

Кинетика и катализ, 2020, T. 61, № 4, стр. 447-468
Гетерогенный катализ и нелинейная динамика
М. М. Слинько a, *, А. Г. Макеев b
a ФГБУН Институт химической физики РАН им. Н.Н. Семенова
119991 Москва, ул. Косыгина, 4, Россия
b ФГБОУ ВО МГУ им. М.В. Ломоносова, Факультет вычислительной математики и кибернетики
119991 Москва, Ленинские Горы, Россия
* E-mail: slinko@polymer.chph.ras.ru
Поступила в редакцию 01.03.2020
После доработки 03.03.2020
Принята к публикации 03.03.2020
Аннотация
В обзоре представлен анализ последних достижений в области изучения нелинейных явлений в гетерогенных каталитических системах. Проанализированы результаты исследований множественности стационарных состояний, колебаний скорости различных реакций и волновых явлений, полученные современными физическими методами. Представлен анализ новых математических моделей, описывающих нелинейные явления в гетерогенных каталитических системах и показана их важность для гетерогенного катализа.
ВВЕДЕНИЕ
Нелинейной динамикой называется междисциплинарная область науки, изучающая нелинейные явления в системах различной природы. К нелинейным явлениям относятся множественность стационарных состояний, автоколебания и волновые режимы. Для их описания обычно используют нелинейные дифференциальные уравнения.
До середины прошлого столетия в области гетерогенного катализа преобладало линейное мышление, в основе которого лежал принцип единственности стационарного состояния протекания гетерогенной каталитической реакции [1]. Наглядным примером таких взглядов являлась теория постоянства удельной каталитической активности [2]. Также считалось, что механизм гетерогенных каталитических реакций одинаков вдали и вблизи от термодинамического равновесия, где возникновение колебаний невозможно, поэтому любое наблюдаемое периодическое изменение скорости реакции считалось ошибочным. Однако скорости гетерогенных каталитических реакций могут описываться нелинейными зависимостями от температуры и от концентрации реагирующих веществ. Нелинейность возникает как в результате взаимодействия частиц на атомно-молекулярном микроуровне, так и за счет влияния реакционной смеси на катализатор на мезо- и макроуровнях. Вследствие этого нелинейные явления широко распространены в гетерогенных каталитических системах. Множественность стационарных состояний была впервые обнаружена в 1953 г. при изучении реакции окисления водорода на никелевом катализаторе [3]. В это же время множественность стационарных состояний наблюдалась в реакции гидрирования оксида углерода на никелевом катализаторе, однако экспериментальные данные смогли быть опубликованы только в 1972 г. [4]. В этом же 1972 г. были впервые обнаружены колебания скорости реакции окисления СО на Pt [5] и реакции окисления водорода на Ni [6], а также впервые отмечались волновые явления в ходе протекания гетерогенной каталитической реакции [7]. За прошедшие годы было опубликовано много работ, посвященных изучению данных нелинейных явлений, их результаты представлены в ряде обзоров [8–10]. Последняя обзорная работа была написана около 10 лет назад и была посвящена в основном исследованиям в высоком вакууме, на гранях монокристаллов [11]. За прошедшее десятилетие были обнаружены новые колебательные и волновые явления, произошло бурное развитие физических методов для изучения нелинейных явлений в ходе протекания гетерогенных каталитических реакций при давлении близком к атмосферному. Целью настоящей работы является обсуждение основных научных результатов, полученных в этой области за последнее время, и демонстрация их роли для развития гетерогенного катализа.
1. МНОЖЕСТВЕННОСТЬ СТАЦИОНАРНЫХ СОСТОЯНИЙ
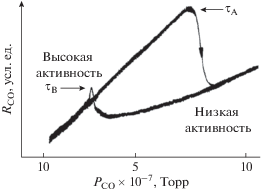
Существует несколько типов множественности стационарных состояний в гетерогенных каталитических реакциях [12]. Каждому типу соответствует гистерезис изменения скорости реакции при вариации концентрации исходного реагента или температуры. Кинетический гистерезис возникает в кинетической области протекания реакции в условиях, когда при одних и тех же внешних параметрах существуют два различных стационарных состояния скорости реакции. Каждое из этих стационарных состояний устанавливается из разных начальных внешних условий, и при варьировании состава реакционной смеси или температуры в реакторе появляются концентрационный или температурный гистерезисы зависимости скорости реакции. Наиболее изучен кинетический гистерезис зависимости скорости реакции окисления СО от концентрации СО на гранях монокристаллов Pt и Pd, наблюдаемый в условиях высокого вакуума [8–10]. На рис. 1 представлен типичный пример гистерезиса скорости реакции окисления СО от концентрации СО, полученный на грани Pt(111) при давлении кислорода 4 × 10–5 Торр [13]. Одновременное измерение сигнала фотоэлектронной эмиссионной спектроскопии (PEEM) и скорости реакции показало, что концентрационный гистерезис в данной системе обусловлен наличием двух устойчивых стационарных состояний протекания реакции: высокореакционного состояния, в котором поверхность покрыта в основном адсорбированным кислородом, и низкореакционного состояния, где преобладают адсорбированные молекулы СО. Такой вывод был сделан на основании PEEM-измерений, т.к. выход фотоэмиссионных электронов определяется работой выхода, которая, в свою очередь, зависит от покрытия поверхности адсорбированными веществами. Покрытие поверхности молекулами СО уменьшает работу выхода, и состояние с низкой активностью можно идентифицировать по светлому цвету на кадрах, полученных с помощью видеокамеры. С другой стороны, адсорбированный кислород повышает работу выхода, и состоянию c высокой скоростью реакции соответствует темный цвет поверхности [14].
Рис. 1.
Кинетический гистерезис зависимости скорости реакции окисления СО на грани Pt(111) от концентрации СО при температуре 413.2 К и давлении кислорода 4 × 10–5 Торр [13].
Достижением последнего времени является изучение кинетической множественности стационарных состояний на поликристаллических поверхностях с помощью усовершенствованного метода PEEM, позволяющего следить за локальным динамическим поведением различных граней монокристалла на поверхности поликристаллического образца в условиях высокого вакуума [15–17]. Таким образом, появилась возможность сравнения кинетических закономерностей одновременного протекания реакции окисления СО на разных гранях монокристалла при абсолютно одинаковых составе реакционного газа и температуре. На рис. 2a и 2б представлены кинетические гистерезисы изменения скорости реакции на поликристаллическом образце Pt от концентрации СО при постоянной концентрации кислорода и температуре, а также гистерезис изменения скорости реакции от температуры при постоянном составе реакционной смеси. Сравнение рис. 1 и 2a показывает, что формы гистерезиса зависимости скорости реакции от концентрации СО на грани Pt(111) и на поликристаллической фольге Pt имеют отличия, в основе которых, как свидетельствуют исследования, проведенные в работах [15, 16], лежат разные условия существования области множественности стационарных состояний для различных граней монокристалла, присутствующих на поликристаллической поверхности. Было продемонстрировано, что переходы между двумя стационарными состояниями на каждой грани происходят при разных концентрациях СО и температуре, поэтому для поликристаллического образца формы гистерезисов от концентрации и от температуры как бы “размываются”.
Рис. 2.
а – Кинетический гистерезис зависимости скорости реакции окисления СО на Pt фольге от концентрации СО при температуре 513 К и давлении кислорода 1.3 × 10–5 мбар; б – кинетический гистерезис зависимости скорости реакции окисления СО на Pt-фольге от температуры при давлении кислорода 1.3 × 10–5 мбар и давлении СО равном 6.6 × 10–6 мбар. Рисунок 2 адаптирован из работы [17].
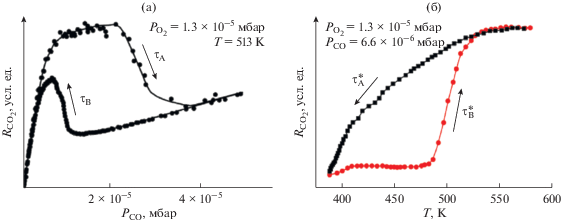
В работе [17] авторы сравнили свойства кинетического гистерезиса зависимости скорости реакции от температуры на поликристаллическом образце Pt двумя различными методами: фотоэлектронной эмиссионной спектроскопии (PEEM) и полевой ионной микроскопии (FIM). В первом случае изучалось динамическое поведение различных граней монокристалла Pt размером в несколько микронов, а во втором случае размер граней монокристалла на поверхности Pt-острия составлял всего лишь несколько нм. Оказалось, что переходы между разными стационарными состояниями скорости реакции окисления СО на различных гранях Pt-острия происходили одновременно в отличие от граней монокристалла поликристаллического образца Pt, где на каждой грани свойства кинетического гистерезиса различались. Авторы [17] объясняют это тем, что в случае нанограней монокристалла диффузия СО эффективна и способна синхронизовать переходы между двумя стационарными состояниями при изменении температуры. В то же время грани монокристалла на поликристаллическом образце разделены более существенными, сложными границами, которые препятствуют эффективной синхронизации переходов из одного состояния в другое с помощью диффузии СО.
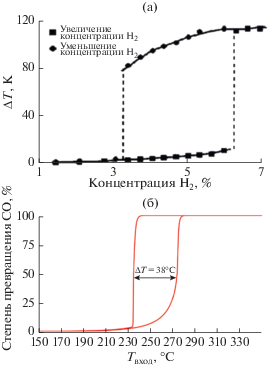
Еще одно интересное явление, которое было обнаружено благодаря изучению множественности стационарных состояний – это влияние различного рода флуктуаций на кинетику гетерогенных каталитических реакций. Кинетический гистерезис зависимости скорости реакции окисления СО от исходной концентрации СО был исследован с использованием молекулярных пучков на модельных Pd-частицах разного размера при достаточно низких давлениях (порядка 10–4 Па) [18]. Было показано, что петля концентрационного гистерезиса сжималась при уменьшении диаметра частиц с 500 до 6 нм, а при размере частиц 1.8 нм гистерезисные явления исчезали. Авторы полагают, что исчезновение концентрационного гистерезиса при уменьшении размера частиц происходит в результате индуцированных быстрых переходов между двумя стационарными состояниями за счет флуктуаций поверхностных покрытий, которые становятся существенными для частиц малого размера. При повышении давления кинетические исследования могут осложняться влиянием процессов переноса вещества и тепла на скорость реакции, и реакция может протекать в диффузионном режиме, где скорость реакции определяется скоростью подвода реагирующих веществ к наружной поверхности катализатора [19]. В таких условиях возможно наблюдение множественности стационарных состояний другого рода, а именно наличие двух стационарных состояний скорости реакции, соответствующих протеканию реакции в кинетической и диффузионной областях. На рис. 3a представлен концентрационный термокинетический гистерезис зависимости перегрева температуры гранулы Pd/Al2O3 катализатора в реакции гидрирования этилена от концентрации Н2 в исходной смеси [20]. На рис. 3б приведен температурный термокинетический гистерезис зависимости степени превращения СО от температуры исходной реакционной смеси на типичном монолитном катализаторе для дожигания выхлопных газов [21]. Сравнение рис. 2б и 3б показывает, что температурные гистерезисы в обоих случаях могут выглядеть практически одинаково. Однако между кинетическими и термокинетическими гистерезисами существует большая разница. Если на рис. 2б гистерезисная зависимость от температуры обусловлена наличием множественности стационарных состояний в кинетической области, то на рис. 3б низкореакционное состояние соответствует протеканию реакции в кинетической области, а высокореакционное состояние представляет собой диффузионный режим, где существует разница температур между катализатором и газовой фазой, и скорость реакции определяется диффузией реагирующих веществ к наружной поверхности катализатора.
Рис. 3.
а – Термокинетический гистерезис зависимости перегрева температуры гранулы катализатора Pd/Al2O3 в реакции гидрирования этилена при изменении концентрации Н2 в исходной смеси [20]; б – термокинетический гистерезис зависимости степени превращения СО от температуры исходной реакционной смеси на типичном монолитном катализаторе для дожигания выхлопных газов [21].
Такое сходство гистерезисов различной природы вызывает трудности и многочисленные ошибки в интерпретации экспериментальных данных по изучению процесса окисления СО на металлах Pt-группы при атмосферном давлении. Если в условиях высокого вакуума гистерезисные зависимости всегда имеют кинетическую природу, то при атмосферном давлении природа возникновения множественности стационарных состояний может быть различна. При атмосферном давлении скорость реакции окисления СО на металлах платиновой группы настолько высока, что исключение влияния процессов массопереноса на скорость реакции в высокоактивном состоянии является трудной задачей, и для выяснения природы множественности стационарных состояний необходимо оценивать воздействие процессов переноса на скорость реакции и рассчитывать коэффициенты массопереноса. В работе [22] была создана проточно-циркуляционная установка, позволяющая измерять линейную скорость потока у поверхности Pt-фольги во время реакции окисления СО и определять скорость массопереноса. Авторы показали, что в состоянии с низкой активностью, где поверхность покрыта в основном молекулами СО, процессы массопереноса, не влияют на скорость реакции, однако в высокореакционном состоянии скорость реакции полностью определяется процессами массопереноса. Позднее эти же авторы проанализировали кинетические данные по изучению реакции окисления СО на различных металлах Pt-группы, как массивных, так и нанесенных [23]. Было продемонстрировано, что даже при больших значениях коэффициента переноса в интервале от 29 до 205 см/с процессы массопереноса по-прежнему влияли на скорость реакции в верхнем стационарном состоянии. Для нанесенных катализаторов диффузионные осложнения в высокоактивном состоянии возникали даже при низких температурах порядка 180°C.
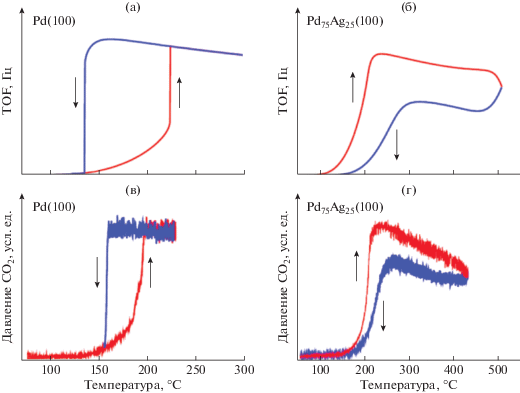
Надо отметить, что наряду с обычными кинетическими и термокинетическими гистерезисами (изменение скорости реакции при варьировании параметра происходит против часовой стрелки, “counterclockwise hysteresis”) в последнее время обнаружены так называемые “обратные” гистерезисы (“reversed hysteresis”), где изменение скорости реакции при варьировании параметра осуществляется по часовой стрелке (“clockwise hysteresis”). На рис. 4 представлены кинетические гистерезисы зависимости скорости реакции от температуры при протекании реакции окисления СО на грани Pd(100) и на той же грани сплава палладия и серебра Pd75Ag25(100). Из рисунка видно, что на грани Pd(100) наблюдается обычный гистерезис зависимости числа оборота (TOF) и скорости образования СО2 от температуры, где температура перехода в активное состояние (температура зажигания) выше, чем температура перехода в нижнее стационарное состояние (точка затухания) [24]. Однако при протекании реакции окисления СО на сплаве с серебром температура затухания становится выше, чем температура зажигания, и наблюдается “обратный” гистерезис. Причина изменения характера гистерезиса связана с динамическим изменением состава поверхности сплава при нагревании. Если при низких температурах на поверхности присутствует в основном Pd, то с ростом температуры увеличивается поверхностная концентрация атомов Ag, повышается энергия активации реакции и возрастает температура, при которой происходит блокировка поверхности молекулами СО.
Рис. 4.
Обычный кинетический гистерезис в реакции окисления СО на грани Pd(100) (а, в) и обратный кинетический гистерезис в этой же реакции на грани Pd75Ag25(100) (б, г). Общее давление 0.7 мбар, отношение O2 /CO равно 10 : 1 [24].
Превращение обычного термокинетического гистерезиса зависимости скорости реакции окисления СО на Pt/Al2O3 в “обратный” обнаружено при добавлении в исходную смесь С3Н6 [25, 26]. В качестве причины такой трансформации авторы видят образование поверхностных соединений, которые являются промежуточными в реакции окисления С3Н6. Блокируя активные места для адсорбции СО, они вызывают снижение скорости реакции окисления СО и повышение температуры затухания процесса. Таким образом, изменение характера и формы гистерезиса с “нормального” на “обратный” свидетельствует о появлении в системе процессов, приводящих к изменению активности катализатора.
Надо отметить, что свойства гистерезиса, а именно ширина петли, могут существенно зависеть от скорости изменения исходной концентрации или температуры. Было показано, что при большой скорости нагрева и охлаждения можно наблюдать петлю термокинетического гистерезиса даже в отсутствии множественности стационарных состояний [27]. Такой “ложный” гистерезис называют динамическим. Как было обнаружено в работе [27], природа динамического гистерезиса связана с запаздыванием изменения температуры катализатора по сравнению с температурой подаваемой реакционной смеси при достаточно быстрой скорости нагрева и/или охлаждения реактора. Согласно работе [28] “ложный” или как его называют авторы “time-dependent hysteresis” может возникать в реакции окисления NO в NO2 на Pt-катализаторе по другой причине. В вышеуказанной системе наблюдалась существенная разница в степени превращения NO при нагреве и охлаждении реакционной смеси за счет того, что при нагреве Pt-катализатор был в восстановленном, а при охлаждении – в окисленном состоянии. Однако при длительном нахождении системы в верхнем активном состоянии происходило уменьшении активности катализатора за счет его окисления, а в нижнем малоактивном состоянии скорость реакции возрастала за счет восстановления Pt. Таким образом, с течением времени или при очень медленной скорости изменения температуры петля гистерезиса полностью исчезала, что указывало на отсутствие множественности стационарных состояний и наличие “ложного” гистерезиса в данной системе.
Приведенные выше данные показывают, как исследование кинетической множественности стационарных состояний в реакции окисления СО на Pt методами PEEM и FIM позволило найти различия протекания этой реакции на мезо- и нано уровнях в условиях высокого вакуума. Изучение зависимости ширины гистерезиса в реакции окисления СО в зависимости от размера частиц Pd-катализатора способствовало выявлению влияния флуктуаций поверхностных покрытий на скорость гетерогенной каталитической реакции, что практически невозможно оценить, исследуя стационарное поведение гетерогенной каталитической реакции. Изучение формы петли гистерезисных зависимостей также позволяет установить наличие процессов или промежуточных соединений, приводящих к снижению активности катализатора.
2. КОЛЕБАНИЯ СКОРОСТИ РЕАКЦИИ
Наряду с гистерезисными явлениями в гетерогенных каталитических реакциях широко наблюдаются колебательные режимы. Перечень известных до 2005 г. гетерогенных каталитических реакций опубликован в приложении к введению в специальном выпуске журнала “Catalysis Today”, который был посвящен колебательным реакциям в гетерогенном катализе [29]. За прошедшие 15 лет были обнаружены и исследованы новые автоколебательные гетерогенные каталитические системы, представленные в табл. 1. Ссылки даны на работы, где впервые упоминается данная колебательная реакция.
Таблица 1.
Автоколебательные гетерогенные каталитические системы, обнаруженные за последние 15 лет
| Реакция | Катализатор [ссылка] |
|---|---|
| CO + O2 | Ni [30]; Co [31]; Cu [32] |
| CH4 + O2 | Rh/H-ZSM-5 [33]; Ru/Al2O3, Ru/SiO2 [34] |
| C2H6 + O2 | Ni [35]; Co [35]; Pd [36] |
| C3H8 + O2 | Pt/YSZ [37]; Pd [38] |
| C4H10 + O2 | Pd [38] |
| C2H4 + O2 | Ni [39]; Co [40] |
| C3H6 + O2 | Ni [40]; Co [40] |
| C6H5CH3 + O2 | Pd–Au [41] |
| C4H5OH + O2 | Pd [42] |
| H2 + O2 | Ru [43] |
| Разложение N2O | Co-ZSM-5 [44]; Fe/Pt-FER [45] |
Все выявленные новые колебательные системы относятся к реакциям окисления, за исключением реакции разложения N2O. Во всех случаях природа колебаний связана с возможностью периодического окисления–восстановления активных центров катализатора. Это относится также и к реакции разложения N2O, т.к. в определенных условиях молекула N2O может одновременно выступать в роли и окислителя, и восстановителя, что приводит к периодическому изменению степени окисления активных центров катализатора [46].
2.1. Колебания в реакции окисления СО на Ni, Co и Cu
В отличие от колебаний скорости реакции окисления СО на Pt, Pd, Ir, Rh, Ru, на Ni, Co и Cu колебательные режимы были обнаружены в избытке СО. Это и являлось основной причиной, почему ранее автоколебания скорости реакции окисления СО на данных катализаторах не наблюдались. По аналогии с металлами платиновой группы колебательные режимы на Ni, Co и Cu пытались найти в избытке кислорода, тогда как эти металлы существенно от них отличаются. Теплоты адсорбции О2 на Ni, Co и Cu значительно больше чем на Pt и Pd, и они намного легче окисляются [47].
Колебания скорости реакции окисления СО на всех трех катализаторах в виде металлических фольг изучали, используя один и тот же состав реакционной смеси, содержащий 40% CO и 10% O2 в гелии [30–32]. На рис. 5a показаны колебания скорости реакции окисления СО на никелевой фольге при 550°С.
Рис. 5.
Периодические изменения концентраций реагирующих веществ, продуктов и температуры катализатора в ходе автоколебаний скорости реакции: а – 40% CО и 10% O2 в гелии при 550°С; б – 40.7% CH4 и 7.3% O2 в гелии при 730°С. Скорость потока реакционной смеси составляла 20 мл/мин. Рис. 5 адаптирован из работы [50] с разрешения Pleiades Publishing.
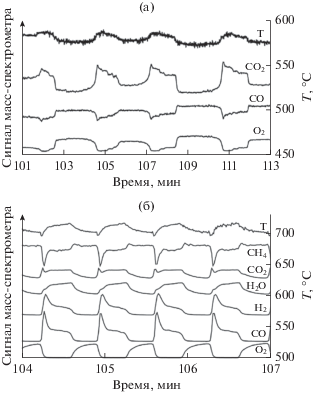
В табл. 2 приведены области температур возникновения колебаний и границы изменения периода колебаний. Наибольший период колебаний наблюдался для Ni, а наименьший для Cu. В случае Co были зарегистрированы только нерегулярные колебания, и период можно было оценить, только используя основную частоту, рассчитанную из спектров Фурье.
Во всех трех системах колебания возникали в результате периодического окисления-восстановления поверхности катализатора. Для всех трех катализаторов металлическая фаза была более активна в реакции окисления СО, чем оксидная. В каждом случае металлическую и оксидную фазы катализатора можно было различить визуально по цвету поверхности в окисленном и в восстановленном состояниях. Визуальные наблюдения за поверхностью катализатора показали, что в ходе колебаний процессам окисления и восстановления соответствовали волновые процессы на поверхности, затрагивающие также объем катализатора.
Наиболее сложная пространственно-временная картина была характерна для Со-катализатора, для которого возможно наличие двух оксидных фаз, а именно СоО и Co3O4. Именно присутствие двух различных оксидных фаз, обладающих достаточно большой активностью в реакции окисления СО, приводило к возникновению сложных пространственных структур на поверхности Со-катализатора [31].
2.2. Колебания в реакциях окисления С1–С4 углеводородов
Первыми реакциями окисления углеводородов, в которых были обнаружены колебательные режимы, являлись реакции окисления метана [48] и пропана [49] на никелевых катализаторах. Как видно из табл. 1, за последние годы были зафиксированы колебания в реакциях окисления этана, бутана, этилена и пропилена. Кроме никелевых катализаторов колебания в реакции окисления углеводородов могут наблюдаться также на Co, Pd, Pt, Rh и Ru. На рис. 5б показано периодическое изменение концентраций реагирующих веществ, продуктов и температуры катализатора в ходе автоколебаний скорости реакции окисления метана на никелевой фольге при 730°С [50].
Причина возникновения колебаний во всех реакциях окисления углеводородов – периодическое окисление–восстановление металлического катализатора, однако для каждой системы есть свои особенности протекания реакции и переключения из окисленного состояния в восстановленное и обратно. Так на Ni, Co, Ru и Rh колебательные режимы протекали с изменением селективности процесса окисления СН4, причем окисленная фаза катализатора была более активна в реакции глубокого окисления с образованием СО2 и Н2О, а восстановленная фаза в виде металла – в реакции парциального окисления с образованием СО и Н2. Исключением являются реакции окисления углеводородов С1–С4 на Pd [38] и реакция окисления пропилена на Pt [37], для которых колебательные режимы наблюдаются при достаточно низкой температуре, когда протекает реакция только глубокого окисления углеводорода [38]. Надо также отметить, что во всех реакциях колебательные режимы были зарегистрированы в избытке углеводорода кроме процесса окисления пропана на катализаторе Pt/YSZ, где колебания возникали при соотношении парциальных давлений ${{{{Р}_{{{{{\text{О}}}_{{\text{2}}}}}}}} \mathord{\left/ {\vphantom {{{{Р}_{{{{{\text{О}}}_{{\text{2}}}}}}}} {{{Р}_{{{{{\text{С}}}_{{\text{3}}}}{{{\text{Н}}}_{{\text{8}}}}}}}}}} \right. \kern-0em} {{{Р}_{{{{{\text{С}}}_{{\text{3}}}}{{{\text{Н}}}_{{\text{8}}}}}}}}}$ в интервале 1.2–2.2 [37]. Это, по-видимому, связано с тем, что коэффициенты прилипания углеводородов на большинстве металлических поверхностей намного меньше, чем коэффициенты прилипания кислорода, и для создания условий попеременного окисления–восстановления металлических катализаторов необходим избыток углеводорода.
Впервые прямое, а не косвенное доказательство того, что механизм колебаний скорости реакции окисления СН4 на никелевых катализаторах обусловлен процессами окисления–восстановления, было получено в работе [51]. Методом термогравиметрии (ТГА) совместно с online масс-спектрометрическим анализом газовой фазы и методом термопрограммируемой реакции (ТПР) было показано, что несколько десятков монослоев кислорода в пористом никелевом катализаторе участвовали в колебательном процессе. Позднее были найдены доказательства изменения концентрации оксида никеля в ходе колебаний методами рентгеновской дифракции и рентгенофазового анализа (РФА) [52]. Данные, полученные методом in situ РФА в ходе колебаний, продемонстировали, что в течение периода низкой активности никелевая фольга была покрыта пленкой оксида никеля NiO, а переход в состояние с высокой активностью сопровождался полным восстановлением оксида никеля до металлического состояния. Применение метода рентгеновской фотоэлектронной спектроскопии (РФЭС) в режиме in situ было возможно только для реакции окисления пропана на Ni, так как до настоящего времени только в этой реакции удалось зафиксировать колебания скорости реакции при давлении порядка 0.5 мбар [53]. In situ РФЭ-спектры показали, что в течение периода низкой активности реакции окисления пропана никелевая фольга покрыта пленкой NiO толщиной по крайней мере 3 нм. Переход в состояние с высокой активностью в ходе одного цикла колебаний так же, как и при окислении метана, сопровождался восстановлением оксида никеля до металлического состояния. Позднее авторы применили in situ метод EXAFS, который использует анализ дальней тонкой структуры рентгеновских спектров поглощения и может зафиксировать процесс окисления на большей толщине приповерхностного слоя [54] по сравнению с методом РФЭС. Было показано, что в ходе колебаний скорости реакции окисления пропана глубина окисления никелевой фольги больше 3 нм и достигает величины в 15 нм.
Надо отметить, что наряду с развитием новых in situ физических методов исследования катализаторов, таких как EXAFS и XANES спектроскопии, требующих дорогих специализированных источников синхротронного излучения, не теряют актуальности и такие традиционные для катализа методы, как ТГА, ТПВ и импульсный метод. Так, с помощью ТГА совместно с online масс-спектрометрическим анализом газовой фазы и методом ТПР удалось выявить существенное отличие колебаний скорости реакции окисления углеводородов на Pd от других металлов. Было показано, что если на Ni-катализаторе изменение веса в ходе колебаний скорости реакции окисления метана происходит за счет изменения количества кислорода в приповерхностных слоях, а концентрация растворенного углерода меняется лишь незначительно, то для Pd-катализаторов наблюдается обратная картина. Изменение веса в ходе колебаний скорости реакции окисления метана на Pd определялось в основном периодическим изменением концентрации углерода в приповерхностном слое катализатора, которое могло достигать нескольких сотен монослоев, а концентрация растворенного кислорода менялась в значительно меньшей степени [55]. Более того, в последнее время было показано, что традиционный импульсный метод может быть применен к изучению автоколебаний скорости реакции [56]. При этом постоянную подачу в реактор реакционной смеси заменяют подачей последовательными отдельными одинаковыми импульсами в течение достаточного времени, что позволяет отследить весь цикл колебания, а по изменению формы импульса-отклика сделать выводы о природе протекающих процессов на различных стадиях колебательного цикла. Расчет баланса по углероду и кислороду в каждом импульсе дает возможность судить о накоплении этих веществ в объеме катализатора. Однако вышеуказанный метод может быть применим только к реакциям, обладающим “эффектом памяти”, т.е. система должна запоминать состояние после каждого импульса и сохранять его в потоке инертного газа до следующего импульса. В настоящее время наиболее информативным оказалось использование импульсного метода для реакции окисления метана на Ni, где существует только одна устойчивая форма оксидной фазы NiO [56]. Было установлено, на каких стадиях колебательного цикла происходит накопление и расходование растворенных в никелевом катализаторе углерода и кислорода. Надо отметить, что применение современных in situ РФЭС [52] и in situ EXAFS [53] не позволило зафиксировать ни одну из форм углерода в ходе автоколебаний скорости реакции окисления метана и пропана на никелевых катализаторах и выявить их роль в данных колебательных процессах.
Кроме наличия периодических процессов окисления–восстановления катализатора, есть еще одно свойство системы, которое объединяет колебания скорости реакции окисления различных углеводородов на разных катализаторах, – это индукционный период, предшествующий зарождению колебаний. Наличие определенного периода времени, необходимого для появления колебательного режима на “свежем” образце, было впервые обнаружено в реакции окисления СН4 на Pd [48], Ni [57], Co [58], Rh [33] и в реакции окисления C3H8 на Ni [59]. Длительность индукционного периода зависит от таких факторов как температура, состав и давление реакционной смеси, а также типa реакции и катализатора. При повышении температуры в реакторе и давления в системе длительность индукционного периода уменьшается. Так при одном и том же составе реакционной смеси и температуре время индукционного периода в реакции окисления метана при давлении 1000 мбар составляло около 45 мин, тогда как для окисления пропана при давлении 0.25 мбар длительность индукционного периода значительно увеличивалась и составляла около 9 ч [60]. Методы РФА и РФЭС не определили никаких изменений в составе катализатора в ходе индукционного периода, однако метод сканирующей электронной микроскопии (СЭМ) зафиксировал необратимое формирование пористой структуры с развитой поверхностью верхнего слоя металлического никеля, которое приводило к росту активности катализатора [60, 61]. На рис. 6 представлено СЭМ-изображение никелевой фольги, которую использовали в качестве катализатора окисления метана при температуре ~680°C в течение часа. Как видно из рис. 6a и 6б, полученных с меньшим увеличением, поверхность катализатора становится пористой, характерный размер пор равен порядка 0.3 мкм [62].

Однако роль индукционного периода состоит не только в том, чтобы повысить активность катализатора, но и в том, чтобы вывести систему на такие условия, когда будет возможна периодическая смена процессов окисления–восстановления. Так, в работах, посвященных изучению реакции окисления метана на Rh [33] и Pd [63], было показано, что если катализатор окисляли перед началом эксперимента, то в ходе индукционного периода для возникновения колебательного режима требуется его частичное восстановление в реакционной смеси, и, наоборот, для восстановленного катализатора необходимо некоторое время для его частичного окисления во время протекания реакции.
Надо отметить, что формирование пористого слоя в приповерхностной области металлического катализатора в ходе колебаний скорости наблюдалось не только для реакций окисления углеводородов на Ni [61] и Pd [63], но также и для реакции окисления СО на Со-фольге [32]. По-видимому, это общее явление для всех колебательных реакций, механизм которых обусловлен периодическим окислением–восстановлением приповерхностного слоя катализатора. Как указывают авторы в работах [60, 62], одной из причин разрыхления поверхности может быть различие мольных объемов металла и окисла. Попеременные процессы окисления и восстановления приводят к образованию пористой структуры и увеличению площади активной поверхности катализатора. Для Pd-катализаторов формирование пористой структуры в ходе колебаний происходит за счет процессов окисления–восстановления, а также вследствие диффузии углерода в приповерхностный слой катализатора и обратно. На рис. 7 показано образование пористой структуры Pd-порошка в результате проведения реакции окисления СН4 в автоколебательном режиме в течении двух часов [64]. Было также установлено: несмотря на то, что свойства колебаний скорости реакции окисления углеводородов С1–С4 примерно одинаковы, они по-разному разрабатывали активную поверхность Pd-катализатора. За одно и то же время степень разработки поверхности в ходе колебательного процесса снижалась в ряду С4Н10 < С3Н8 < < С2Н6 < СН4 симбатно с уменьшением количества растворенного углерода [64]. Более того, было продемонстрировано, что на катализаторе, разработанном колебаниями скорости реакции окисления углерода с количеством атомов углерода N, невозможно наблюдать колебания скорости реакции окисления углеводорода с числом атомов N – 1 (т.е. нельзя изучать колебания скорости реакции окисления этана после метана и т.д.) [38].

Способность колебаний скорости различных реакций окисления разрыхлять поверхность катализатора может быть использована для получения катализаторов с развитой наружной поверхностью и более высокой активностью. Авторы работы [64] показали, что автоколебания скорости реакции разрыхляют поверхность никелевых и палладиевых катализаторов и увеличивают их активности в стационарно протекающих реакциях углекислотной конверсии метана, гидрирования этилена на Ni и реакции глубокого окисления СН4 на Pd.
3. ПРОСТРАНСТВЕННО-ВРЕМЕННЫЕ СТРУКТУРЫ НА ПОВЕРХНОСТИ КАТАЛИЗАТОРА
Пространственно-временные структуры на поверхности катализатора наблюдаются как в изотермических, так и в неизотермических условиях. В высоком вакууме на различных монокристаллах Pt и Rh методом фотоэмиссионной электронной микроскопии (PEEM) были обнаружены изотермические волновые явления [8, 11]. В частности, с помощью PEEM была подробно изучена реакция CO + O2 на Pt(110) при низких давлениях. В зависимости от давлений реагентов и температуры катализатора система представляла собой колебательную, возбудимую или бистабильную среду. Было зафиксировано большое разнообразие пространственно-временных структур: однородные колебания, реакционные фронты, бегущие и стоячие волны, спиральные волны с различной частотой вращения, “химическая турбулентность”. В 2007 г. эти результаты вошли в Нобелевскую лекцию лауреата по химии Герхарда Эртла [65].
Применение электронных микроскопов, работающих при околоатмосферных давлениях (near ambient pressure, NAP), является новым способом изучения пространственно-временных структур на поверхности катализатора. Например, сканирующая электронная микроскопия (scanning electron microscopy, SEM) дает возможность наблюдать образование пространственных структур на поверхности при температурах до 1500°С и в широком интервале давлений, от 10–5 до 103 Пa. С помощью SEM в ходе реакции гидрирования NO2 на Pt-фольге при давлениях 10–5–102 Пa удалось увидеть бегущие реакционные фронты и спиральные волны, зафиксировать процессы спилловера и активирование неактивных участков поверхности за счет протекания реакции на соседних участках [66]. С помощью электронной микроскопии низких энергий (low energy electron microscopy, LEEM) была изучена реакция CO + O2 на Pt при общем давлении до 10 Пa [67]. Применение NAP-LEEM позволило увидеть появление локальных возмущений размером менее 3 мкм на структурных дефектах поверхности Pt(110), а также формирование обрывков спиралей на поликристаллической платине. По сравнению с результатами, ранее полученными при низких давлениях с помощью PEEM, характерные размеры пространственных структур при более высоких давлениях уменьшились приблизительно в десять раз. Авторы объяснили эти наблюдения снижением эффективной скорости диффузии адсорбированных молекул CO на сильно заполненной адсорбатом поверхности при высоких давлениях, поскольку именно диффузия CO определяет характерные пространственные размеры бегущих волн.
При атмосферном давлении до недавнего времени наиболее успешно применен метод ИК-термографии, с помощью которого в различных реакциях окисления были зафиксированы неизотермические бегущие волны, “горячие пятна” и “дышащие” структуры [7, 9, 10]. В последнее время для изучения пространственных структур при атмосферном давлении был успешно использован метод визуального наблюдения за состоянием поверхности металлических катализаторов в ходе колебательных реакций [30, 35, 36, 39]. Этот метод оказался наиболее информативен и полезен при наблюдении пространственных структур в ходе протекания колебательных процессов на никелевых и кобальтовых катализаторах, для которых существует значительное различие в цвете восстановленной и окисленной поверхности. Визуальное наблюдение за цветом поверхности позволило впервые зафиксировать состояние поверхностной турбулентности на поверхности катализатора при атмосферном давлении. Это было сделано в ходе реакции окисления этана на кобальтовой фольге [35]. Также было показано, что скорость реакции, рассчитанная по изменению концентраций в газовой фазе, была постоянна, несмотря на сложный, динамический режим на поверхности катализатора.
Наиболее изучены волновые явления на Ni- и Co-фольгах в реакциях окисления СН4 [68, 69] и СО при атмосферном давлении [50, 30 ], окисления пропана при давлении 0.5 мбар [70]. Волновые явления в реакциях окисления СН4 и СО на никелевой фольге удивительно похожи, хотя автоколебательные режимы, представленные на рис. 5, сильно различаются по составу продуктов и температуре катализатора. В обеих реакциях в ходе колебаний скорости реакции происходила периодическая смена цвета поверхности. При этом волна темного цвета, соответствующая окислению образца Ni, двигалась сверху вниз в направлении потока реакционной смеси, как показано на рис. 8 для случая реакции окисления СО на Ni. После прохождения волны окисления весь Ni находился в окисленном состоянии с малой активностью. При этом продуктами реакции окисления СН4 в газовой фазе были в основном СО2 и Н2О. Для реакции окисления СО на окисленной поверхности также наблюдался минимум скорости реакции образования СО2. Во время процесса восстановления цвет поверхности никеля менялся с темного на светлый. При этом волна восстановления двигалась снизу вверх против потока реакционной смеси. На восстановленной поверхности наряду с небольшими количествами СО2 и Н2О резко возрастали концентрации продуктов парциального окисления метана СО и Н2, а в реакции окисления СО достигалось максимальное образование СО2 [30 , 50]. Во время колебаний скорости реакции окисления пропана на Ni-фольге при давлении 0.5 мбар волны восстановления зарождались в центре образца и распространялись к периметру границы катализатора, а волна окисления двигалась в обратном направлении [70]. По-видимому, такое различие в характере распространения волн связано с меньшей скоростью реакции и отсутствием градиента концентраций по газовой фазе при более низком общем давлении.
Рис. 8.
Фотографии поверхности никелевой фольги во время распространения волны восстановления (светлый цвет) и окисления (более темные цвета побежалости) наряду с колебаниями концентрации СО2 в газовой фазе в указанные моменты времени. Реагирующая смесь, содержащая 40% CO и 10% O2 в He, подается сверху вниз со скоростью 30 мл/мин при температуре 570°C.
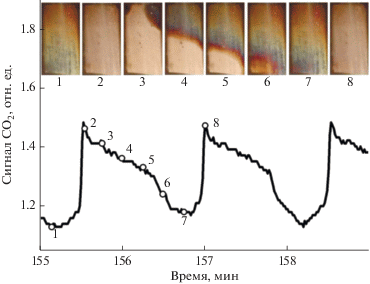
Скорость движения волн окисления в ходе колебаний скорости реакции окисления СО и СН4 зависела от температуры, причем с повышением температуры скорость распространения фронта волны восстановления увеличивалась, а скорость распространения волны окисления падала. При этом скорость распространения волны восстановления всегда превышала скорость движения волны окисления. Замедление движения волны окисления с ростом температуры можно объяснить большей глубиной проникновения кислорода вглубь катализатора и большей толщиной образующейся оксидной пленки при более высокой температуре [30]. На основе сходства свойств автоколебаний скорости реакций окисления СО и СН4 можно сделать вывод о том, что основной общей причиной возникновения колебательных режимов являются периодические процессы окисления и восстановления Ni, которые протекают практически одинаково как для “простой”, малостадийной реакции окисления СО, так и для “сложной”, многостадийной реакции окисления метана. Это означает, что участие углерода в восстановлении никелевого катализатора не играет ключевой роли в механизме возникновения колебаний скорости реакции окисления СН4 на Ni [50].
Как видно из рис. 8, во время прохождения волны окисления в ходе реакций окисления СО и СН4 происходит сложное изменение цвета Ni-катализатора, которое можно наблюдать при просмотре видеофильмов, представленных на сайте ИХФ РАН [71]. Это обусловлено тем, что пленка NiO с изменяющейся толщиной представляет собой дифракционную решетку. Наблюдаемый цвет никелевой фольги зависит от интерференции между светом, отраженным от низа и верха оксидной пленки [72]. По мере увеличения толщины пленки никеля цвет меняется от соломенного к кирпично-коричневому, а потом голубому и темно-синему [71]. Наиболее красивые изменения цвета в ходе протекания колебательного процесса были обнаружены для реакции окисления СО на фольге Cu [32]. На рис. 9 и видеофильме, представленном в дополнительном материале к статье [32], можно заметить во время прохождения волны окисления всевозможные цвета побежалости меди, включая розовый, малиновый, сиреневый, синий, голубой и серый.
Рис. 9.
Изменение цвета поверхности медной фольги при распространении волны восстановления (a–е) и волны окисления (ж–м) в ходе реакции окисления СО на Cu фольге при 540°C. Скорость подачи реакционной смеси, содержащей 40% CO, 10% O2 и 3.85% Ar в He, составляла 20 мл/мин (дополнительный материал к статье [32]).

Самые сложные пространственные структуры, включая режим поверхностной турбулентности, были зафиксированы в реакции окисления СО на Co [31]. Как уже указывалось выше, колебания скорости реакции окисления на Со всегда были нерегулярными, что обусловлено наличием двух оксидных форм, а именно СоО и Co3O4, активность которых была ненамного меньше чем у металлического Co. Это приводило к возможности зарождения волн в разных местах поверхности и к появлению кольцевых и полукольцевых волн из одного центра зарождения. Небольшое различие в активности разных фазовых состояний кобальта являлось, по-видимому, причиной нарушения синхронизации колебаний на различных местах поверхности и возникновения режима поверхностной турбулентности [31].
4. МАТЕМАТИЧЕСКОЕ МОДЕЛИРОВАНИЕ НЕЛИНЕЙНЫХ ЯВЛЕНИЙ В ГЕТЕРОГЕННЫХ КАТАЛИТИЧЕСКИХ СИСТЕМАХ
Анализ причин возникновения нелинейных явлений в гетерогенных каталитических системах невозможен без построения кинетических моделей и без их нелинейного анализа. Далее будет приведен ряд примеров, когда математическое моделирование помогло понять и осмыслить ряд наблюдаемых экспериментальных данных.
4.1. Математическое моделирование автоколебаний в реакции окисления CO на металлах Pt-группы при атмосферном давлении
В последнее время в теории колебаний скорости гетерогенных каталитических реакций остро стоит вопрос о возможности установления наиболее активной фазы металла (окисленное или восстановленное состояние) из анализа свойств автоколебательных режимов, которые возникают в результате периодического окисления–восстановления катализатора. За последние 10–12 лет были созданы методы in situ слежения за составом и структурой поверхности катализатора в ходе колебательных режимов при около- или атмосферном давлении, включая сканирующую туннельную микроскопию при высоком давлении (high pressure, STM), рентгеновскую спектроскопию поглощения (X-ray absorption spectroscopy, XAS) и метод поверхностной дифракции рентгеновских лучей (Surface X-Ray Diffraction, SXRD). Эти физические методы стали применяться для изучения состава поверхности и степени окисления катализатора одновременно с измерением скорости реакции окисления СО на металлах Pt-группы в ходе колебательных режимов при атмосферном давлении. Так в работе [73] использовали in-situ метод дальней тонкой структуры рентгеновских спектров поглощения (EXAFS) одновременно с масс-спектрометрическим анализом газовой фазы для изучения колебаний скорости реакции окисления СО на Pt/γ-Al2O3. Было обнаружено, что во время колебаний максимум скорости реакции совпадал с максимумом XAS-сигнала от окисла Pt. На основании полученных данных был сделан вывод о том, что наиболее активной фазой в реакции окисления СО является окисел Pt. К аналогичным заключениям пришла голландская группа Френкена (J.W.M. Frenken) в работах [74–76], где реакцию окисления СО при околоатмосферном давлении на гранях монокристаллов Pt и Pd изучали с помощью сканирующей туннельной микроскопии и методом поверхностной дифракции рентгеновских лучей. Также было обнаружено, что в ходе колебаний максимум сигнала SXRD соответствовал максимуму концентрации СО2 [76]. Авторы утверждали, что широко используемая ранее математическая модель S.–T.–M. (Sales–Turner–Maple) [77], описывающая колебания в реакции окисления СО на металлах Pt-группы за счет процессов окисления-восстановления, ошибочна, т.к. именно окисленные узлы решетки металла являются наиболее активными каталитическими центрами. Тем самым подтверждался механизм Марс-ван Кревелена и отрицался традиционный механизм Ленгмюра–Хиншельвуда для реакции окисления CO на металлах платиновой группы.
Против данной точки зрения высказывался профессор Гудмен (D.W. Goodman) с сотр. [78, 79], которые использовали обычные кинетические измерения и инфракрасную абсорбционную спектроскопию с поляризационной модуляцией (polarization modulation infrared absorption spectroscopy, PM-IRAS). Ими критиковались выводы, сделанные в работах группы Френкена по поводу высокой активности окисленного катализатора. Было высказано предположение, что небольшого количества (~0.01%) активных узлов “чистого” металла достаточно, чтобы обеспечить наблюдаемую скорость реакции [79]. Позднее появились и другие экспериментальные работы, показывающие, что восстановленная фаза катализатора является наиболее активной в реакции окисления СО [80–82].
Интерпретация результатов экспериментальных измерений является важной задачей, не всегда имеющей единственное и очевидное решение. Помочь здесь могут методы математического моделирования, позволяющие избежать неверных выводов, которые кажутся очевидными лишь на первый взгляд. В наших работах [83, 84] было показано, каким образом математическое моделирование может помочь в интерпретации данных и разрешить спорные вопросы о наиболее активном состоянии катализатора в реакции окисления СО.
В работе [83] были представлены результаты моделирования реакции окисления CO на монокристаллах платиновой группы в условиях избытка кислорода при атмосферном давлении. Математическое моделирование дает достаточно простое объяснение различным нелинейным эффектам, таким как множественность стационарных состояний и автоколебания скорости реакции. Стандартная кинетическая модель реакции окисления СО на металлах платиновой группы выглядит следующим образом:
(1)
$\begin{gathered} {{{\text{d}}{{\theta }_{{\text{O}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{\theta }_{{\text{O}}}}} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = 2{{k}_{1}}{{s}_{1}}{{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}\theta _{*}^{2} - 2{{k}_{3}}\theta _{{\text{O}}}^{2} - {{k}_{5}}{{\theta }_{{\text{O}}}}{{\theta }_{{{\text{CO}}}}}, \\ {{{\text{d}}{{\theta }_{{{\text{CO}}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{\theta }_{{{\text{CO}}}}}} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = {{k}_{2}}{{s}_{2}}{{P}_{{{\text{CO}}}}}\theta {\text{*}} - {{k}_{4}}{{\theta }_{{{\text{CO}}}}} - {{k}_{5}}{{\theta }_{{\text{O}}}}{{\theta }_{{{\text{CO}}}}}. \\ \end{gathered} $Здесь ${{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}$ и ${{P}_{{{\text{CO}}}}}$ – парциальные давления реагентов; ${{k}_{1}}$ и ${{k}_{2}}$ – константы скоростей адсорбции, ${{s}_{1}}$ и ${{s}_{2}}$ – коэффициенты прилипания, а ${{k}_{3}}$ и ${{k}_{4}}$ – константы скоростей десорбции (для CO и O2, соответственно); ${{k}_{5}}$ – константа скорости бимолекулярной реакции. Значения ${{k}_{i}}$ вычисляются из уравнения Аррениуса: ${{k}_{i}} = k_{i}^{0}\exp \left( {{{ - {{E}_{i}}} \mathord{\left/ {\vphantom {{ - {{E}_{i}}} {(R{{T}_{{{\text{cat}}}}})}}} \right. \kern-0em} {(R{{T}_{{{\text{cat}}}}})}}} \right)$, где $k_{i}^{0}$ – предэкспонента, ${{E}_{i}}$ – энергия активации, Тcat – температура катализатора, $R$ – универсальная газовая постоянная. Модель основана на механизме Ленгмюра–Хиншельвуда. Переменными модели являются поверхностные концентрации θО и θСО. Концентрация свободных узлов θ* вычисляется по формуле: θ* = 1 – θО – θСО. Если скоростями десорбции можно пренебречь, то стационарные решения системы (1) определяются из уравнений:
(2)
$\begin{gathered} 2{{k}_{1}}{{s}_{1}}{{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}\theta _{*}^{2} - {{k}_{5}}{{\theta }_{{\text{O}}}}{{\theta }_{{{\text{CO}}}}} = 0, \\ {{k}_{2}}{{s}_{2}}{{P}_{{{\text{CO}}}}}\theta {\text{*}} - {{k}_{5}}{{\theta }_{{\text{O}}}}{{\theta }_{{{\text{CO}}}}} = 0. \\ \end{gathered} $Отсюда следует, что в стационарных условиях значение $\theta {\text{*}}$ определяется по формуле:
(3)
$\theta * = \frac{{{{k}_{2}}{{s}_{2}}{{P}_{{{\text{CO}}}}}}}{{2{{k}_{1}}{{s}_{1}}{{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}}}.$Такое решение имеет физический смысл при $2{{k}_{1}}{{s}_{1}}{{P}_{{{{{\text{O}}}_{{\text{2}}}}}}} > {{k}_{2}}{{s}_{2}}{{P}_{{{\text{CO}}}}}$ (в избытке кислорода). Решение соответствует высокой скорости реакции ${{R}_{{{\text{C}}{{{\text{O}}}_{2}}}}}$, вычисляемой по формуле:
(4)
${{R}_{{{\text{C}}{{{\text{O}}}_{{\text{2}}}}}}} = \frac{{{{{({{k}_{2}}{{s}_{2}}{{P}_{{{\text{CO}}}}})}}^{2}}}}{{2{{k}_{1}}{{s}_{1}}{{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}}}.$Кроме высокореакционного решения, всегда существуют два тривиальных стационарных решения вида (θО = 0, θСО = 1) и (θО = 1, θСО = 0), которые соответствуют нулевой скорости реакции. Обычно десорбцией кислорода можно пренебречь при температурах меньше 700 K, а десорбция CO становится существенной при температурах выше 300 K. Если скорость десорбции CO мала по сравнению со скоростью бимолекулярной реакции, то высокореакционное решение приближенно описывается формулой (4), в то время как низкореакционное решение соответствует поверхности, заполненной молекулами СО (0 $ \ll $ θСО < 1). Эти два стационарных решения дают хорошо известное для реакции окисления CO свойство бистабильности, которое обычно наблюдается в диапазоне температур 300–600 K в зависимости от давлений реагентов, типа реактора и свойств катализатора. Основная причина появления множественности стационарных решений в модели (1) обусловлена различием в механизмах адсорбции CO и O2: для диссоциативной адсорбции O2 требуется два соседних свободных узла на поверхности, в то время как для адсорбции CO требуется один свободный узел. Другим важным условием возникновения множественности является достаточно высокая скорость реакции по сравнению со скоростями адсорбции и десорбции. Устойчивое решение системы (1) с CO-заполненным состоянием поверхности существует при низких температурах катализатора, а высокореакционное решение вида (4) становится устойчивым при высоких температурах. При повышении температуры, начиная с низких значений, осуществляется переход от низко- к высокореакционному состоянию. Такой переход в другое состояние системы может соответствовать либо области множественности (точке поворота), либо быстрому изменению скорости реакции от температуры без области множественности.
Таким образом, бистабильность и гистерезис зависимости скорости реакции от температуры являются характерными свойствами реакции окисления CO. Они могут наблюдаться как при низких, так и при высоких давлениях. Однако система (1) не допускает решений в виде автоколебаний (предельного цикла) ни при каких значениях параметров. Автоколебания могут быть получены в модели S.–T.–M. [77], которая учитывает процессы окисления–восстановления катализатора и является расширением модели (1). В этом случае предполагается, что некоторые узлы поверхности катализатора окислены и практически не участвуют в реакции; их концентрация обозначена как θox, а концентрация свободных узлов определяется как θ* = 1 – θО – θСО – θox. Уравнения модели S.–T.–M. получаются, если к модели (1) добавить следующее обыкновенное дифференциальное уравнение (ОДУ) для переменной θox:
Слагаемые в правой части уравнения описывают процессы окисления и восстановления поверхности катализатора с константами скоростей ${{k}_{6}}$ и ${{k}_{7}}$, соответственно. В работе [83] был предложен другой вариант записи ОДУ для θox, который дополнительно учитывает непосредственную декомпозицию оксида металла (с константой скорости ${{k}_{8}}$) при высоких температурах. Уравнения модели выглядят следующим образом:
(5)
$\begin{gathered} {{{\text{d}}{{\theta }_{{\text{O}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{\theta }_{{\text{O}}}}} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = 2{{k}_{1}}{{s}_{1}}{{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}\theta _{*}^{2} - 2{{k}_{3}}\theta _{{\text{O}}}^{2} - \\ - \,\,{{k}_{5}}{{\theta }_{{\text{O}}}}{{\theta }_{{{\text{CO}}}}} - {{k}_{6}}{{\theta }_{{\text{O}}}} + {{k}_{8}}{{\theta }_{{{\text{ox}}}}}, \\ {{{\text{d}}{{\theta }_{{{\text{CO}}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{\theta }_{{{\text{CO}}}}}} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = {{k}_{2}}{{s}_{2}}{{P}_{{{\text{CO}}}}}\theta {\text{*}} - {{k}_{4}}{{\theta }_{{{\text{CO}}}}} - \\ - \,\,{{k}_{5}}{{\theta }_{{\text{O}}}}{{\theta }_{{{\text{CO}}}}} - {{k}_{7}}{{\theta }_{{{\text{ox}}}}}{{\theta }_{{{\text{CO}}}}}, \\ {{{\text{d}}{{\theta }_{{{\text{ox}}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{\theta }_{{{\text{ox}}}}}} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = {{k}_{6}}{{\theta }_{{\text{O}}}} - {{k}_{7}}{{\theta }_{{{\text{ox}}}}}{{\theta }_{{{\text{CO}}}}} - {{k}_{8}}{{\theta }_{{{\text{ox}}}}}. \\ \end{gathered} $Значения параметров модели приведены в работе [83]. Если считать θox заданным параметром модели, то можно наглядно объяснить механизм автоколебаний в системе (5). Заметим, что формулы (3) и (4) дают хорошее приближение к стационарному решению, поскольку скорости десорбции, окисления и восстановления малы по сравнению со скоростями адсорбции и реакции. Скорость реакции в (4) явно не зависит от значений θox, но решение (3), (4) не является физически допустимым, когда условие $\theta {\text{*}}$ ≤ 1 нарушается при больших значениях θox. На рис. 10 показаны зависимости стационарной скорости реакции от θox для первых двух уравнений системы (5), где значение θox считается параметром. Красная прямая линия на рис. 11 определяется формулой (4) и показывает верхнюю границу скорости реакции (при $k_{5}^{0} \to \infty $). Две другие зависимости построены для $k_{5}^{0} = {{10}^{{13}}}$ и $k_{5}^{0} = {{10}^{{14}}}$ с–1. Стационарная скорость реакции бистабильна: в определенном диапазоне параметра θox существует два устойчивых стационарных состояния. Одно из них является высокореакционным (где поверхность покрыта Oads и оксидом), а другое – низкореакционным (где поверхность покрыта COads и оксидом). Стрелки показывают один цикл колебаний, который осуществляется, если θox является переменной модели. В верхнем высокореакционном состоянии поверхность покрыта Oads, и здесь происходит медленное увеличение θox за счет реакции окисления до тех пор, пока не достигается правая точка поворота. После этого система быстро переключается в низкореакционное состояние, где поверхность преимущественно покрыта СOads. Здесь θox медленно уменьшается за счет реакции восстановления до тех пор, пока не достигается левая точка поворота. Затем система быстро переходит в высокореакционное состояние, и цикл автоколебаний повторяется снова. Рассмотренный циклический процесс описывает достаточно универсальный механизм релаксационных колебаний типа окисление–восстановление, впервые предложенный для модели S.–T.–M. в работе [77]. Этот механизм может быть основой для математического моделирования автоколебаний скорости других реакций окисления (водорода, углеводородов, метанола и др.) на различных катализаторах.
Рис. 10.
Множественность в зависимости скорости реакции от θох для системы (5), где значение θох считается фиксированным параметром, при Tcat = 515 К, ${{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}$ = 2 × 104 Па, PСО = 103 Па. Значения предэкспоненты скорости реакции: $k_{5}^{0} = {{10}^{{13}}}$ с–1 (жирная линия), $k_{5}^{0} = {{10}^{{14}}}$ с–1 (тонкая линия), $k_{5}^{0} \to \infty $ (уравнение (4), прямая линия. Вычисленные устойчивые (неустойчивые) стационарные решения показаны с помощью сплошных (пунктирных) линий. При $k_{5}^{0} = {{10}^{{13}}}$ с–1 точки поворота типа седло–узел показаны кружками. Стрелки (точечный пунктир) демонстрируют переход от высокореакционного к низкореакционному состоянию и обратно.
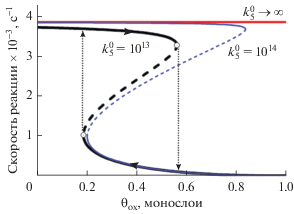
Рис. 11.
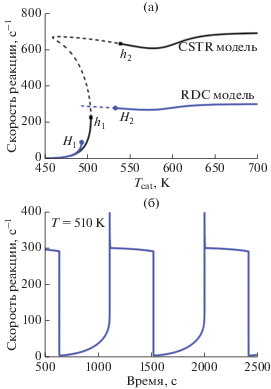
a – Влияние температуры катализатора на стационарные решения при $P_{{{{{\text{O}}}_{2}}}}^{{{\text{in}}}}$ = 100 мбар и $P_{{{\text{CO}}}}^{{{\text{in}}}}$ = 10 мбар. Показаны скорости реакции, вычисленные с помощью процедуры продолжения по параметру для моделей CSTR и 3D-RDC. Устойчивые (неустойчивые) стационарные решения показаны с помощью сплошных (пунктирных) линий. Точки h1 и h2 (H1 и H2) обозначают бифуркации Андронова–Хопфа в случае модели CSTR (3D-RDC). б – Колебания скорости реакции в модели 3D-RDC при Tcat = 510 K. Рис. 11 адаптирован из работы [84] с разрешения Elsevier.
Для моделирования протекания реакции на уровне реактора система (5) была дополнена дифференциальными уравнениями для концентраций CO, O2 и CO2 в газовой смеси [83, 84]. В работе [83] была рассмотрена модель проточного реактора идеального смешения (модель CSTR), а в работе [84] – распределенная модель типа реакция–диффузия–конвекция с учетом диффузионного и конвективного переноса частиц в объеме реактора (модель 3D-RDC). Результаты расчетов сравнивали с экспериментальными данными для реакции окисления CO на Pd(100), представленными в работах [76, 78]. Модель CSTR получается из системы (5) если добавить следующие ОДУ:
(6)
$\begin{gathered} {{{\text{d}}{{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = \left( {P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}} - {{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}} \right)F{{V}^{{ - 1}}} - \\ - \,\,\sigma \left( {{{k}_{1}}{{P}_{{{{{\text{O}}}_{{\text{2}}}}}}}\theta _{*}^{2} - {{k}_{3}}\theta _{{\text{O}}}^{2}} \right), \\ {{{\text{d}}{{P}_{{{\text{CO}}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{P}_{{{\text{CO}}}}}} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = \left( {P_{{{\text{CO}}}}^{{{\text{in}}}} - {{P}_{{{\text{CO}}}}}} \right)F{{V}^{{ - 1}}} - \\ - \,\,\sigma \left( {{{k}_{2}}{{P}_{{{\text{CO}}}}}\theta {\text{*}} - {{k}_{4}}{{\theta }_{{{\text{CO}}}}}} \right), \\ {{{\text{d}}{{P}_{{{\text{C}}{{{\text{O}}}_{{\text{2}}}}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{P}_{{{\text{C}}{{{\text{O}}}_{{\text{2}}}}}}}} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = - {{P}_{{{\text{C}}{{{\text{O}}}_{{\text{2}}}}}}}F{{V}^{{ - 1}}} + \sigma {{R}_{{{\text{C}}{{{\text{O}}}_{{\text{2}}}}}}}, \\ \end{gathered} $Модель типа реакция–диффузия–конвекция для трехмерной области (3D-RDC) предполагает, что каталитический проточный реактор представляет собой прямоугольный параллелепипед размеров Lx × Ly × Lz; его внутренняя область обозначена как $\Omega $, а его граница – $\partial \Omega $. Плоский прямоугольный катализатор размеров $L_{x}^{{{\text{cat}}}} \times L_{y}^{{{\text{cat}}}}$ расположен, согласно экспериментальным данным, в центре нижней стенки ($z = 0$) камеры реактора; область катализатора обозначена как ${{\Omega }_{{{\text{cat}}}}} \subset \partial \Omega $. Газовая смесь поступает через левую сторону реактора ($x = 0$) и покидает его через противоположную правую сторону (х = Lx). Изменения локальных концентраций газов описываются уравнениями в частных производных:
(7)
$\frac{{\partial {{C}_{i}}}}{{\partial t}} = - {\mathbf{u}}\nabla {{C}_{i}} + \nabla \cdot \left[ {{{D}_{i}}\nabla {{C}_{i}}} \right] + W_{i}^{{{\text{reac}}}},$При изучении нелинейных явлений в гетерогенном катализе бифуркационный анализ и продолжение по параметру стационарных решений становятся основными методами анализа поведения системы. Для систем малой размерности, например, для модели CSTR, такие методы хорошо известны и применяются более 50 лет. Однако для систем большой размерности проведение бифуркационного анализа сопряжено со значительными вычислительными трудностями, поскольку стандартные методы для решения больших задач становятся практически непригодными. Для таких задач, возможно применение так называемых безматричных методов. Стационарные решения для модели 3D-RDC вычислялись с помощью алгоритма продолжения по параметру “по длине дуги” (“pseudo-arclength continuation”), основанному на безматричном методе Ньютона–Крылова [85]. Алгоритм позволяет продолжать по параметру решения систем большой размерности (>106 уравнений), при этом вычисляются как устойчивые, так и неустойчивые стационарные решения, а также определяются точки различных бифуркаций, например, точки бифуркаций типа седло–узел и Андронова–Хопфа. При моделировании также использовалось непосредственное интегрирование систем ОДУ большой размерности с помощью неявных безматричных методов.
На рис. 11а сравниваются зависимости от температуры стационарных решений в моделях CSTR и 3D-RDC в условиях избытка кислорода. При низких температурах обе модели дают одно и тоже низкореакционное решение, соответствующее поверхности, заполненной CO. При повышении температуры, сразу после “зажигания”, обе модели предсказывают появление автоколебаний. Колебания появляются и исчезают в результате бифуркаций Андронова–Хопфа. При высоких температурах, в области слабой зависимости скорости реакции от температуры, модели CSTR и 3D-RDC дают разные результаты – скорость реакции отличается почти в 2.5 раза. Сильные отличия вызваны тем, что модель CSTR предполагает полное перемешивание в реакторе и, следовательно, не описывает диффузионный режим, при котором скорость реакции лимитируется скоростью диффузии молекул CO в объеме реактора. Пример автоколебаний скорости реакции показан на рис. 11б, где изображена зависимость скорости реакции от времени при фиксированной температуре.
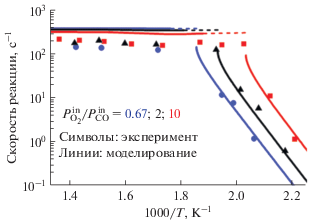
Зависимости стационарной скорости реакции от обратной температуры, вычисленные с помощью модели 3D-RDC для трех значений ${{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} \mathord{\left/ {\vphantom {{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} {P_{{{\text{CO}}}}^{{{\text{in}}}}}}} \right. \kern-0em} {P_{{{\text{CO}}}}^{{{\text{in}}}}}}$, представлены на рис. 12. Для тех же значений парциальных давлений реагентов экспериментальные данные показаны символами. Расчеты дают хорошее описание экспериментальных зависимостей, полученных в работе [76]. При низких температурах реализуется низкореакционное состояние, где катализатор “отравлен” адсорбированными молекулами CO. При промежуточных температурах (пунктирные линии на рис. 12) модель предсказывает автоколебательный режим. При высоких температурах значения ${{R}_{{{\text{C}}{{{\text{O}}}_{{\text{2}}}}}}}$ почти не зависят и от температуры, и от отношения ${{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} \mathord{\left/ {\vphantom {{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} {P_{{{\text{CO}}}}^{{{\text{in}}}}}}} \right. \kern-0em} {P_{{{\text{CO}}}}^{{{\text{in}}}}}}$. Независимость ${{R}_{{{\text{C}}{{{\text{O}}}_{{\text{2}}}}}}}$ от температуры вызвана выходом системы в диффузионный режим. Слабая зависимость ${{R}_{{{\text{C}}{{{\text{O}}}_{{\text{2}}}}}}}$ от ${{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} \mathord{\left/ {\vphantom {{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} {P_{{{\text{CO}}}}^{{{\text{in}}}}}}} \right. \kern-0em} {P_{{{\text{CO}}}}^{{{\text{in}}}}}}$ объясняется добавлением аргона в газовую смесь, что делается для поддержания общего давления постоянным. При этом, скорость диффузии CO, определяющая скорость реакции, сохраняется при изменении ${{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} \mathord{\left/ {\vphantom {{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} {P_{{{\text{CO}}}}^{{{\text{in}}}}}}} \right. \kern-0em} {P_{{{\text{CO}}}}^{{{\text{in}}}}}}$.
Рис. 12.
Зависимости скорости реакции окисления CO на Pd(100) от обратной температуры при трех значениях ${{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} \mathord{\left/ {\vphantom {{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} {P_{{{\text{СО}}}}^{{{\text{in}}}}}}} \right. \kern-0em} {P_{{{\text{СО}}}}^{{{\text{in}}}}}},$ равных 0.67, 2 и 10, при фиксированном $P_{{{\text{СО}}}}^{{{\text{in}}}}$ = 10 мбар. Символами показаны экспериментальные данные из работы [76]. Вычисленные устойчивые (неустойчивые) стационарные решения показаны с помощью сплошных (пунктирных) линий. Рис. 12 адаптирован из работы [84] с разрешения Elsevier.
Одним из новых методов изучения свойств каталитических реакций является метод плоскостной лазерно-индуцированной флуоресценции (Planar Laser-Induced Fluorescence, PLIF), позволяющий регистрировать мгновенное распределение концентраций различных веществ в плоском сечении потока газа в реакторе [86–88]. Метод используется для количественной визуализации состава смеси при изучении реакций на поверхности монокристаллов. С помощью PLIF молекулы газа возбуждаются плоским лазерным лучом и затем релаксируют c излучением фотонов, что фиксируется с помощью фотокамеры. В работах [86–88] метод PLIF применялся при изучении автоколебаний в реакции (CO + O2)/Pd(100), при этом визуализировались мгновенные пространственные распределения молекул CO и CO2 в объеме реактора. Было показано, что в высокореакционном состоянии наблюдается диффузионный режим протекания реакции, которая лимитируется подводом молекул CO к поверхности катализатора.
В работе [84] для моделирования экспериментальных данных, полученных с помощью PLIF при изучении реакции окисления CO на Pd(100), применялась модель 3D-RDC. В соответствии с экспериментальными условиями использовались следующие параметры реактора: $V = 3 \times 3 \times 2 = 18$ см3; $F = 1.1 \times {{10}^{{ - 5}}}$ м3/с; катализатор размеров $4 \times 4$ мм2; суммарное давление равно 150 мбар. Подробная пространственная дискретизация уравнений модели 3D-RDC приводила к необходимости решать более трех миллионов ОДУ.
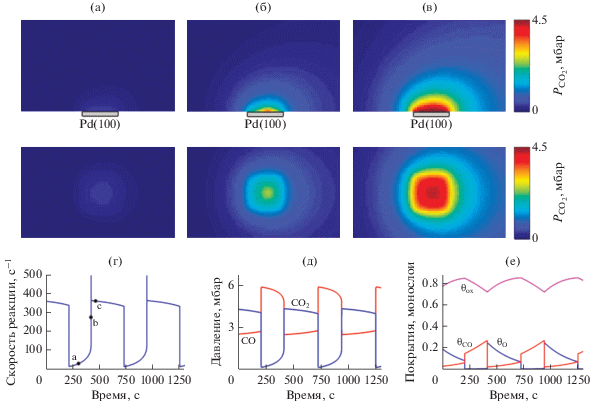
Результаты расчетов хорошо описывают экспериментальные данные, полученные с помощью метода PLIF (см. рис. 6 в [87]). На рис. 13 показаны пространственные распределения концентрации молекул CO2 в различные моменты времени. По причине высокой скорости реакции скорость диффузии в газе оказывается недостаточной для полного перемешивания молекул газа, из-за этого вокруг катализатора образуется пограничный слой. Рассчитанный по модели период колебаний составляет ~520 с, что близко к экспериментальному значению (~500 с [87]). Время нахождения системы в высокореакционном диффузионном режиме почти в два раза превышает время нахождения системы в низкореакционном кинетическом режиме. Общая конверсия CO колеблется в пределах от 7.5 до 0.35%. Колебания происходят на частично окисленной поверхности, при этом концентрация поверхностного оксида остается довольно высокой (см. рис. 13е).
Рис. 13.
Результаты расчетов для модели 3D-RDC при $P_{{{{{\text{O}}}_{2}}}}^{{{\text{in}}}}$ = 120 мбар, $P_{{{\text{СО}}}}^{{{\text{in}}}}$ = 6 мбар, $P_{{{\text{Ar}}}}^{{{\text{in}}}}$ = 24 мбар, Tcat = 513 K. Вверху показаны пространственные распределения концентрации CO2 в моменты времени, отмеченные точками a, б и в на рис. 13г. Первая строка: вертикальные разрезы (вид сбоку), показывающие фрагмент размеров 1.7 × 1.0 см2 около поверхности катализатора. Вторая строка: горизонтальные разрезы (вид сверху), показывающие фрагмент размеров 1.7 × 1.0 см2 около поверхности катализатора. a – Низкореакционное состояние; б – состояние в момент процесса “зажигания”; в – высокореакционное состояние; г – временная зависимость общей скорости реакции; д – временные зависимости средних парциальных давлений CO и CO2 вблизи поверхности; е – временные зависимости средних заполнений θО, θСО и θох. Рис. 13 адаптирован из работы [84] с разрешения Elsevier.
Благодаря сравнительной простоте рассмотренной модели становятся понятны физико-химические механизмы, объясняющие природу автоколебаний и активного состояния катализатора, снимки PLIF, особенности протекания диффузионного режима. Слабая зависимость скорости реакции от степени окисления катализатора может приводить к неверной интерпретации результатов экспериментальных измерений. Утверждение о том, что модель S.–T.–M. и механизм Ленгмюра–Хиншельвуда противоречат результатам измерений [74–76], основывалось на прямой зависимости между появлением оксида и увеличением скорости реакции. Однако моделирование показало, что экспериментальные результаты, включая колебания скорости реакции, могут быть описаны на основе модели (5) [83, 84]. При этом константа скорости бимолекулярной реакции (${{k}_{5}}$) на много порядков превышает константу скорости реакции восстановления оксида (${{k}_{7}}$), что подтверждает действие механизма Ленгмюра–Хиншельвуда, а не механизма Марс-ван Кревелена. Согласно результатам моделирования, частичное окисление катализатора (увеличение θox), не обязательно приводит к существенному уменьшению скорости реакции в высокореакционном состоянии. Физически это означает, что высокая реакционная способность катализатора может поддерживаться за счет лишь небольшого количества узлов “чистого” металла, в то время как окисленные узлы металла практически не участвуют в реакции. Это свойство сохраняется и при протекании реакции в диффузионном режиме, когда скорость реакции пропорциональна скорости диффузии CO в газовой фазе.
4.2. Математическое моделирование автоколебаний в реакции окисления CO на Ni
Как обсуждалось выше, недавно были обнаружены автоколебания скорости реакции окисления CO на никелевой фольге в проточном реакторе при атмосферном давлении. Примечательно, что эти автоколебания появляются при избытке CO, а не O2, как было в случае металлов Pt-группы. Известно, что модель S.–T.–M. дает колебания только в условиях избытка кислорода, а точнее при $2{{k}_{1}}{{s}_{1}}{{P}_{{{{{\text{O}}}_{{\text{2}}}}}}} > {{k}_{2}}{{s}_{2}}{{P}_{{{\text{CO}}}}}$ (см. уравнение (3)). Поскольку константы ${{k}_{1}}$ и ${{k}_{2}}$ имеют близкие значения, то условие $2{{k}_{1}}{{s}_{1}}{{P}_{{{{{\text{O}}}_{{\text{2}}}}}}} > {{k}_{2}}{{s}_{2}}{{P}_{{{\text{CO}}}}}$можно обеспечить, если выбрать ${{s}_{2}} \ll {{s}_{1}}$. Однако экспериментальные данные дают практически одинаковые значения для коэффициентов прилипания у O2 и CO на Ni. Поэтому в работах [89, 90] использовали экспериментально подтвержденное предположение о том, что адсорбция CO происходит через предадсорбционное состояние, для чего применяли известную модель Кислюка [91]. Модель реакции окисления CO на Ni состоит из уравнений (5) и (6), при этом скорость адсорбции CO вычисляется по формуле:
(7)
$R_{{{\text{CO}}}}^{{{\text{ads}}}} = {{{{k}_{2}}{{s}_{2}}{{P}_{{{\text{CO}}}}}\theta {\text{*}}} \mathord{\left/ {\vphantom {{{{k}_{2}}{{s}_{2}}{{P}_{{{\text{CO}}}}}\theta {\text{*}}} {\left( {\theta {\text{*}} + {{K}_{{{\text{CO}}}}}(1 - \theta {\text{*}})} \right)}}} \right. \kern-0em} {\left( {\theta {\text{*}} + {{K}_{{{\text{CO}}}}}(1 - \theta {\text{*}})} \right)}},$(8)
${{{\text{d}}{{T}_{{{\text{cat}}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{T}_{{{\text{cat}}}}}} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = {{a}_{1}}({{T}_{{{\text{gas}}}}} - {{T}_{{{\text{cat}}}}}) + {{a}_{2}}{{R}_{{{\text{C}}{{{\text{O}}}_{{\text{2}}}}}}},$Рис. 14.
a – Двухпараметрические бифуркационные диаграммы на плоскости (Tgas, ${{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} \mathord{\left/ {\vphantom {{P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}} {P_{{{\text{СО}}}}^{{{\text{in}}}}}}} \right. \kern-0em} {P_{{{\text{СО}}}}^{{{\text{in}}}}}}$) при $P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}$ + + $P_{{{\text{CO}}}}^{{{\text{in}}}}$ = 500 мбар и различных значениях параметра $k_{6}^{0}$ [с–1]. Области колебаний ограничены линиями бифуркаций Андронова–Хопфа. б – Сравнение вычисленных и экспериментальных значений периода колебаний в зависимости от температуры газа для системы (CO + + O2)/Ni. Рис. 14 адаптирован из работы [90] с разрешения Springer Nature.
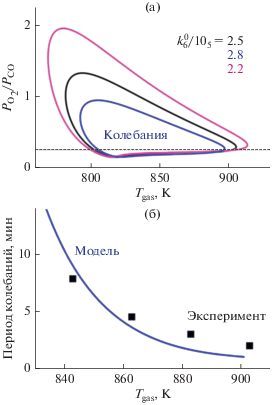
Рис. 15.
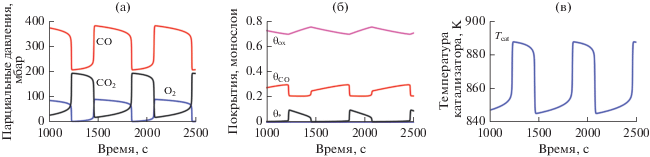
Автоколебания в реакции CO + O2 на Ni при $P_{{{{{\text{O}}}_{{\text{2}}}}}}^{{{\text{in}}}}$ = 10 мбар, $P_{{{\text{CO}}}}^{{{\text{in}}}}$ = 40 мбар, Tgas = 840 K: a – парциальные давления O2, CO, и CO2; б – покрытия θСО и θох и θ*; в – температура катализатора. Рис. 15 адаптирован из работы [90] с разрешения Springer Nature.
4.3. Моделирование нелинейной динамики в других реакциях окисления
В последние годы механизм возникновения автоколебаний за счет процессов типа окисление–восстановление использовался для моделирования реакции окисления не только CO, но и углеводородов CxHy. В работах [92, 93] рассматривались математические модели для реакций окисления метана и пропана на никелевых катализаторах. В этом случае подробная кинетическая схема содержит большое количество реакций, связанных в основном с разложением CxHy на углерод и водород. Если считать, что разложение CxHy происходит моментально, а водород быстро десорбируется либо удаляется с поверхности в виде H2O, то можно выделить малый параметр и провести редукцию системы дифференциальных уравнений [92, 93]. Упрощенная кинетическая модель окисления CxHy содержит всего три поверхностных концентрации: атомарного кислорода (θО), углерода (θС) и поверхностного оксида (θох). Вместе с тем, уравнение для переменной θС в упрощенной модели окисления CxHy имеет схожую структуру с уравнением для θСО в модели реакции окисления CO. Поэтому механизм окисления–восстановления может применяться и при изучении автоколебаний в реакциях окисления углеводородов. Присутствие дополнительных особенностей конкретной реакции, несомненно, играет определенную роль в поведении системы, однако основная “движущая сила” колебаний, связанная с окислением–восстановлением катализатора, не изменяется.
Реакция окисления углеводородов может осложняться диффузией кислорода вглубь катализатора. Однако можно предположить, что основную роль играют процессы, происходящие на поверхности катализатора, в то время как наличие растворенного кислорода не играет существенной роли. Важно отметить, что при атмосферных давлениях в процессе колебаний скорость реакции обычно высока (>103 монослоев/с), а период колебаний составляет несколько минут, следовательно, заполнение нескольких десятков подповерхностных слоев атомами кислорода может практически не влиять на общую скорость реакции [89].
Экспериментальные данные, приведенные в части 2.2, показывают, что если углерод не играет большой роли в механизме реакции окисления СН4 на Ni, то в ходе колебаний скорости реакции окисления СН4 на Pd-катализаторе в виде порошка происходит накопление в объеме и удаление сотен монослоев углерода. В отличие от реакции окисления СН4 на Ni, только продукты глубокого окисления СО2 и Н2О были зафиксированы в ходе реакции окисления СН4 на Pd, притом скорости образования этих двух единственных продуктов колебались со сдвигом по фазе [55]. Такое явление наблюдалось впервые, его природа была не понятна, и только с помощью математического моделирования удалось выяснить, при каких условиях может возникнуть такой сдвиг по фазе. В работе [94] была создана математическая модель типа S.–T.–M., в которой колебания возникали в результате периодического окисления–восстановления Pd-катализатора. Учитывалась также диффузия углерода в глубину катализатора и обратно. Было показано, что при реальных значениях параметров сдвиг по фазе между единственными продуктами реакции СО2 и Н2О можно получить, если предположить, что порядка 100 слоев растворенного углерода участвуют в образовании СО2 [94].
В последние годы проводилось также моделирование нелинейной динамики в реакции окисления водорода на Rh-катализаторах при низких давлениях. В работе [95] были изучены автоколебания скорости реакции окисления Н2 на поверхности Rh в виде острия иглы нанометровых размеров с использованием метода полевой ионной микроскопии (field ion microscope, FIM). Была разработана подробная математическая модель, показывающая, что колебания скорости реакции возникают из-за воздействия внешнего электрического поля, которое влияет на парциальные давления реагентов и на энергии активации элементарных стадий. Модель содержит много нелинейностей и учитывает предадсорбционное состояние при адсорбции O2, формирование подповерхностного кислорода, латеральные взаимодействия, изменяющие энергии активации элементарных процессов. В дальнейшем эта модель применялась при изучении реакции H2 + O2 на поликристаллическом Rh также при низких давлениях [96], но уже без учета воздействия электрического поля. Для этой реакции в экспериментах, использующих PEEM, наблюдались спиральные волны и другие сложные пространственные структуры.
ЗАКЛЮЧЕНИЕ
Приведенные выше данные показывают, какую большую роль играют нелинейные явления для понимания механизма гетерогенных каталитических реакций. Изучение множественности стационарных состояний в реакции СО на Pd-катализаторе позволило выявить роль флуктуаций при проведении реакции на частицах малого размера. Форма колебаний скорости реакции окисления на металлических катализаторах, возникающих в результате процессов окисления–восстановления металла, позволяет делать выводы о состоянии катализатора [97]. Наблюдаемый сдвиг по фазе между скоростями образования двух единственных продуктов реакции может указывать на большую роль растворенных веществ в механизме реакции. Исследование волновых процессов на поверхности катализатора свидетельствует, что скорость реакции, рассчитанная по концентрации веществ в газовой фазе, может быть постоянна, в то время как на поверхности происходит сложный процесс поверхностной турбулентности.
Множество интересных явлений в гетерогенных каталитических системах возникает исключительно из-за того, что в системе присутствуют нелинейные связи. Так в работе [98] было показано, что колебания на Pt/Rh биметаллическом катализаторе могут возникать в условиях, когда отдельные монометаллические образцы Pt и Rh проявляют только бистабильность, но не колебания. Основной причиной таких “синергетических автоколебаний” являются нелинейная кинетика реакции окисления СО и сильное отличие в энергиях связи адсорбированного кислорода на поверхностях Rh и Pt, что приводит к эффекту спилловера атомарного кислорода. Из-за нелинейных свойств кинетики реакции окисления метана на Pd-катализаторе может наблюдаться такой интересный эффект, когда динамическое поведение Pd-фольги – целой и разрезанной на две равные половины – будет отличаться [99]. При определенных условиях, при полном перемешивании в газовой фазе только одна часть Pd-фольги будет активна, а активность второй половины Pd-фольги будет мала.
Надо отметить, что изучение и понимание нелинейных явлений в гетерогенных каталитических системах немыслимо без математического моделирования. Отличительной особенностью нелинейного анализа является то, что рассматриваются решения системы при всех возможных параметрах, поэтому результаты бифуркационного анализа позволяют не только объяснять, но и делать прогнозы возможного поведения каталитических систем [1]. Так, в работах [100, 101] с помощью математического моделирования было обнаружено, что с увеличением площади активной поверхности Pt степень конверсии NO в важной с экологической точки зрения реакции CO + NO + O2 может уменьшаться, причем это не связано с изменением свойств самого катализатора или с проблемами доставки газа к его активной поверхности. Моделирование показало, что существует оптимальное значение величины площади катализатора, при котором достигается наибольшая степень конверсии NO в N2, при этом степень конверсии CO в CO2 близка к 100%. Была установлена важная роль обратимости реакции разложения NO на N и O, т.е. когда катализатора становилось “слишком много”, то начинала возрастать скорость реакции образования NO. Таким образом, наличие нелинейных зависимостей приводит к тому, что не всегда скорость реакции пропорционально увеличивается с ростом активной поверхности катализатора.
В заключении надо отметить, что нелинейные явления особенно важны для гетерогенного катализа, т.к. они должны учитываться при определении оптимальных условий эксплуатации химических реакторов [1]. Наличие множественности стационарных состояний, волновые явления на поверхности катализатора и наличие горячих пятен может приводить к потере устойчивости работы реактора [10]. Для анализа устойчивости химических реакторов необходимо детальное знание механизма реакции, кинетических особенностей протекания реакции, включая такие нелинейные свойства системы, как множественность стационарных состояний и колебательные режимы.
Список литературы
Слинько М.Г. Основы и принципы математического моделирования каталитических процессов“, Новосибирск: Издание ИК СО АН РАН, 2004.
Боресков Г.К. Гетерогенный катализ в химической промышленности. Материалы Всесоюзного совещания 1953 г. Москва: Госхимиздат, 1955.
Боресков Г.К., Слинько М.Г., Филиппова А.Г. // ДАН СССР. 1953. Т. 92. № 2. С. 293.
Слинько М.Г., Бесков В.С., Дубяга Н.А. // ДАН СССР. 1972. Т. 204. № 4. С. 1174.
Beusch H., Fieguth P., Wicke E. // Chem. Ing. Tech. 1972. V. 44. P. 445.
Беляев В.Д., Слинько М.М., Тимошенко В.И., Слинько М.Г. // Кинетика и катализ. 1973. Т. 14. С. 810.
Барелко В.В., Мержанов А.Г. Проблемы кинетики и катализа. Нестационарные и неравновесные процессы в гетерогенном катализе. 1973. Т. 17. Москва: Наука, С. 182.
Imbihl R., Ertl G. // Chem. Rev. 1995. V. 95. P. 697.
Slinko M.M., Jaeger N.I. Oscillating heterogeneous catalytic systems, V. 86. Eds. Delmon B. and Yates J.T., Elsevier, 1994.
Luss D, Sheintuch M. // Catal. Today. 2005. V. 105. P. 254.
Imbihl R. Non-linear Dynamics in Catalytic Reactions. Handbook of Surface Science. Amsterdam: Elseviers, 2008.
Raj R., Harold M.P., Balakotaiah V. // Chem. Eng. J. 2015. V. 281. P. 322.
Berdau M., Karpowicz A., Yelenin G.G., Christmann K., Block J.H. // J. Chem. Phys. 1997. V. 106. P. 4291.
Rotermund H.H. // J. Elec. Spectr. and Rel. Phenom. 1999. V. 98–99. P. 41.
Suchorski Y., Spiel C., Vogel D., Drachsel W., Schlőgl R., Rupprechter G. // ChemPhysChem. 2010. V. 11. P. 3231.
Vogel D., Spiel C., Suchorski Y., Urich A., Schlögl R., Rupprechter G. // Surf. Sci. 2011. V. 605. P. 1999.
Spiel C., Vogel D., Schlögl R., Rupprechter G., Suchorski Y. // Ultramicroscopy. 2015. V. 159. P. 178.
Johánek V., Laurin M., Grant A.W., Kasemo B., Henry C.R., Libuda J. // Science. 2004. V. 304. P. 1639.
Франк-Каменецкий Д.А. Диффузия и теплопередача в химической кинетике. Москва: Наука, 1967.
Bos A.N.R., Hof E., Kuper W., Westerterp K.R. // Chem. Eng. Sci. 1993. V. 48. P. 1959.
Koutoufaris I., Koltsakis G. // Can. J. Chem. Eng. 2014. V. 92. P. 1561.
Fuchs S., Hahn T. // Chem. Eng. and Proc. 1993. V. 32. P. 225.
Fuchs S., Hahn T., Lintz H.G. // Chem. Eng. and Proc. 1994. V. 33. P. 363.
Fernandes V.R., Bossche M., Knudsen J., Farstad M.H., Gustafson J., Venvik H.J., Grönbeck H., Borg A. // ACS Catal. 2016. V. 6. P. 4154.
Abedi A., Hayes R., Votsmeier M., Epling W.S. // Catal. Lett. 2012. V. 142. P. 930.
Kota A.S., Dadi R.K., Luss D., Balakotaiah V. // Chem. Eng. Sci. 2017. V. 166. P. 320.
Raj R., Harold M. P., Balakotaiah V. // Chem. Eng. J. 2015. V. 281. P. 322.
Li J., Kumar A., Kamasamudram K., Currier N., Yezerets A. // Catal. Today. 2015. V. 258. P. 167.
Slinko M.M., Jaeger N. // Appendix – Oscillatory Heterogeneous Catalytic Systems // Catal. Today. 2005. V. 105. P. I-II.
Bychkov V.Yu., Tulenin Yu.P., Slinko M.M., Gordienko Yu.A., Korchak V.N. // Catal. Lett. 2018. V. 148. P. 653.
Bychkov V.Yu., Tulenin Yu.P., Slinko M.M., Gorenberg A.Ya., Shashkin D.P., Korchak V.N. // Reac. Kin. Mech. Cat. 2019. V. 128. P. 587.
Bychkov V.Yu., Tulenin Yu.P., Gorenberg A.Ya., Korchak V.N. // Reac. Kin. Mech. Cat. 2020. V. 129. P. 57.
Liu Y., Fang W.P., Weng W.Z., Wan H.L. // J. Mol. Catal. A. Chem. 2005. V. 239. P. 193.
Wang M., Weng W., Zheng H., Yi X., Huang C., Wan H. // J. Nat. Gas Chem. 2009. V. 18. P. 300.
Bychkov V.Y., Tyulenin Y.P., Slinko M.M., Korchak V.N. // Catal. Lett. 2007. V. 119. P. 339.
Bychkov V.Y., Tyulenin Y.P., Slinko M.M., Korchak V.N. // Surf. Sci. 2009. V. 603. P. 1680.
Kokkofitis C., Stoukides M. // J. Catal. 2006. V. 243. P. 428.
Bychkov V.Y., Tyulenin Y.P., Slinko M.M., Korchak V.N. // Catal. Lett. 2011. V. 141. P. 602.
Bychkov V.Y., Tyulenin Y.P., Slinko M.M., Lomonosov V.I., Korchak V.N. // Catal. Lett. 2018. V. 148. P. 3646.
Bychkov V.Yu., Tyulenin Yu.P., Slinko M.M., Korchak V.N. // Proc. of the IX Int. Conf. “Mechanisms of catalytic reactions”, St. Petersburg, Russia, 2012, P. 165.
Barakat T., Rooke J.C., Chlala D., Cousin R., Lamonier J.-F., Giraudon J.-M., Casale S., Massiani P., Su B.-L., Siffert S. // Catalysts. 2018. V. 8. P. 574.
Ouariach O., Kacimi M., Ziyad M. // Appl. Catal. A: General. 2015. V. 503. P. 84.
Кипнис М.А., Волнина Э.А. // Кинетика и катализ. 2013. Т. 54. № 2. С. 263.
Abu-Zied B.M., Schwieger W. // Appl. Catal. B: Environ. 2009. V. 85. P. 120.
Kaucký D., Jíŝa K., Vondrová A., Nováková J., Sobalík Z. // J. Catal. 2006. V. 242. P. 270.
Ciambelli P., Garufi E., Pirone R., Russo G., Santagata F. // Appl. Catal. B: Environ. 1996. V. 8. P. 333.
Toyoshima I., Somorjai G.A. // Catal. Rev. Sci. Eng. 1979. V. 19. P. 105.
König D., Weber W.H., Poindexter B.D., McBride J.R., Graham G.W., Otto K. // Catal. Lett. 1994. V. 29. P. 329.
Gladky A.Y., Ermolaev V.K., Parmon V.N. // Catal. Lett. 2001. V. 77. P. 103.
Бычков В.Ю., Тюленин Ю.П., Слинько М.М., Втюрина Д.Н., Корчак В.Н. // Хим. физика. 2018. Т. 37. № 9. С. 10.
Bychkov V.Y., Tyulenin Y.P., Korchak V.N., Aptekar E.L. // Appl. Catal. A: General. 2006. V. 304. P. 21.
Saraev A.A, Vinokurov Z.S, Kaichev V.V, Shmakov A.N, Bukhtiyarov V.I. // Catal. Sci. Technol. 2017. V. 7. P. 1646.
Kaichev V.V., Gladky A.Y., Prosvirin I.P., Saraev A.A., Hävecker M., Knop-Gericke A., Schlögl R., Bukhtiyarov V.I. // Surf. Sci. 2013. V. 609. P. 113.
Kaichev V.V., Gladky A.Y., Saraev A.A., Kosolobov S.S., Sherstyuk O.V., Bukhtiyarov V.I. // Top. Catal. 2020. V. 63. P. 24.
Bychkov V.Y., Tyulenin Y.P., Slinko M.M., Shashkin D.P., Korchak V.N. // J. Catal. 2009. V. 267. P. 181.
Bychkov V.Y., Tyulenin Y.P., Slinko M.M., Gorenberg A.Ya., Korchak V.N. // Catal. Lett. 2017. V. 147. P. 2664.
Zhang X., Hayward D.O., Mingos D.M.P. // Catal. Lett. 2002. V. 83. P. 149.
Zhang X., Lee C.S.M., Mingos D.M.P., Hayward D.O. // Appl. Catal. A: General. 2003. V. 248. P. 129.
Gladky A.Y., Ermolaev V.K., Parmon V.N. // Catal. Lett. 2001. V. 77. P. 103.
Сараев А.А. Дисс. … канд. физ.-мат. наук. Новосибирск: Институт катализа, 2016.
Kaichev V.V., Teschner D., Saraev A.A., Kosolobov S.S., Gladky A.Y., Prosvirin I.P., Rudina N.A., Ayupov A.B., Blume R., Hävecker M., Knop-Gericke A., Schlögl R., Latyshev A.V., Bukhtiyarov V.I. // J. Catal. 2016. V. 334. P. 23.
Сараев А.А., Косолобов С.С., Каичев В.В., Бухтияров В.И. // Кинетика и катализ. 2015. Т. 56. № 5. С. 606.
Bychkov V.Y., Tyulenin Y.P., Gorenberg A.Ya., Sokolov S., Korchak V.N. // Appl. Catal. A: General. 2014. V. 485. P. 1.
Bychkov V.Y., Tyulenin Y.P., Slinko M.M., Sokolov S., Korchak V.N. // Catal. Lett. 2017. V. 147. P. 1019.
Ertl G. Noble Lecture, 2007. https://www.nobelprize.org/prizes/chemistry/2007/ertl/lecture/
Barroo C., Wang Z.-J., Schlögl R., Willinger M.G. // Nature Catal. 2020. V. 3. P. 30.
Franz T., von Boehn B., Marchetto H., Borkenhagen B., Lilienkamp G., Daum W., Imbihl. R. // Ultramicroscopy. 2019. V. 200. P. 73.
Bychkov V.Y., Tyulenin Y.P., Slinko M.M., Korchak V.N. // Surf. Sci. 2009. V. 603. P. 1680.
Bychkov V.Y., Tyulenin Y.P., Slinko M.M., Korchak V.N. // Appl. Catal. A: General. 2007. V. 321. P. 180.
Kaichev V.V., Saraev A.A., Gladky A.Y., Prosvirin I.P., Blume R., Teschner D., Hävecker M., Knop-Gericke A., Schlögl R., Bukhtiyarov V.I. // Phys. Rev. Lett. 2017. V. 119. P. 026001.
http://center.chph.ras.ru/files/vb/
McAdam D.J., Geil J.W., Geil G.W. // J. Res. Nat. Bur. Stand. 1942. V. 28. P. 593.
Singh J., Nachtegaal M., Alayon E.M.C., Stotzel J., van Bokhoven J.A. // ChemCatChem. 2010. V. 2. P. 653.
Hendriksen B.L.M., Bobaru S.C., Frenken J.W.M. // Catal. Today. 2005. V. 105. P. 234.
van Rijn R., Balmes O., Felici R., Gustafson J., Wermeille D., Westerstrom R., Lundgren E., Frenken J.W.M. // J. Phys. Chem. C. 2010. V. 114. P. 6875.
van Rijn R., Balmes O., Resta A., Wermeille D., Westerstrom R., Gustafson J., Felici R., Lundgren E., Frenken J.W.M. // Phys. Chem. Chem. Phys. 2011. V. 13. P. 13167.
Sales B.C., Turner J.E., Maple M.B. // Surf. Sci. 1982. V. 114. P. 381.
Gao F., Wang Y., Cai Y., Goodman D.W. // J. Phys. Chem. C. 2009. V. 113. P. 174.
Gao F., Wang Y., Goodman D.W. // J. Phys. Chem. C. 2010. V. 114. P. 6874.
Zorn K., Giorgio S., Halwax E., Henry C.R., Grönbeck H., Rupprechter G. // J. Phys. Chem. C. 2011. V. 115. P. 1103.
Gänzler A.M., Casapu M., Boubnov A., Müller O., Conrad S., Lichtenberg H., Frahm R., Grunwaldt J.-D. // J. Catal. 2015. V. 328. P. 216.
Dann E.K., Gibson E.K., Catlow C.R.A., Celorrio V., Collier P., Eralp T., Amboage M., Hardacre C., Stere C., Kroner A., Raj A., Rogers S., Goguet A., Wells P.P. // J. Catal. 2019. V. 373. P. 201.
Makeev A.G., Slinko M.M., Luss D. //Appl. Catal. A: General. 2019. V. 571. P. 127.
Makeev A.G., Slinko M.M. // Surf. Sci. 2020. V. 691. P. 121488.
Makeev A.G., Semendyaeva N.L. // Comp. Math. and Math. Phys. 2009. V. 49. P. 623.
Blomberg S., Brackmann C., Gustafson J., Aldén M., Lundgren E., Zetterberg J. // ACS Catal. 2015. V. 5. P. 2028.
Blomberg S., Zhou J., Gustafson J., Zetterberg J., Lundgren E. // J. Phys.: Condens. Matter. 2016. V. 28. P. 453002.
Zhou J., Pfaff S., Lundgren E., Zetterberg J. // Appl. Phys. B. 2017. V. 123. P. 87.
Makeev A.G., Peskov N.V., Semendyaeva N.L., Slinko M.M., Bychkov V.Yu., Korchak V.N. // Chem. Eng. Sci. 2019. V. 207. P. 644.
Makeev A.G., Peskov N.V., Slinko M.M., Bychkov V.Yu., Korchak V.N. // Top. Catal. 2020. V. 63. P. 49.
Kisliuk P. // Chem. Solids. 1957. V. 3. P. 95.
Lashina E.A., Kaichev V.V., Saraev A.A., Vinokurov Z.S., Chumakova N.A., Chumakov G.A., Bukhtiyarov V.I. // J. Phys. Chem. A. 2017. V. 121. P. 6874.
Lashina E.A., Kaichev V.V., Saraev A.A., Vinokurov Z.S., Chumakova N.A., Chumakov G.A., Bukhtiyarov V.I. // Top. Catal. 2020. In press. https://doi.org/10.1007/s11244-019-01219-5
Peskov N.V., Slinko M.M., Bychkov V.Yu., Korchak V.N. // Chem. Eng. Sci. 2012. V. 84. P. 684.
McEwen J.-S., Gaspard P., de Bocarmé T.V., Kruse N. // J. Phys. Chem. C. 2009. V. 113. P. 17045.
Suchorski Y., Datler M., Bespalov I., Zeininger J., Stöger-Pollach M., Bernardi J., Grönbeck H., Rupprechter G. // J. Phys. Chem. C. 2019. V. 123. P. 4217.
Slinko M.M. // Catal. Today. 2010. V. 153. P. 38.
Makeev A.G., Semendyaeva N.L., Slinko M.M. // Chem. Eng. J. 2015. V. 282. P. 3.
Lysak T.M., Peskov N.V., Slinko M.M., Tyulenin Yu.P., Bychkov V.Yu., Korchak V.N. // Chem. Eng. Sci. 2016. V. 144. P. 7.
Makeev A.G., Peskov N.V. // Appl. Catal. B: Environ. 2013. V. 132. P. 151.
Makeev A.G., Peskov N.V., Hiromichi Y. // Appl. Catal. B: Environ. 2012. V. 119. P. 273.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ