

Кинетика и катализ, 2020, T. 61, № 4, стр. 585-592
Синтез мелкокристаллического цеолита типа SAPO-11 и исследование его физико-химических и каталитических свойств
М. Р. Аглиуллин a, *, А. В. Файзуллин a, А. Н. Хазипова a, Б. И. Кутепов a
a ФГБУН Институт нефтехимии и катализа УФИЦ РАН
450075 Уфа, просп. Октября, 141, Россия
* E-mail: Maratradikovich@mail.ru
Поступила в редакцию 20.11.2019
После доработки 27.02.2020
Принята к публикации 03.03.2020
Аннотация
Сопоставлены результаты кристаллизации двух силикоалюмофосфатных гелей состава 1.0Al2O3 ⋅ · 1.0P2O5 ⋅ 0.4SiO2 ⋅ 1.0ДПА ⋅ 50H2O, при приготовлении которых применяли в одном случае псевдобемит, в другом – изопропоксид Al. Показано, что выбор источника алюминия существенного влияет на фазовый состав формирующихся силикоалюмофосфатных гелей, а также на физико-химические и каталитические свойства кристаллизующихся из них образцов SAPO-11 в парофазной изомеризации циклогексаноноксима в капролактам.
ВВЕДЕНИЕ
В настоящее время в крупнотоннажных процессах нефтехимии все шире используются каталитические системы на основе цеолитсодержащих материалов [1]. Столь широкий интерес к этим материалам обусловлен наличием в них сильных Бренстедовских кислотных центров (БКЦ) и молекулярно-ситового эффекта.
Одним из примеров успешного промышленного применения цеолитов является процесс парофазной изомеризации циклогексаноноксима в капролактам (перегруппировка Бекмана) – важная стадия в процессе производства полиамида-6 [2]. В большинстве случаев указанная технология основана на использовании в качестве катализатора концентрированной серной кислоты, что вызывает усиленную коррозию оборудования и образование кислотных стоков. Для решения этой проблемы фирмой “Sumitomo” (Япония) был разработан промышленный процесс парофазной изомеризации циклогексаноноксимав капролактам с применением гетерогенного катализатора на основе высокомодульного цеолита ZSM-5 и растворителя метанола [3]. Из-за быстрой дезактивации катализатора технология реализована в системе реактор–регенератор. Кроме того, в качестве гетерогенных катализаторов предложены каталитические системы с использованием цеолитов LTL [4], Beta [5], аморфного алюмосиликата [6] и мезопористых мезоструктурированных материалов H-MCM-41 [7] и H-FSM-16 [8].
Как правило, цеолиты по химическому составу являются алюмосиликатами, однако за последние 20 лет нашли применение каталитические системы на основе силикоалюмофосфатных молекулярных сит SAPO-n. На основе SAPO-11 фирмой “Chevron” (США) реализован процесс изодепарафинизации масел и дизельного топлива [9], на основе SAPO-34 фирмой “UOP” (США) и Далянским институтом физической химии – процесс получения низших олефинов из метанола (MTO) [10]. SAPO-n характеризуются большим разнообразием структур и различаются как по размеру пор (SAPO-18 – 3.8 × 3.8 Å; VPI-5 – 12.7 × 12.7 Å), так и по размерности каналов (1D-SAPO-11, 2D-SAPO-40, 3D-SAPO-37) [11]. Бренстедовские кислотные центры в SAPO-n формируются в результате внедрения атомов Si в алюмофосфатную решетку [12]. Следует отметить, что БКЦ в SAPO-n по силе слабее, чем в алюмосиликатах, и такие кислотные центры принято называть “умеренными”.
Среди большого разнообразия силикоалюмофосфатных молекулярных сит SAPO-n особый интерес вызывает структура SAPO-11 (AEL), благодаря наличию “умеренных” по силе кислотных центров и одномерной канальной системы эллиптических пор размером 4.0 × 6.5 Å. Из-за указанных особенностей SAPO-11 могут стать перспективными каталитическими системами для парофазной изомеризации циклогексаноноксима в капролактам [13]. Следует отметить, что, несмотря на значительное количество публикаций [14] в области синтеза и применения SAPO-11 в катализе, по-прежнему не решены вопросы управления морфологией кристаллов и, как следствие, характеристиками вторичной пористой структуры, которые, на наш взгляд, во многом обуславливает селективность и стабильность разрабатываемых каталитических систем.
В предыдущих наших работах было показано [15–17], что одним из параметров, который определяет морфологию и дисперсность кристаллов AlPO4-11, является реакционная способность соединения алюминия, взятого для приготовления цеолита. Мы предположили, что использование различных источников алюминия при кристаллизации силикоалюмофосфата SAPO-11 также позволит нам влиять на морфологию и дисперсность кристаллов и характеристики вторичной пористой структуры SAPO-11 и, как следствие, его каталитические свойства в парофазной изомеризации циклогексаноноксима.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез силикоалюмофосфатов SAPO-11
Силикоалюмофосфаты готовили гидротермальным синтезом из реакционных гелей состава: 1.0Al2O3 ⋅ 1.0P2O5 ⋅ 0.4SiO2 ⋅ 1.0ДПА ⋅ 50H2O. Силикоалюмофосфатные гели получали следующим образом: к 10.0 г ортофосфорной кислоты (H3PO4, 85%, “Реахим”) приливали 35.6 г дистиллированной воды, в полученный раствор добавляли в зависимости от источника Al или 5.6 г псевдобемита (PB, 77% Al2O3, “Plural Sasol SB”), или 17.2 г изопропоксида Al (AI, 98%, “Acros”) и интенсивно перемешивали в течение 1 ч, затем приливали 4.4 г темплата ди-н-пропиламина (ДПА, 99%, “Acros”) и 3.5 г тетраэтилортосиликата (ТЭОС, 99%, “Acros”). Выбор ТЭОС в качестве источника Si обусловлен лучшим его внедрением в решетку алюмофосфата с целью формирования кислотных центров [18].
Приготовленные с использованием различных источников алюминия силикоалюмофосфатные гели кристаллизовали при 200–205°C в течение 12–14 ч в автоклавах из нержавеющий стали со специальным фторопластовым покрытием. Ранее нами было показано [15–17], что указанные условия позволяют синтезировать AlPO4-11 высокой фазовой чистоты и степени кристалличности. После проведения кристаллизации полученные продукты отмывали в дистиллированной воде до нейтрального рН, сушили при 90°C в течение 48 ч и затем прокаливали в атмосфере воздуха при 600°C в течение 6 ч для удаления темплата.
Просушенные исходные силикоалюмофосфатные гели, приготовленные на основе псевдобемита и изопропоксида алюминия, обозначены SAPO(PB) и SAPO(AI), а продукты их дальнейшей кристаллизации – SAPO-11(PB) и SAPO-11(AI) соответственно.
Методы анализа материалов
Химический состав полученных силикоалюмофосфатов анализировали методом рентгенофлуоресцентной спектрометрии на приборе EDX 720/900HS (“Shimadzu”, Япония).
Рентгенофазовый анализ (РФА) непрокаленных образцов осуществляли на дифрактометре D8 Advance (“Bruker ”, Германия) в CuKα-излучении со сканированием в области углов 2θ от 3° до 50° с шагом 1 град/мин, время накопления в точке – 2 с. Фазовый анализ проводили в программе Eva с привлечением базы данных PDF2. Абсолютную степень кристалличности определяли в программе TOPAS по относительному содержанию аморфной фазы через гало в области 2θ от 20° до 30°.
Координационное окружение атомов алюминия и фосфора оценивали по спектрам ВМУ ЯМР 27Al и 31P. Спектры получали на ЯМР-спектрометре Avance-400 (“Bruker”, Германия) в условиях одноимпульсного эксперимента при вращении образцов под магическим углом (ВМУ) (~7 кГц) в роторах из диоксида циркония (Ø 4 мм). Частота Лармора – 104 МГц (27Al), 162 MГц (31Р), длительность импульса π/2 – 2 мкс (31P) и 2.5 мкc (27Al).
Значения рН приготовленных алюмофосфатных гелей, продуктов кристаллизации измеряли на ионометре Нитрон 01 (“БИОМЕД”, Россия), снабженном электродом ЭСК-10601 и автоматическим термокомпенсатором ТК1000 при ~25°C.
Морфологию и размер кристаллов для силикоалюмофосфатов исследовали методом сканирующей электронной микроскопии (СЭМ) на электронном микроскопе JSM-6490LV (“JEOL”, Япония). Съемку изображений вели в режиме регистрации вторичных электронов при ускоряющем напряжении 20 кВ и рабочем расстоянии 10 мм. Перед съемкой образцы помещали на поверхность алюминиевого столика диаметром 25 мм, фиксировали при помощи проводящей липкой ленты.
Характеристики пористой структуры изучали методом низкотемпературной адсорбции–десорбции азота на сорбтометреNova 1200e (“Quantachrome”, США). Удельную поверхность (SБЭТ) рассчитывали в интервале относительных парциальных давлений Р/Р0 от 0.05 до 0.35. Объем микропор и их вклад в удельную поверхность в присутствии мезо- и макропор находили t-методом Хэлси. Суммарный объем пор оценивали при достижении относительно относительного давления 0.99. Расчет объема мезо- и макропор проводили в программном пакете выше описанного прибора.
ИК-спектры регистрировали на Фурье-спектрометре Protégé 460 (“Nicolet Technologies”, США) с разрешением 4 см–1 в диапазоне 400–4000 см–1. Диаметр таблетки для регистрации ИК-спектров составлял 10 мм. Адсорбцию пиридина осуществляли при 423 К в течение 30 мин, затем физически адсорбированный пиридин удаляли вакуумированием при 423 К в течение 30 мин. Количество Бренстедовских кислотных центров (БКЦ) оценивали путем интегрирования пика при 1545 см–1, Льисовских кислотных центров (ЛКЦ) – пика при 1454 см–1, исходя из известных в литературе интегральных мольных коэффициентов экстинкции пиридина для центров каждого типа [19]. Термопрограммированную десорбцию (ТПД) аммиака проводили по методике, описанной в [20].
Тестирование катализаторов
Каталитические свойства образов SAPO-11 исследовали в парофазной изомеризации циклогексаноноксима. Реакцию осуществляли в проточном реакторе при температуре 330°C и давлении 1 атм с загрузкой катализатора на 1.3 г (фракционный состав 0.1–0.2 мм). Смесь оксима, ацетонитрила (растворитель) и азота (газ-носитель) с мольным соотношением 1 : 52 : 17 пропускали через реактор при объемной скорости подачи сырья 0.15 ч–1 в пересчете на оксим. До начала реакции катализатор предварительно нагревали при 350°С в течение 3 ч в потоке сухого азота (3 мл/мин) с целью удаления адсорбированной влаги. Продукты реакции анализировали методом газожидкостной хроматографии на хроматографе GS-2014 (“Shimadzu”, Япония) с пламенно-ионизационным детектором, фаза SE-30, температура анализа – 50–280°C с программируемым нагревом 8°C/мин. Температура детектора – 275°C, температура испарителя – 300°C, расход гелия в качестве газа-носителя – 30 мл/мин.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
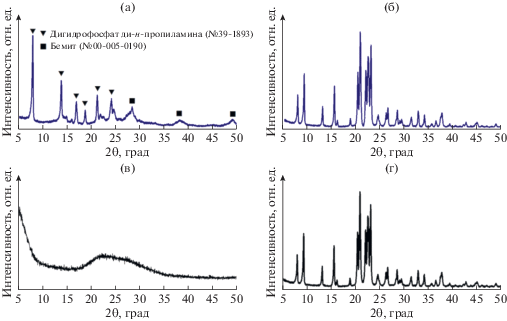
На рис. 1а и 1в приведены рентгенограммы просушенных силикоалюмофосфатных гелей. Видно, что образец геля SAPO(PB) состоит из смеси фаз фосфата ди-н-пропиламина, нерастворенного бемита и аморфной фазы.
Рис. 1.
Рентгенограммы исходных силикоалюмофосфатных гелей и продуктов их кристаллизации: а – SAPO(PB); б – SAPO-11(PB); в – SAPO(AI); г – SAPO-11(AI).
Для более детальной оценки взаимодействия источников алюминия с фосфорной кислотой на начальной стадии приготовления исходных гелей использовали ВМУ ЯМР 27Al и 31P. На рис. 2а, 2в, 2д и 2ж приведены спектры ВМУ ЯМР 27Al и 31P для исходных гелей и продуктов их кристаллизации. В спектрах 27Al образца SAPO(PB) наблюдаются сигналы при 42, 7 и –14 м. д. Первый сигнал связан с атомами Al с тетраэрическим окружением по кислороду [AlO4] в аморфном алюмофосфате, сигнал при 7 м. д. – с атомами Al с октаэрическим окружением по кислороду [AlO6] в непрореагировавшем источнике алюминия, а сигнал при –14 м. д. – с атомами Al с октаэрическим окружением по кислороду [AlO6] в аморфномалюмофосфате [21–23]. В спектре образца SAPO(AI) кроме сигналов при 42 и 7 м. д. присутствует сигнал при –9 м. д., который соответствует [AlO6] в аморфном алюмофосфате [21–23].
Рис. 2.
Спектры ВМУ ЯМР27 Al и 31P для силикоалюмофосфатных гелей и продуктов их кристаллизации: спектры 27Al образцов SAPO(PB) (а), SAPO-11(PB) (б), SAPO(AI) (д) и SAPO-11(AI) (е); спектры 31P образцов SAPO(PB) (в), SAPO-11(PB) (г), SAPO(AI) (ж) и SAPO-11(AI) (з).
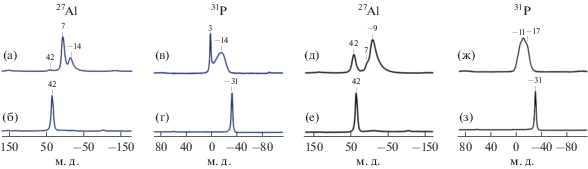
В спектре 31P образца SAPO(PB) наблюдаются сигналы при 3 и –14 м. д. Сигнал при 3 м. д. относится к химическим соединениям, в которых атомы P не связаны с атомами Al [21–23]. Сопоставляя полученные результаты ВМУ ЯМР 27Al и 31P с данными РФА (рис. 1), можно сделать вывод о содержании в образце SAPO(PB) фосфатади-н-пропиламина. Широкий сигнал при –14 м. д. согласно [21–23] соответствует алюмофосфатам P(OAl)nOH, где n = 2–4. В спектре образца SAPO(AI) имеются широкие сигналы при –11 и –17 м. д. Первый, как было отмечено выше, связан с алюмофосфатом P(OAl)2(H2O)2. Сигнал при –17 м. д. согласно [21–23] относится к алюмофосфатам состава P(OAl)3(H2O).
В табл. 1 приведены данные рН исходных гелей. Видно, что рН образца геля SAPO(PB) до и после добавления амина значительно ниже, чем рН SAPO(AI). Наблюдаемое различие в значениях рН обусловлено более низкой реакционной способностью псевдобемита.
Таблица 1.
Химический и фазовый состав, рН и характеристики пористой структуры исходных силикоалюмофосфатных гелей и продуктов кристаллизации на их основе
| Образец | pH | Химический состав |
Фазовый состав | Степень кристалличности,% |
| SAPO(PB) | 3.5 | Al1.0P1.06Si0.19 | Ph. DPA + SAPO + PB | н/д |
| SAPO(AI) | 8.2 | Al1.0P0.98Si2.01 | SAPO | н/д |
| SAPO-11(PB) | 9.2 | Al1.0P0.91Si0.16 | SAPO-11 | 96 |
| SAPO-11(AI) | 9.9 | Al1.0P0.92Si0.17 | SAPO-11 | 92 |
Таким образом, из полученных результатов исследования образцов гелей SAPO(PB) и SAPO-11(AI) с помощью физико-химических методов следует, что различие в реакционной способности источника алюминия существенно влияет на состав силикоалюмофосфатных гелей уже на начальной стадии их приготовления. Так, менее реакционноспособный псевдобемит слабо взаимодействует с фосфорной кислотой, формируя лишь незначительную часть аморфного силикоалюмофосфата. В то же время более реакционноспособный изопропоксид алюминия практически полностью превращается в аморфный силикоалюмофосфат уже на начальной стадии приготовления геля.
На рис. 1б и 1г приведены рентгенограммы продуктов кристаллизации силикоалюмофосфатных гелей SAPO(PB) и SAPO(AI). Видно, что в обоих случаях в результате кристаллизации при 200°C через 12 ч формируется фаза SAPO-11 высокой фазовой чистоты и степени кристалличности. Образец SAPO-11(PB) имеет более бóльшую степень кристалличности по сравнению с SAPO-11(AI). В спектрах ВМУ ЯМР 27Al для обоих цеолитов SAPO-11 присутствует основной сигнал при 42 м. д., который характерен для [AlO4] в молекулярных ситах AlPO4-11 и SAPO-11. В спектре ЯМР 27Al для образца SAPO-11(AI) дополнительно наблюдается слабый сигнал при –9 м. д., относящийся к [AlO6] в аморфной фазе, содержание которой в SAPO-11(AI) незначительно (рис. 1г). В спектрах ВМУ ЯМР 31P для обоих образцов имеется единственный сигнал при –31 м. д., обусловленный наличием [PO4] в молекулярном сите SAPO-11.
Следует отметить, что содержание кремния в образцах кристаллических силикоалюмофосфатов ниже, чем в исходных гелях (табл. 1). Полученные результаты объясняются неполным внедрением атомов кремния в алюмофосфатную решетку.
На рис. 3 приведены снимки СЭМ для SAPO-11. Видно, что кристаллы образца SAPO-11(PB) представляют собой псевдосферические агрегаты диаметром от 8 до 10 мкм, состоящие из более мелких первичных кристаллов размером от 0.5 до 1 мкм. SAPO-11(PB) имеет площадь удельной поверхности SBET = 210 м2/г, объем микропор Vмикро = = 0.07 см3/г и объем мезопор Vмезо = 0.09 см3/г. Образец SAPO-11(AI) также характеризуется кристаллами псевдосферической морфологии диаметром от 2 до 8 мкм, которые состоят из нанодисперсных кристаллов размером от 0.1 до 0.3 мкм, и имеет SBET = 273 м2/г, Vмикро = 0.06 см3/г и Vмезо = = 0.16 см3/г. Известно [24], что размер первичных кристаллов и их вторичных агрегатов сильно зависит от степени пересыщения по зародышам кристаллизации. По-видимому, в SAPO-11(AI) благодаря использованию более реакционноспособного источника алюминия в индукционный период кристаллизации формируется большее количество зародышей, которые в дальнейшем превращаются в нанокристаллы и их сростки.
Рис. 3.
Снимки СЭМ кристаллических силикоалюмофосфатов: а, б – образец SAPO-11(PB); в, г – образец SAPO-11(AI).

На рис. 4 приведены изотермы низкотемпературной адсорбции–десорбции азота, а в табл. 2 – характеристики пористой структуры для образцов SAPO-11. Видно, что для обоих образцов наблюдаются изотермы IV типа с петлей гистерезиса, которые свидетельствуют о наличии в материалах мезопор. Для SAPO-11(PB) отмечается резкий подъем изотермы в области относительного давления P/P0 ≈ 1.0, что обусловлено присутствием макропор. Расчет размера пор по модели BJH (Barrett–Joyner–Halenda) показывает, что образец SAPO-11(PB) обладает более крупными по размеру мезопорами по сравнению с SAPO-11(AI). При сопоставлении характеристик пористой структуры образцов (табл. 2) видно, что для цеолита SAPO-11(AI) внешняя удельная поверхность и объем мезопор вдвое выше, чем для SAPO-11(PB). В тоже время SAPO-11(PB) характеризуется бóльшим объемом макропор. Полученные результаты объясняются размерами первичных кристаллов, сростки которых формируют мезо- и макропоры. У образца SAPO-11(AI) размеры первичных кристаллов в 2–3 раза меньше, чем у SAPO-11(PB), поэтому у него формируется преимущественно микро-мезопористая структура, а у цеолита SAPO-11(PB) – микро-макропористая структура.
Рис. 4.
Изотермы адсорбции–десорбции N2 и распределение пор по размеру, найденное по методу BJH (на вставках): a – SAPO-11(PB); б – SAPO-11(AI).
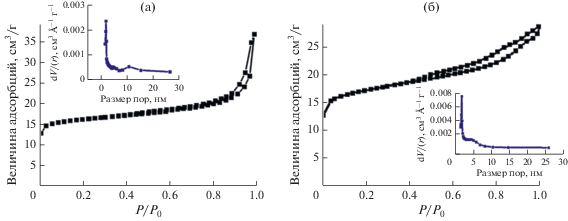
Таблица 2.
Характеристики пористой структуры и кислотные свойства кристаллических силикоалюмофосфатов
| Образец | SБЭТ, м2/г | SВН, м2/г | Vмикро, см3/г | Vмезо, см3/г | Vмакро, см3/г |
|---|---|---|---|---|---|
| SAPO-11(PB) | 210 | 78 | 0.07 | 0.08 | 0.10 |
| SAPO-11(AI) | 273 | 143 | 0.06 | 0.16 | 0.03 |
На рис. 5 приведены спектры ОН-групп и адсорбированного пиридина на образцах SAPO-11(PB) и SAPO-11(AI). Для обоих образцов наблюдаются полосы поглощенияпри 3744, 3677, 3627 и 3533 см–1, связанные с колебаниями ОН-групп. Согласно [25] полосу при 3744 см–1 принято относить к силанольным группам (SiOH), полосу при 3677 см–1 − к РОН-группам, полосы при 3627 и 3533 см–1 − к мостиковым ОН-группам. В спектрах адсорбированного пиридина имеются полосы поглощения (п. п.) при 1452, 1490 и 1550 см–1. Полоса поглощения при 1452 см–1 соответствует пиридину, адсорбированному на Льюисовских кислотных центрах, п. п. при 1490 см–1 – Брентедовским кислотным центрам, появление полосы при 1490 см–1 обусловлено вкладом обоих типов центров – БКЦ и ЛКЦ [25]. В табл. 3 представлены результаты расчета концентрации кислотных центров. Видно, что у образца SAPO-11(AI) концентрация и ЛКЦ, и БКЦ выше, чем у образца SAPO-11(РВ), несмотря на близкое содержание кремния. Следует отметить, что по данным ТПД NH3 для обоих образцов SAPO-11 значения концентраций кислотных центров близки, при этом значительная их часть приходиться кислотные центры “умеренной” силы (табл. 2). Полученные результаты объясняются тем, что образец SAPO-11(AI) имеет бóльшую поверхности мезопор, которая обеспечивает лучшую доступность кислотных центров для молекул пиридина по сравнению с молекулой аммиака, имеющей меньший размер.
Рис. 5.
ИК-спектры ОН-групп и адсорбированного пиридина: a, б – образец SAPO-11(BM); в, г – образец SAPO-11(AI).
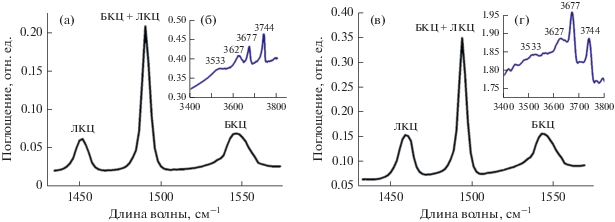
Таблица 3.
Характеристики пористой структуры и кислотные свойства кристаллических силикоалюмофосфатов
| Образец | *“Слабые” кислотные центры |
**“Cредние” кислотные центры |
***“Cильные” кислотные центры |
БКЦ, мкмоль/г | ЛКЦ, мкмоль/г |
|---|---|---|---|---|---|
| SAPO-11(PB) | 178 | 240 | 72 | 70 | 40 |
| SAPO-11(AI) | 244 | 262 | 77 | 91 | 56 |
Главной особенностью силикоалюмофосфатного молекулярного сита SAPO-11, как уже отмечалось ранее, является наличие “умеренных” по силе кислотных центров и одномерной канальной структуры, которые оказывают специфическое влияние на каталитические свойства материала.
Ранее было показано [2], что в присутствии гетерогенных кислотных катализаторов, в том числе цеолитов, парофазная изомеризации циклогексаноноксима протекает с образованием капролактама (I) и побочных продуктов – циклогексанона (II), гексеннитрила (III), циклогексен-1-она (IV), гексаннитрила (V) и анилина (VI) (схема 1 ). При этом для SAPO-11 в работе [13] было установлено, что за селективную изомеризацию оксима в капролактам отвечают Бренстедовские кислотные центры умеренной силы, локализованные на внешней поверхности кристаллов.
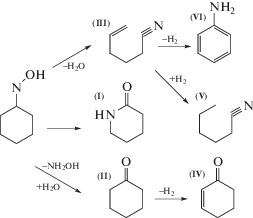
Схема 1 . Схема превращений циклогексаноноксима на кислотных гетерогенных катализаторах.
На рис. 6а приведены результаты изомеризации циклогексаноноксима в присутствии образцов SAPO-11. Видно, что на образце SAPO-11(AI) наблюдается более высокая конверсия оксима, чем на SAPO-11(РВ). Полученный результат объясняется более развитой системой транспортных пор, которая обеспечивает доступность большего количества кислотных центров для молекул оксима, а также увеличивает скорость его диффузии к кислотным центрам и скорость обратной диффузию продуктов реакции. На рис. 6б представлены кривые селективности образования капролакатама. Видно, что образец SAPO-11(AI) характеризуется большей селективностью по капролактаму (~95%) по сравнению с SAPO-11(РВ) (~85%). Это может быть связано с уменьшением времени пребывания продуктов реакции в каналах молекулярного сита SAPO-11(AI) с более развитой системой транспортных пор, что снижает вероятность протекания побочных реакций.
Рис. 6.
Изомеризация циклогексаноноксима на SAPO-11: а – конверсия оксима на катализаторах SAPO-11(PB) (1) и SAPO-11(AI) (2); б – селективность образования капролактама в присутствии SAPO-11(PB) (1) и SAPO-11(AI) (2).
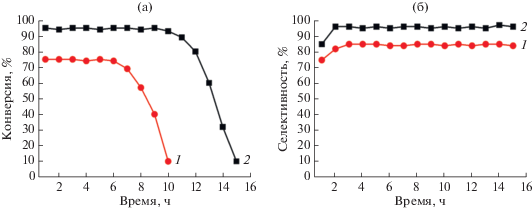
Следует отметить, что в начальный период работы катализаторов наблюдается некоторый рост селективности образования капролактама, который, вероятно, объясняется дезактивацией самых сильных кислотных центров, неселективных в изомеризации оксима. Аналогичные результаты были получены ранее в работе [26].
Как отмечалось выше, изомеризация циклогексаноноксима характеризуется потерей активности катализаторов в процессе его эксплуатации из-за дезактивации кислотных центров в результате блокировки продуктами конденсации капролактама. Из рис. 6 видно, что стабильность работы образца SAPO-11(AI) с меньшим размером кристаллов и более развитой вторичной микро-мезопористой структурой втрое выше, чем у микропористогоцеолита SAPO-11(РВ), что обусловлено лучшей диффузией, снижающей вклад побочных реакций конденсации капролактама.
Таким образом, из представленных результатов следует, что в парофазной реакции изомеризации циклогексаноноксима в капролактам эффективность работы катализатора на основе силикоалюмофосфатного молекулярного сита SAPO-11 сильно зависит от размеров кристаллов и наличия вторичной пористой структуры, которая обеспечивает эффективность диффузии оксима и обратную диффузию продуктов реакции изомеризации. Можно предположить, что для создания более стабильных каталитических систем на основе SAPO-11 необходимо синтезировать материалы с еще меньшим размером кристаллов и более развитой вторичной микро-мезопористой структурой.
ЗАКЛЮЧЕНИЕ
В работе с применением методов рентгенофлюоресцентной спектроскопии, РФА, ВМУ ЯМР 27Al31P, СЭМ и адсорбции–десорбции азота исследовано влияние источника алюминия на формирование силикоалюмофосфатных гелей и продуктов кристаллизации на их основе.
Обнаружено, что использование в качестве источника алюминия псевдобемита приводит к формированию геля, состоящего из фазы фосфата ди-н-пропиламина, нерастворенного псевдобемита и аморфного силикоалюмофосфата. Применениежеизопропоксидаалюминия способствует формированию аморфного силикоалюмофосфата.
Установлено, что для образца SAPO-11, синтезированного с использованием псевдобемита, кристаллы имеют форму сферических агрегатов диаметром от 8 до 10 мкм, состоящих из первичных кристаллов размером от 0.5 до 1 мкм. Образец SAPO-11, полученный на основе изопропоксида алюминия, обладает кристаллами в виде псевдосферических агрегатов размером от 2 до 8 мкм, состоящих из нанокристаллов размером от 0.1 до 0.3 мкм.
Впервые показано, что активность, селективность и стабильность работы молекулярного SAPO-11 в парофазной изомеризации циклогексаноноксима в капролактам существенно зависят от размеров кристаллов и вторичной пористой структуры. Установлено, что SAPO-11, полученный с использованием изопропоксида алюминия, стабильно работает более 19 ч и позволяет при 95% конверсии циклогексаноноксима достигать селективности по капролактаму более 95%. Для образца SAPO-11, синтезированного с применением псевдобемита, конверсия оксима составляет ~75%, а селективность ~85%.
Список литературы
Martinez C., CormaA. // Coord. Chem. Rev. 2011. V. 255. P. 1558.
Dahlhoff G., Niederer J.P.M., Hoelderich W.F. // Catal. Rev. 2001. V. 43. I. 4. P. 381.
Izumi Y., Ichihashi H., Shimazu Y., Kitamura M., Sato H. // Bull. Chem. Soc. Jpn. 2007. V. 80. I. 7. P. 1280.
Dai L.-X., Iwaki Y., Koyama K., Tatsumi T. //. Appl. Surf. Sci. 1997. V. 121–122. P. 335.
Heitmann G.P., Dahlhoff G., Hölderich W.F // Appl. Catal. A: Gen. 1999. V. 185. I. 1. P. 99.
Gutierrez E., Aznar A.J., Ruiz-Hitzky E. // Stud. Surf. Sci. Catal. 1991. V. 59. P. 539.
Conesa T.D., Mokaya R., Yang Z., Luque R., Campelo J.M., Romero A.A. // J. Catal. 2007. V. 252. I. 1. P. 1.
Dai L.X., Koyama K., Tatsumi T. // Catal. Lett. 1998. V. 53. I. 3–4. P. 211.
Miller S.J. // Micropor. Mater. 1994. V. 2. I. 5. P. 439.
Tian P., Wei Y., Ye M., Liu Z. // ACS Catal. 2015. V. 5. P. 1922.
Baerlocher C., McCusker L.B., Olson D.H. Atlas of Zeolite Framework Types. Amsterdam: Elsevier, 6th edn, 2007. 404 p.
Barthomeuf D. // Zeolites. 1994. V. 14. I. 6. P. 394.
Singh P.S., Bandyopadhyay R., Hegde S.G., Rao B.S. // Appl. Catal. A: Gen. 1996. V. 136. P. 249.
Yadav R., Sakthivel A. // Appl. Catal. A: Gen. 2014. V. 481. P. 143.
Agliullin M.R., Khairullina Z.R., Faizullin A.V., Petrov A.I., Badretdinova A.A., Talzi V.P., Kutepov B.I. // Catal. Indust. 2019. V. 11. I. 2. P. 87.
Agliullin M.R., Khairullina Z.R., Faizullin A.V., Petrov A.I., Badretdinova A.A., Talzi V. P., Kutepov B.I. // Catal. Indust. 2019. V. 11. I. 1. P. 1.
Agliullin M.R., Khairullina Z.R., Faizullin A.V., Kutepov B.I. // Petrol. Chem. 2019. V. 59. I. 3. P. 349.
Yang Z., Li J., Liu Y., Liu C. // J. Energy Chem. 2017. V. 26. I. 4. P. 688.
Emeis C.A.//. J. Catal. 1993. V. 141. P. 347.
Yushchenko V.V. // Russ. J. Phys. Chem. A. 1997. V. 71. P. 628.
Zhang B., Xu J., Fan F., Guo Q., Tong X., Yan W., Yu J., Deng F., Li C., Xu R. // Micropor. Mesopor. Mater. 2012. V. 147. P. 212.
Chen B., Huang Y. // J. Phys. Chem. 2007. V. 111. P. 15236.
Xu R., Zhang W., Xu J., Tian Z., Deng F., Han X., Bao X. // J. Phys. Chem. C. 2013. V. 117. P. 5848.
Cundy C.S., Cox P.A. // Micropor. Mesopor. Mater. 2005. V. 82. P. 1.
Yang L., Aizhen Y., Qinhua X. // Appl. Catal. 1991. V. 67. P. 169.
Dai L.-X., Koyama K., Tatsumi T. // Catal. Lett. 1998. V. 53. P. 211.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ