поверхностного метила | |
|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | |||
| Pt6 (Pt) CH3COOC2H5 координируется по C=O и H Ac-группы на соседних атомах Pt |
–20.9 | 8.0 | –14.1 |
| CH3COOC2H5 координируется по O= на Pt | –16.5 | 17.9 | –26.8 |
| Pt9Sn4 (Pt3Sn) CH3COOC2H5 координируется по H Ac-группы на Pt CH3COOC2H5 координируется по H Ac-группы на Pt (вариант) |
–6.1 –5.1 |
12.9 9.3 |
–11.0 –13.7 |
| Pt6Sn5 (PtSn) (CH3COOC2H5 координируется по O= на Pt) |
–5.2 | 31.2 | 12.7 |
| Pt6Sn8 (Pt2Sn3, PtSn2) (CH3COOC2H5 координируется по O= на Pt) |
–3.5 | 34.8 | 21.5 |
| Pt2Sn6 (PtSn4) (CH3COOC2H5 координируется по O= на Sn) |
–2.7 | 27.2 | 7.9 |
Наконец, реакция пиролиза сложных эфиров, упомянутая выше, рассчитана как в некаталитическом газофазном варианте (включая реакцию через 4-членное переходное состояние, с существенно более высоким активационным барьером), так и при различных вариантах координации молекулы этилацетата с модельными кластерами. Энтальпии активации, а также стандартные изменения энтальпии адсорбции и реакции приведены в табл. 13. Структуры, соответствующие стационарным точкам поверхности потенциальной энергии, показаны на рис. 15. Платина за счет активации атома водорода снижает активационный барьер данной реакции, однако по мере роста содержания олова такая координация становится невозможной. В случае координации этилацетата по атому кислорода (на атоме олова или атоме платины) влияние катализатора на процесс становится незначительным. Также отметим, что реакционный путь через 4-членное переходное состояние при адсорбции на любом из исследованных кластеров получить не удалось вообще (в отличие от некаталитической реакции в газовой фазе).
Таблица 13.
Пиролиз этилацетата на модельных кластерах
| Модельный кластер и начальная координация этилацетата | Образование предреакцион-ного комплекса (адсорбция CH3COOC2H5) | Пиролиз этилацетата через 6-членное переходное состояние |
Пиролиз этилацетата через 4-членное переходное состояние | ||
|---|---|---|---|---|---|
| ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | $\Delta H_{{{\text{673}}}}^{ \ne }$ | ΔH673 | |
| ккал/моль | |||||
| Некаталитическая реакция | – | 37.7 | 14.2 | 56.1 | 14.2 |
| Pt6 (Pt) CH3COOC2H5 на Pt по H CH3COOC2H5 на Pt по –O– |
–10.1 –15.8 |
23.8 36.1 |
–23.9 11.4 |
– – |
– – |
| Pt9Sn4 (Pt3Sn)* CH3COOC2H5 на Pt по H CH3COOC2H5 на Pt по –O– CH3COOC2H5 на Pt по O= |
–6.2/–5.8/–5.6 –16.8/–16.0 –9.6/–9.0 |
23.1/21.7/36.6. 38.8/38.5 30.9/30.4 |
–17.4/–0.7/13.9 20.2/19.7 7.0/6.9 |
– – – |
– – – |
| Pt6Sn5 (PtSn) CH3COOC2H5 на Pt по H CH3COOC2H5 на Pt по –O– CH3COOC2H5 на Pt по O= CH3COOC2H5 на Sn по O= |
0.9 –1.2 –4.3 –1.6 |
29.6 34.0 37.7 38.4 |
–3.9 10.7 16.9 15.2 |
– – – – |
– – – – |
| Pt6Sn8 (Pt2Sn3, PtSn2) CH3COOC2H5 на Pt по –O– CH3COOC2H5 на Pt по O= CH3COOC2H5 на Sn по O= |
1.1 –1.5 –0.1 |
35.5 38.0 37.6 |
11.4 16.4 13.8 |
– – – |
– – – |
| Pt2Sn6 (PtSn4) CH3COOC2H5 на Pt по –O– CH3COOC2H5 на Sn по O= |
0.5 –1.8 |
36.4 38.4 |
12.4 16.2 |
– – |
– – |
Рис. 15.
Структуры, соответствующие предреакционному комплексу (а), переходному состоянию (б) и постреакционному комплексу (в) в реакции пиролиза этилацетата на кластере Pt9Sn4.
Нами были изучены механизмы гидродеоксигенации сложных эфиров на Pt- и Pt–Sn-катализаторах. Оказалось, что реакция может протекать в двух основных стадиях с образованием альдегида и спирта, в трех основных стадиях с промежуточным образованием полуацеталя и в трех основных стадиях с промежуточным образованием поверхностного этилового эфира (причем канал через полуацеталь оказывается наименее вероятным). В ходе реакции на интерметаллидах Pt–Sn возможна активация связи C=O как атомом Sn, так и атомом Pt, при этом только для интерметаллида Pt–Sn возможны оба варианта координации исходных веществ. В двухстадийной реакции, как и в трехстадийной, через поверхностный этиловый эфир, лимитирующей стадией является первая. Также возможны и синхронные реакции переноса водорода и разрыва связи C–O, однако на всех рассмотренных кластерах они сопровождаются сопоставимыми или бóльшими активационными барьерами.
Во всех случаях также возможно протекание различных реакций без участия водорода: пиролиза сложного эфира, разрыва связей C–H, C–C и C–O, в том числе синхронных реакций. При этом в случае чистой Pt или при небольшом содержании Sn энергетические барьеры многих из этих реакций сравнительно невелики, что объясняет низкую селективность Pt/Al2O3-катализатора в реакции деоксигенации. При переходе от Pt к интерметаллидам Pt–Sn энергетические барьеры реакций крекинга и дегидрирования возрастают, в то время как барьеры реакций гидродеоксигенации, а также разрыва связей C–H и C–O, не приводящих к потере углеродной массы, остаются достаточно невысокими.
Таким образом, наблюдаемая высокая селективность катализатора Pt : Sn = 1 : 5 объясняется существенным повышением активационного барьера реакций крекинга по мере роста содержания Sn в катализаторе при сохранении относительно невысокими барьеров реакций, не приводящих к потере углеродной массы.
Список литературы
Sarker S., Lamb J.J., Hjelme D.R., Lien K.M. // Fuel. 2018. V. 226. P. 686.
Rothermel J. Renewable Raw Materials in the EU Chemical Industry. http://dechema.de/Datei_Download-p-20014215-dateityp-ap-tagung-757-file-7206.html
Knothe G. // Prog. Energ. Combust. Sci. 2010. V. 36. № 3. P. 364
Mohammad M., Hari T.K., Yaakob Z., Sharma Y.C., Sopian K. // Renew. Sustain. Energy Rev. 2013. V. 22. P. 121.
Knothe G., Razon L.F. // Prog. Energ. Combust. Sci. 2017. V. 58. P. 36.
Bezergianni S., Dimitriadis A. // Renew. Sustain. Energy Rev. 2013. V. 21. P. 110.
Kordulis C., Bourikas K., Gousi M., Kordouli E., Lycourghiotis A. // Appl. Catal. B. 2016. V. 181. P. 156.
Seifi H., Sadrameli S.M. // J. Anal. Appl. Pyrol. 2016. V. 121. P. 1.
Palanisamy S., Gevert B.S. // Appl. Therm. Eng. 2016. V. 107. P. 301.
DePuy C.H., King R.W. // Chem. Rev. 1960. V. 60. № 5. P. 431.
Kubátová A., Luo Y., Štávová J., Sadrameli S.M., Aulich T., Kozliak E., Seames W. // Fuel. 2011. V. 90. № 8. P. 2598.
Kozliak E., Mota R., Rodriguez D., Overby P., Kubátová A., Stahl D., Niri V., Ogden G., Seames W. // Ind. Crop. Prod. 2013. V. 43. P. 386.
Адкинс Г. Органические реакции. Сб. 8. М.: Изд-во иностр. лит., 1956. С. 7.
Gutsche B., Röẞler H., Würkert S. Handbook of Heterogeneous Catalysis. V. 1. Weinheim: Wiley-VCH, 2008. P. 3329.
Normann W. // Z. Angew. Chem. 1931. V. 44. № 35. P. 714.
Wojcik B., Adkins H. // J. Am. Chem. Soc. 1933. V. 55. № 3. P. 1293.
Hosman B.B.A., van Steenis J., Waterman H.I. // Recueil. 1949. V. 68. P. 939.
Santillan-Jimenez E., Morgan T., Loe R., Crocker M. // Catal. Today. 2015. V. 258. P. 284.
Chen L., Zhang F., Li G., Li X. // Appl. Catal. A. 2017. V. 529. P. 175.
Itthibenchapong V., Srifa A., Kaewmeesri R., Kidkhunthod P., Faungnawakij K. // Energ. Convers. Manage. 2017. V. 134. P. 188.
Zhang H., Lin H., Wang W., Zheng Y., Hu P. // Appl. Catal. B. 2014. V. 150–151. P. 238.
Liang J., Ding R., Wu Y., Chen Y., Wu K., Meng Y., Yang M., Wang Y. // J. Mol. Catal. A. 2016. V. 411. P. 95.
Ding R., Wu Y., Chen Y., Liang J., Liu J., Yang M. // Chem. Eng. Sci. 2015. V. 135. P. 517.
Orozco L.M., Echeverri D.A., Sánchez L., Rios L.A. // Chem. Eng. J. 2017. V. 322. P. 149.
Peroni M., Mancino G., Baráth E., Gutiérrez O.Y., Lerchera J.A. // Appl. Catal. B. 2016. V. 180. P. 301.
Wang H.-Y., Jiao T.-T., Li Z.-X., Li C.-S., Zhang S.-J., Zhang J.-L. // Fuel Process. Technol. 2015. V. 139. P. 91.
Onyestyák G., Harnos S., Štolcová M., Kaszonyi A., Kalló D. // Catal. Commun. 2013. V. 40. P. 32.
Zhou L., Lawal A. // Appl. Catal. A. 2017. V. 532. P. 40.
Wang C., Liu Q., Liu X., Yan L., Luo C., Wang L., Wang B., Tian Z. // Chinese J. Catal. 2013. V. 34. P. 1128.
Chen N., Gong S., Shirai H., Watanabe T., Qian E.W. // Appl. Catal. A. 2013. V. 466. P. 105.
Sun K., Schulz T.C., Thompson S.T., Lamb H.H. // Catal. Today. 2016. V. 269. P. 93.
Wang W.-C., Thapaliya N., Campos A., Stikeleather L.F., Roberts W.L. // Fuel. 2012. V. 95. P. 622.
Raut R., Banakar V.V., Darbha S. // J. Mol. Catal. A. 2016. V. 417. P. 126.
Chen H., Wu Y., Qi S., Chen Y., Yang M. // Appl. Catal. A. 2017. V. 529. P. 79.
Bhattacharjee S., Tan C.-S. // J. Cleaner Production. 2017. V. 156. P. 203.
Janampelli S., Darbha S. // Mol. Catal. 2018. V. 451. P. 125.
Janampelli S., Darbha S. // Catal. Today. 2018. V. 309. P. 219.
Choi I.-H., Lee J.-S., Kim C.-U., Kim T.-W., Lee K.-Y., Hwang K.-R. // Fuel. 2018. V. 215. P. 675.
Chen N., Ren Y., Qian E.W. // J. Catal. 2016. V. 334. P. 79.
Yeh T.M., Hockstad R.L., Linic S., Savage P.E. // Fuel. 2015. V. 156. P. 219.
Pouilloux Y., Autin F., Guimon C., Barrault J. // J. Catal. 1998. V. 176. P. 215.
Цодиков М.В., Чистяков А.В., Нетрусов А.И. Продукты биомассы и их превращения в компоненты топлив и мономеры. LAMBERT Academic Publishing RU, 2017. С. 181.
Zharova P.A., Chistyakov A.V., Shapovalov S.S., Pasynskii A.A., Tsodikov M.V. // Mendeleev Commun. 2018. V. 28. P. 91.
Чистяков А.В., Жарова П.А., Цодиков М.В., Шаповалов С.С., Пасынский А.А., Мурзин В.Ю., Гехман А.Е., Моисеев И.И. // ДАН. 2015. Т. 460. № 1. С. 57.
Шаповалов С.С., Пасынский А.А., Торубаев Ю.В., Скабицкий И.В., Шеер М., Боденштайнер М. // Координац. химия. 2014. Т. 40. № 3. С. 131.
Hachemi I., Murzin D.Yu. // Chem. Eng. J. 2018. V. 334. P. 2201.
Lu J., Faheem M., Behtash S., Heyden A. // J. Catal. 2015. V. 324. P. 14.
Завелев Д.Е., Жидомиров Г.М., Цодиков М.В. // Кинетика и катализ. 2018. Т. 59. № 4. С. 404.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865.
Stevens W.J., Basch H., Krauss M. // J. Chem. Phys. 1984. V. 81. № 12. P. 6026.
Stevens W.J., Krauss M., Basch H., Jasien P.G. // Can. J. Chem. 1992. V. 70. P. 612.
Laikov D.N. // Chem. Phys. Lett. 1997. V. 281. P. 151.
Лайков Д.Н., Устынюк Ю.А. // Изв. АН. Сер. хим. 2005. № 3. С. 804.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ