

Кинетика и катализ, 2020, T. 61, № 5, стр. 725-740
Влияние условий синтеза “сжиганием в растворе” на свойства блочных Pt–MnOx-катализаторов глубокого окисления углеводородов
Н. В. Шикина a, , С. А. Яшник a, А. А. Гаврилова a, О. А. Николаева a, Л. С. Довлитова a, А. В. Ищенко a, З. Р. Исмагилов a, b
a ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
b ФИЦ угля и углехимии Институт углехимии и химического материаловедения СО РАН
650000 Кемерово, просп. Советский, 18, Россия
Поступила в редакцию 14.02.2020
После доработки 10.03.2020
Принята к публикации 18.03.2020
Аннотация
Катализаторы на основе оксидов Mn с добавлением Pt, нанесенных на керамические блоки сотовой структуры, были приготовлены методами пропитки и “сжигания в растворе” (“solution combustion synthesis”, SCS). Для SCS-метода использовали глицин в качестве топливной добавки, обеспечивая различные условия реакции горения обедненных (φ < 1) и обогащенных (φ > 1) топливных смесей. Катализаторы исследованы методами ТГА, РФА, ПЭМВР, ТПВ-Н2, БЭТ и дифференцирующего растворения. Каталитические свойства образцов изучены в глубоком окислении бутана и метана. Показано, что в условиях SCS активные компоненты формируются в виде высокодисперсных частиц металлической Pt и оксидов марганца Mn3O4, расположенных в приповерхностных слоях носителя. В отличие от этого при термической обработке пропиточного катализатора образующиеся оксиды марганца обогащены катионами Mn(IV) и преимущественно локализованы в объеме носителя, формируя с ним фазу взаимодействия. Высокая активность SCS-катализаторов в реакциях окисления бутана и метана определяется в основном наличием восстановленных форм марганца и доступностью активных компонентов для реактантов в приповерхностных слоях носителя.
ВВЕДЕНИЕ
Каталитические системы на структурированных носителях эффективны для решения экологических проблем в различных промышленных процессах, связанных со сжиганием углеводородов [1], где катализатор одновременно обеспечивает комплексную очистку отработанных газов от СО, NOx и СН при относительно низких температурах и высоких объемных скоростях газовых потоков [1, 2]. Активность катализаторов во многом определяется условиями их синтеза, от которых зависят структурные и текстурные характеристики образцов, включая химическую природу активных центров, фазовый состав, дисперсность активных компонентов, наличие в структуре микронапряжений и дефектов, морфологию поверхности и др. Поиск новых методов приготовления катализаторов ведется постоянно. Одним из перспективных способов является самораспространяющийся высокотемпературный синтез (СВС) [3], в котором для формирования активных компонентов катализатора используется энергия экзотермических окислительно-восстановительных реакций между их массивными предшественниками [4–6]. Однако ограничения СВС-метода для получения катализаторов связаны с развитием чрезвычайно высоких температур и трудностями контроля над экстремальными температурами процесса. Из-за высоких температур СВС-волны (>2000 K) практически невозможно получить наноразмерные частицы, и конечным продуктом синтеза являются массивные материалы с низкой удельной поверхностью.
Низкотемпературной альтернативой самораспространяющемуся высокотемпературному синтезу каталитических систем является метод “сжиганием в растворе”, называемый в англоязычной литературе “solution combustion synthesis” (SCS). SCS-метод, предложенный Kingsley и Patil [7], заключается в сжигании окислительно-восстановительной смеси, состоящей из насыщенных растворов требуемых солей металлов (восстанавливаемый агент) и подходящего органического топлива (восстанавливающий агент), к примеру: мочевины, карбогидразида, гидразида малеиновой кислоты и др. Максимальная температура, развиваемая в распространяемой тепловой волне, зависит от природы и количества топливной добавки и, как правило, не превышает 500°С. Это предотвращает спекание активного компонента и обеспечивает формирование нанокристаллических оксидных материалов [8–13]. Быстрое образование большого объема газов во время сгорания топливной добавки рассеивает тепло, ограничивает повышение температуры, уменьшает возможность спекания за счет снижения вероятности контакта между частицами, приводит к диспергированию частиц [14]. Среди веществ, используемых в качестве топливной добавки для SCS-метода, глицин (NH2CH2COOH) является одним из наиболее востребованных в силу его доступности, невысокой стоимости и устойчивости. Молекула глицина, имеющая биполярный характер благодаря карбоксильной группе и аминогруппе в ее составе, может выступать комплексоном для ионов металлов. Карбоксильная группа образует комплексы со щелочными или щелочно-земельными металлами, аминогруппа – с переходными металлами. Присутствие хелатных комплексов повышает гомогенность раствора, что в дальнейшем предотвращает сегрегацию и выпадение осадков при упаривании (сушке) [15].
SCS-методом можно готовить нанесенные катализаторы. В этом случае экзотермическая реакция протекает на поверхности материала носителя и в его порах, где как раз и распределены соли предшественников активного компонента и топливная добавка. После нагрева воздушно-сухого образца (содержащего соли-предшественники активного компонента и топливную добавку) до температур инициирования сжигания топливной добавки, фронт самораспространяющегося беспламенного горения проходит по образцу и затухает после полного сжигания топливной добавки [16–19]. В процессе сжигания соль-предшественник активного компонента разлагается до оксида, сульфида или другого соединения активного компонента, частицы которого имеют размер в нанометровом диапазоне и механически прочно сцеплены с носителем, что также является стабилизирующим дисперсность фактором [20–22].
Возможности SCS-синтеза исследуются преимущественно для получения нанодисперсных материалов на основе простых [23, 24] и сложных [25, 26] оксидов, сульфидов, сплавов переходных металлов [27], но для сжигания углеводородов (как с целью получения энергии, так и для очистки отработанных газов) представляют интерес нанесенные каталитические системы [28]. Среди каталитических систем, приготовленных с помощью SCS, наиболее изучены оксиды переходных металлов, такие как MnOx [23], Co3O4 [20–22, 24], CuO [24], FeOx [24, 29, 30], NiO [20–22, 24, 26], и их смеси с редкоземельными металлами [31, 32]. SCS-синтезу каталитических систем на основе благородных металлов посвящены единичные работы, преимущественно обсуждающие Pt и Pd, нанесенные на CeO2 [33–38], Al2O3 [37], кордиерит [39]. Общеизвестным считается [40, 41], что активность нанесенных катализаторов на основе Pt и/или Pd, полученных методами пропитки по влагоемкости, в реакциях окисления углеводородов и монооксида углерода превосходит каталитические характеристики систем на основе оксидов переходных металлов. Кроме того, для систем оксид переходного металла–благородный металл зачастую наблюдается неаддитивное увеличение каталитической активности в реакциях окисления углеводородов и СО [42–50]. Систематические данные об активности катализаторов оксид переходного металла–благородный металл, синтезированных с помощью SCS, в литературе отсутствуют.
В настоящей работе изучена возможность формирования в Pt–Mn-катализаторе нанодисперсных частиц Pt с узким распределением частиц по размерам с использованием для этого метода синтеза “сжиганием в растворе” при варьировании количества топливной добавки. Для выявления роли Pt и MnOх в каталитическом окислении бутана и метана катализаторы были охарактеризованы современными физико-химическими методами: РФА, ПЭМВР, ДР, ТПВ-Н2.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление Pt–MnOx-катализаторов
В качестве носителя использовали керамический блок сотовой структуры из алюмосиликатной керамики с удельной поверхностью 12.5 м2/г и влагоемкостью 0.2 см3/г. На стадии проведения поисковых работ активные компоненты наносили на фракцию носителя размером 0.5–1.0 мм для исключения влияния концентрационного градиента по высоте блока на свойства катализаторов.
Пропиточный Pt–MnOx-катализатор получали методом пропитки носителя по влагоемкости раствором смеси азотнокислых солей Mn(NO3)2 и Pt(NO3)4. Концентрацию пропиточного раствора рассчитывали с учетом влагоемкости носителя и количества активного компонента, который требовалось нанести на носитель. Для приготовления катализатора, содержащего 5 мас. % MnO2 и 0.5 мас. % Pt, к смеси 1.15 мл 5.3 М раствора Mn(NO3)2 (приготовленного растворением Mn(NO3)2 · 6Н2О в дистиллированной воде), 1.55 мл 0.164 М раствора Pt(NO3)4 (свежеприготовленного растворением H2Pt(OH)6 в азотной кислоте) и 5.3 мл дистиллированной воды добавляли 10 г фракции носителя и тщательно перемешивали до полного впитывания раствора носителем, затем пропитанный носитель высушивали под лампой и прокаливали при 450°С в течение 4 ч. Образец обозначен как Pt–Mn/AlSi.
Синтез “сжиганием в растворе” Pt–MnOx-катализаторов (далее SCS-катализаторов) осуществляли пропиткой носителя раствором соответствующих азотнокислых солей, в который был добавлен глицин. Условия дальнейших сушки и прокаливания были аналогичны таковым для пропиточного катализатора. Содержание активных компонентов в SCS-катализаторах, также как и в пропиточном образце, составляло 5 мас. % MnO2 и 0.5 мас. % Pt. Согласно [12, 24] реакцию горения азотнокислых солей с глицином можно описать следующей схемой:
Для выявления роли оксида марганца в каталитической активности смешанных Pt–Mn-катализаторов аналогичными способами были приготовлены Mn-катализаторы, содержащие 5 мас. % MnOx: пропиточный Mn/AlSi и SCS-катализаторы Mn/AlSi(0.7), Mn/AlSi(2), Mn/AlSi(3.5).
Исследование катализаторов
Химический анализ образцов носителя и катализаторов проводили рентгеноспектральным флуоресцентным методом (РФлА) на анализаторе ARL PERFORM’X (“Thermo Fisher Scientific”, США) c Rh-анодом рентгеновской трубки.
Термический анализ образцов осуществляли с использованием прибора синхронного термического анализа STA 449 С Jupiter (“NETZSCH”, Германия), совмещая дифференциальную сканирующую калориметрию (ДСК) и термогравиметрию (ТГ) в одном измерении. Для исследования образцов применяли корундовые тигли без крышки. Скорость подачи воздуха в камеру образца составляла 30 см3/мин. В весовой блок подавали инертный газ (гелий) со скоростью 20 см3/мин. Образец нагревали со скоростью 2°С/мин от комнатой температуры до 50°С и выдерживали при данной температуре 60 мин. Далее проводили программируемый температурный нагрев со скоростью 10°С/мин до 700°С.
Рентгенофазовый анализ (РФА) образцов осуществляли с помощью дифрактометра HZG-4C (“Freiberger Prazisionsmechanik”, Германия) с монохроматизированным излучением CoKα (λ = = 1.79021 Å) в области углов 20°–80° по 2θ со скоростью сканирования 1 град/мин. Фазы идентифицировали по рентгенографической базе данных JCPDS.
Морфологию поверхности и состояние активных компонентов исследовали методом просвечивающей электронной микроскопии высокого разрешения (ПЭМВР) на электронном микроскопе JEM-2200FS (“JEOL”, Япония), ускоряющее напряжение – 200 кВ, разрешение по решетке – 1 Å. Микроскоп оснащен EDX-спектрометром для локального элементного анализа (локальность электронного зонда до 1 нм, разрешение по энергии порядка 130 эВ) и картирования состава образца по элементам. Также реализован сканирующий режим с детектором высокоугловых рассеянных электронов (HAADF-STEM). Образцы для ПЭМ-исследования готовили напылением их взвеси в этиловом спирте на медные подложки с помощью ультразвукового диспергатора.
Окислительно-восстановительные свойства катализаторов изучали методом термопрограммируемого восстановления водородом (ТПВ-H2) на установке, оборудованной проточным реактором и детектором по теплопроводности. Восстановление проводили в интервале температур от 25 до 950°C со скоростью подъема температуры 10°C/мин, пропуская смесь 10% H2 в Ar со скоростью 30 см3/мин через навеску образца. Перед проведением эксперимента образец подвергали тренировке в потоке Ar, продуваемом через реактор со скоростью 30 см3/мин при 450°С в течение 30 мин. Навеска образца составляла 100 мг, размер частиц – 250–500 мкм, для снятия экзотермических эффектов образец смешивали со 100 мг кварца с аналогичным размером частиц. Воду, образующуюся в ходе восстановления, удаляли из газовой смеси вымораживанием в ловушке при температуре –70°С. Количество поглощенного водорода калибровали относительно водорода, потребляемого на восстановление CuO при аналогичных условиях, предполагая полное одностадийное восстановление CuO до Cu.
Текстурные свойства носителя и катализаторов – удельная поверхность (SБЭТ, м2/г), суммарный объем пор (VΣ, см3/г) и средний диаметр пор (Dпор, нм) – были исследованы методом низкотемпературной адсорбции азота при 77 К на установке ASAP 2400 (“Micrometrics”, США).
Распределение элементов на внешней поверхности носителя и в объеме, а также количественное содержание фаз и их стехиометрический состав в анализируемых образцах определяли методом дифференцирующего растворения (ДР) на атомно-эмиссионном спектрофотометре с индуктивно связанной плазмой (“BAIRD”, США) с использованием стехиографа. Для оптимизации условий ДР-анализа образцов с различной макроструктурой использовали режим стехиографического титрования [52], при котором растворение навески в 10 мг начинали с H2О и затем последовательно переходили к растворам HCl (1.2 M и 3 M), смеси царской водки с водой (с объемным соотношением 4 : 1) и HF (3.6 М). Процесс растворения сопровождался постепенным повышением температуры от 22 до 90°С. Поток образующегося раствора пробы со скоростью 3.6 см3/мин направляли непосредственно в детектор-анализатор стехиографа. Детектор-анализатор позволяет одновременно определять в образующемся растворе все элементы из состава образца каждые 3 с по спектральным линиям элементов (нм): 308.2 для Al, 288.1 для Si, 293.3 для Mn, 265.9 для Pt с чувствительностью на уровне n × 10–3 мкг/мл и 5 отн. % погрешностью. Конечная информация выдается в виде кинетических кривых растворения найденных фаз.
Каталитическую активность образцов измеряли в реакции глубокого окисления бутана и метана в проточном кварцевом реакторе при температурах от 100 до 500°С. Для измерения конверсии бутана катализатор загружали в объеме 1 см3 в виде фракции с размером зерна 0.5–1.0 мм, смешанной с кварцем при соотношении 1 : 1. Исходную реакционную смесь, содержащую 0.1 об. % С4Н10 в воздухе, подавали в реактор при объемной скорости потока 12 000 ч–1. Для измерения конверсии метана в реактор загружали 1 см3 катализатора (без кварца) и подавали реакционную смесь 1 об. % CH4 в воздухе с объемной скоростью 1000 ч–1. Для определения концентраций реагентов и продуктов реакции (C4H10, СH4, O2/СO и CO2) использовали газовый хроматограф ЛХМ-8Д (Россия), оснащенный детектором по теплопроводности и разделительными колонками с Porapak Q и NaX (для бутана), АГ-3 и NaX (для метана). Каталитическую активность оценивали по температуре 50%-ной конверсии бутана и метана.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Механизм формирования активных компонентов по данным термического анализа катализаторов
Исследование воздушно-сухих Pt–Mn-катализаторов методом термического анализа показало существенное различие в картинах ТГ и ДСК образцов, полученных методом “сжигания в растворе” с различным соотношением топливной добавки к исходным солям-предшественникам активного компонента.
При нагревании воздушно-сухого образца Pt–Mn/AlSi, приготовленного методом пропитки без использования глицина, наблюдаются два пика потери веса на кривой ДТГ при температурах 96 и 205°С, их температуры совпадают с температурами эндотермических пиков на кривой ДСК (рис. 1а). Первый пик соответствует потере веса образца в результате удаления адсорбированной воды и воды, входящей в состав кристаллогидрата Mn(NO3)2 · · 6Н2О. Дальнейшее резкое снижение веса образца в области 180–230°С указывает на интенсивное разложение азотнокислых солей. Затем процесс разложения солей замедляется и завершается при температуре около 420°С с образованием соответствующих оксидов. Незначительное уменьшение веса при температуре выше 420°С может быть связано с термическим восстановлением Pt.
Рис. 1.
Кривые ТГ (1), ДТГ (2), ДСК (3) по данным термического анализа образцов: а – Pt–Mn/AlSi; б – Pt–Mn/AlSi(0.7); в – Pt–Mn/AlSi(2); г – Pt–Mn/AlSi(3.5).
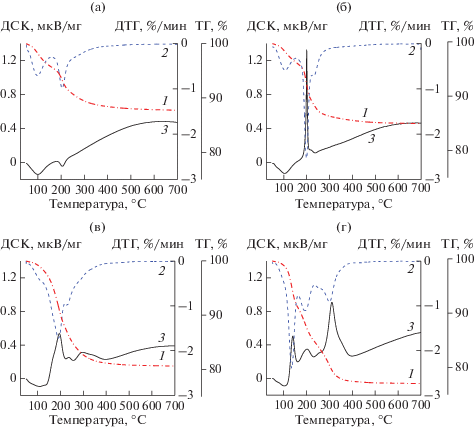
Аналогичные процессы удаления воды и разложения азотнокислых солей наблюдаются и при нагреве образца Pt–Mn/AlSi(0.7), синтезированного при значении φ < 1 с использованием глицина (рис. 1б). Однако на кривой ДСК вместо эндотермического пика при температуре 200°С наблюдается сильный экзотермический эффект, совпадающий с пиком потери веса на кривой ДТГ. Упомянутые эффекты связаны с разложением нитратов Mn и Pt и реакцией горения между нитратами и топливной добавкой (глицином) в одном температурном интервале. Кроме того, на кривой ДТГ имеется небольшой отрицательный пик при температуре 231°С, который может быть обусловлен частичным восстановлением оксидов Mn и, возможно, Pt. Дальнейший нагрев образца Pt–Mn/AlSi(0.7) не приводит к каким-либо значительным изменениям в картине термического анализа по сравнению с образцом Pt–Mn/AlSi. Таким образом, в ходе SCS при недостатке топливной добавки формирование активных компонентов происходит в узком температурном интервале в результате протекания двух процессов: термического разложения нитратов и реакции горения с глицином.
Иная картина наблюдается при термическом анализе образцов Pt–Mn/AlSi(2) (рис. 1в) и Pt–Mn/AlSi(3.5) (рис. 1г), полученных в условиях избытка топливной добавки. Кроме пика потери веса, относящегося к удалению воды, на кривой ДТГ присутствуют четыре отрицательных пика при температурах 193, 230, 288, 330°С для образца Pt–Mn/AlSi(2) и 135, 194, 252, 311°С для Pt–Mn/AlSi(3.5). Им соответствуют экзотермические эффекты на кривой ДСК в близких температурных областях. Первые пики при температурах 193 и 135°С свидетельствуют о начинающемся разложении азотнокислых солей марганца и платины. Экзотермические эффекты указывают на протекание реакции SCS между нитратами Mn, Pt и глицином. Наличие разделенных пиков на ДСК- и ДТГ-кривых, вероятно, связано со ступенчатым восстановлением катионов Mnn+ (Mn2O3 → Mn3O4 → → MnO), и Pt4+ (PtO2 → PtOx → Pt (где x < 2)), поскольку глицин выступает не только как топливо, но и как восстановитель в SCS-реакции. Следует отметить, что при увеличении количества топливной добавки начало разложения азотнокислых солей сдвигается в область меньших температур, а температурный интервал формирования фаз расширяется. Первое обусловлено повышением температуры на поверхности катализатора из-за инициирования горения топливной добавки и самораспространяющегося синтеза, частным случаем которого является SCS-реакция. Второе можно объяснить большей устойчивостью хелатных комплексов Mn–глицин и Pt–глицин к разложению в сравнении с их нитратными предшественниками. Следовательно, активные компоненты в SCS-катализаторах, приготовленных в условиях избытка топлива, формируются в результате протекания совокупности процессов: разложения исходных солей и их комплексов с глицином, термической реакции между солями и глицином и реакций восстановления образовавшихся оксидов марганца и платины глицином. Подобный эффект наблюдался в работе [31] при исследовании Mn–La- и Mn–Ce-катализаторов, полученных пропиткой и методом SCS в условиях избытка и недостатка глицина, и в работе [53] при синтезе магнетита (Fe3O4) с использованием глицина в качестве топливной добавки. При избытке топлива авторы наблюдали полимодальный пик на кривой ДСК, который объяснили синергетическим эффектом реакции горения и реакции восстановления ионов металлов.
Структурные свойства катализаторов
Фазовый состав и структурные свойства катализаторов были исследованы методами РФА и ПЭМ. Однако метод РФА оказался малоинформативным для определения фазового состояния активных компонентов. Дифрактограммы всех катализаторов, независимо от метода приготовления, были идентичными и содержали только рефлексы, соответствующие фазам носителя: α-Al2O3 и α-SiO2 (табл. 1). Отсутствие на дифрактограммах пиков, относящихся к фазам соединений платины, связано с низким содержанием Pt в катализаторе и, возможно, с ее высокой дисперсностью. Трудность обнаружения оксидов марганца в сложных марганецсодержащих каталитических системах на различных типах носителей является известным фактом, описанным в литературе. В работах [31, 32] идентифицировать методом РФА структуру активного компонента в катализаторах на основе MnOx, MnOx–La2O3, MnOx–CeO2 и CuO–MnOx, нанесенных на блочные алюмосиликатные носители, не удалось даже при проведении дополнительных съемок с длительным накоплением сигнала в каждой точке. В [54, 55] методом РФА были изучены марганецсодержащие одно- и многокомпонентные катализаторы на кордиерите. На дифракционных картинах авторы также не обнаружили фаз оксидов Mn, объяснив данный факт возможным растворением Mn2O3 в решетке кордиерита с образованием твердого раствора.
Таблица 1.
Физико-химические свойства носителя и катализаторов
| Носитель и катализаторы | Содержание активного компонента (мас. %) | SБЭТ, м2/г | VΣ, cм3/г | Dпор, нм | Фазовый состав |
|---|---|---|---|---|---|
| AlSi (носитель) | – | 12.5 | 0.068 | 21.7 | α-Al2O3-корунд; α-SiO2-кварц |
| Pt–Mn/AlSi | 5.02 MnOx*, 0.52 Pt | 15.3 | 0.056 | 15.0 | α-Al2O3, α-SiO2 |
| Pt–Mn/AlSi(0.7) | 5.29 MnOx, 0.52 Pt | 22.4 | 0.050 | 9.0 | α-Al2O3, α-SiO2 |
| Pt–Mn/AlSi(2) | 4.95 MnOx, 0.51 Pt | 20.8 | 0.053 | 10.3 | α-Al2O3, α-SiO2 |
| Pt–Mn/AlSi(3.5) | 4.74 MnOx, 0.48 Pt | 18.5 | 0.058 | 12.6 | α-Al2O3, α-SiO2 |
Исследования катализаторов методом ПЭМВР показали, что в пропиточном катализаторе Pt–Mn/AlSi оксиды марганца образуют фазу взаимодействия с компонентами носителя, а также присутствуют в виде Mn3O4 в приповерхностных слоях катализатора (рис. 2а), платина находится в металлическом состоянии и хорошо окристаллизована (рис. 2б). В катализаторах SCS активный компонент также представлен фазами Mn3O4 и Pt0 (рис. 2в и 2г). Влияние метода приготовления на дисперсность платины демонстрируют микрофотографии ПЭМ и снимки HAADF-STEM. Показано, что использование метода пропитки приводит к образованию высокодисперсных частиц металлической платины со средним размером частиц около 2 нм (рис. 2д и 2ж), а в условиях SCS-синтеза формируются частицы еще меньшего размера – до 1 нм (рис. 2е и 2з).

Окислительно-восстановительные свойства катализаторов
На рис. 3а приведены кривые поглощения водорода катализаторами Pt–Mn/AlSi, приготовленными методами пропитки и SCS при варьировании количества глицина. Свойства некоторых Mn/AlSi-катализаторов детально обсуждались нами в работах [31, 32, 56]. В настоящей работе профили ТПВ-Н2 однокомпонентных Mn/AlSi-катализаторов (рис. 3б), полученных при аналогичных условиях синтеза, показаны для удобства сопоставления окислительно-восстановительных свойств и роли Pt в них.
Рис. 3.
Профили ТПВ-Н2 для пропиточных (1) и SCS (2–4) катализаторов Pt–Mn/AlSi (а) и Mn/AlSi (б). Количество топливной добавки φ, использованное для SCS-синтеза, составляет 0.7 (2), 2 (3) и 3.5 (4). H2/Mn и H2/(Pt + Mn) соответствуют мольному соотношению количеств поглощенного за эксперимент водорода к нанесенному каталитически активному металлу/металлам.
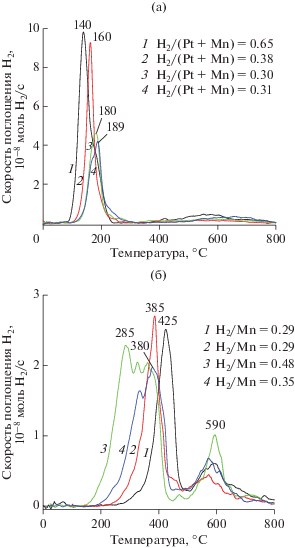
Профили ТПВ-Н2 как Pt–Mn/AlSi, так и Mn/AlSi содержат два пика поглощения водорода. Один из этих пиков находится в области температур 500–700°С и соответствует, согласно результатам наших предыдущих работ [31, 32], восстановлению высокодисперсных частиц оксидов Fe(II,III), входящих в состав алюмосиликатной матрицы. Пики в низкотемпературной (140–190°С) и среднетемпературной (280–425°С) областях обусловлены восстановлением оксидов Pt и марганца. Сопоставление температурных интервалов восстановления водородом активного компонента в катализаторах Pt–Mn/AlSi и Mn/AlSi показывает, что независимо от способа приготовления катализатора Mn/AlSi добавление в его состав Pt способствует значительному понижению температур полного восстановления активного компонента, а также сужению температурных интервалов восстановления. Так, для пропиточных образцов Pt–Mn/AlSi и Mn/AlSi максимумы низкотемпературного поглощения водорода наблюдались при 140 и 425°С соответственно. Максимум поглощения водорода SCS-катализатором Pt–Mn/AlSi(3.5) был при ~190°С против 335–380°С, наблюдаемых для образца Mn/AlSi(3.5). Улучшение способности к восстановлению водородом MnOx в присутствии Pt [49, 50] и/или Pd [46, 49] отмечалось нами ранее для нанесенных катализаторов MnOx/Al2O3 после их модифицирования добавками Pt–Pd, а также другими авторами, изучавшими каталитические системы на основе оксидов переходных металлов (MnOx, CuO) и благородных металлов [42, 43]. M. Ferrandon с соавт. [43] связали наблюдаемый синергетический эффект в восстановлении систем Pt(Pd)–оксид переходного металла со спилловером водорода с металлических частиц благородного металла на MnOx(CuO), что возможно при сильном взаимодействии между указанными частицами благородных металлов и оксидов переходных металлов.
Состав оксида марганца в образцах Mn/AlSi можно предположить из мольного соотношения количеств поглощенного образцом водорода за ТПВ-эксперимент и марганца в нем, а также косвенно из температурных интервалов поглощения водорода. Значения H2/Mn, рассчитанные из экспериментальных данных ТПВ-Н2 и РФлА, приведены на рис. 3а и 3б. Пропиточный катализатор Mn/AlSi (рис. 3б, кривая 1) восстанавливается в интервале температур 350–480°С и имеет H2/Mn равное 0.29. Оба этих параметра, согласно литературным источникам [57, 58], соответствуют восстановлению Mn3O4 до MnO. Доказанным считается, что окристаллизованный массивный Mn3O4 восстанавливается в относительно узком интервале температур 400–470°С, указывающем на высокую однородность кислорода в нем [57], тогда как все другие оксиды марганца – MnO2, Mn2O3 и Mn5O8 – восстанавливаются в две стадии [59] с увеличением стехиометрии восстановления (H2/Mn) до 0.5 (для Mn2O3) и выше (0.57 для Mn5O8 и 1 для MnO2).
В сравнении с пропиточным катализатором профили SCS-катализаторов Mn/AlSi (рис. 3б, кривые 2–4) имеют тенденцию к сдвигу максимумов поглощения водорода в низкотемпературную область и уширению температурного интервала поглощения, но эти эффекты нелинейно зависят от количества топливной добавки, использованной для синтеза. Максимальная величина сдвига пиков наблюдалась для катализатора Mn/AlSi(2) – 285°С против 425°С для Mn/AlSi. Нелинейно от количества топливной добавки изменяется и соотношение H2/Mn. При ее недостатке (φ < 1), экспериментальное значение соотношения H2/Mn близко к стехиометрии, характерной восстановлению Mn3O4 (0.29 против 0.33). При 2-кратном избытке топливной добавки (φ = 2) экспериментальное значение соотношения H2/Mn составляет 0.48, что ближе к стехиометрии восстановления Mn2O3. При 3.5-кратном избытке глицина соотношение H2/Mn = 0.35 указывает на формирование в катализаторе Mn/AlSi(3.5), аналогично образцу Mn/AlSi(0.7), частиц Mn3O4, но их дефектность и доступность для водорода выше, чем в пропиточном катализаторе. Об этом говорит тот факт, что SCS-катализатор восстанавливается при меньших температурах: 330–380°С вместо 425°С (рис. 3б, кривые 1 и 4). Плечо при 230–250°С в профиле ТПВ-Н2 катализатора Mn/AlSi(3.5) свидетельствует о наличии в образце сверхстехиометрического кислорода, связанного с катионами Mn(III) в тетрагональных позициях дефектного высокоповерхностного Mn3O4 [57, 58]. Таким образом, причиной наиболее важного отличия окислительно-восстановительного поведения SCS-образцов, приготовленных в избытке глицина, от свойств пропиточного катализатора Mn/AlSi является наличие сверхстехиометрического кислорода в высокодисперсных частицах Mn3O4. Считается также [58], что тетраэдрические катионы Mn(III) ослабляют связь между катионом Mn(III) в октаэдрических позициях и кислородом решетки Mn3O4. На это указывает снижение температуры восстановления Mn3O4 до MnO, составляющей 380 и 425°С для катализаторов Mn/AlSi(3.5) и Mn/AlSi соответственно. Большая реакционная способность кислорода, связанного как с тетраэдрическими, так и с октаэдрическими катионами Mn(III) в дефектных частицах Mn3O4, может обеспечивать эффективность SCS-катализаторов в реакциях окисления углеводородов.
Проанализировать состав каталитически активных частиц Pt/PtOx и MnOx в бикомпонентных катализаторах Pt–Mn/AlSi оказалось сложнее, поскольку в них эти частицы восстанавливаются вместе и при температурах существенно более низких, чем те, что характерны для индивидуальных оксидов марганца [31, 32, 56–59] и окристаллизованного PtO [60]. Мольное соотношение Mn/Pt в катализаторах Pt–Mn/AlSi достаточно велико (Mn/Pt = 23), поэтому соотношение H2/(Pt + Mn), рассчитываемое из экспериментов и РФлА, определяется преимущественно состоянием MnOx в катализаторе, и однозначно установить состояние платины с помощью данных ТПВ-Н2 становится невозможным. Максимальная величина H2/(Pt + Mn) = 0.65 наблюдалась для пропиточного катализатора (рис. 3а, кривая 1). Она ниже стехиометрического значения для восстановления MnO2 до MnO, но выше такового для восстановления Mn5O8 и Mn2O3 до MnO. Из чего следует, что образец Pt–Mn/AlSi содержит смешанный оксид Mn5O8, обогащенный катионами Mn(IV). Для SCS-катализаторов соотношение H2/(Pt + Mn) было в 1.7–2.2 раза ниже, чем для пропиточного и составляло 0.30–0.38, что свидетельствует о преобладании Mn3O4 в составе этих образцов независимо от количества глицина, использованного для синтеза. Отличительной особенностью Pt–Mn/AlSi-катализаторов от Mn/AlSi является некоторое ухудшение их способности к восстановлению при переходе от пропиточных к SCS-катализаторам. На это указывает сдвиг низкотемпературного пика в область бóльших температур: для катализатора Pt–Mn/AlSi(0.7) максимум смещается от 140 к 160°С. Эта тенденция усиливается для катализаторов Pt–Mn/AlSi(2) и Pt–Mn/AlSi(3.5), приготовленных SCS-методом в избытке глицина (рис. 3а, кривые 3 и 4). Причиной изменения окислительно-восстановительных свойств SCS-катализаторов, по-видимому, является снижение размера частиц Pt, регистрируемое на снимках ПЭМВР (рис. 2), а также усиление взаимодействия Pt и/или PtOx с Mn3O4 и носителем. Известно, что температурный интервал восстановления PtOx зависит не только от дисперсности частиц PtOx, но и силы их взаимодействия с носителем, составляя 150–220 и 220–250°C для окристаллизованных [60] и дисперсных частиц PtOx [60–62] соответственно, но по мере усиления взаимодействия между PtOx и носителем (например, Al2O3) восстановление сдвигается в высокотемпературную область, иногда до 375–550°C [50, 61–63].
Текстурные свойства катализаторов
Основные текстурные характеристики носителя и катализаторов, такие как значения удельной поверхности, суммарного объема пор и среднего диаметра пор, приведены в табл. 1. Распределение пор по размерам, построенное по данным BJH, представлено на рис. 4.
Рис. 4.
Дифференцированное распределение пор по размерам в образцах носителя и катализаторов: 1 – носитель; 2 – Pt–Mn/AlSi; 3 – Pt–Mn/AlSi(0.7); 4 – Pt–Mn/AlSi(2); 5 – Pt–Mn/AlSi(3.5).
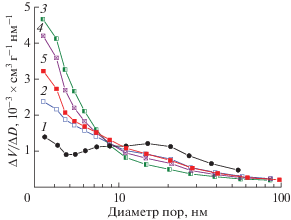
Как видно из табл. 1, катализаторы имеют бóльшую величину удельной поверхности, чем носитель. Нанесение активных компонентов методом пропитки приводит к незначительному снижению суммарного объема пор и резкому уменьшению среднего диаметра пор. Это может свидетельствовать о частичном заполнении пор среднего размера (10–50 нм) оксидами марганца и соединениями Pt. В результате заполнения средних пор их доля в пористой структуре носителя сокращается, и увеличивается доля мелких пор с преобладающим диаметром 3.5 нм, что продемонстрировано на рис. 4 (кривая 1 – носитель, кривая 2 – пропиточный катализатор). В результате роста количества тонких пор повышается удельная поверхность и уменьшается средний диаметр пор. Нанесение активных компонентов методом SCS способствует формированию более тонкопористой структуры по сравнению с пропиточным образцом (рис. 4, кривая 3). Это происходит за счет процесса спонтанного порообразования при кратковременном бурном выделении газообразных продуктов реакции горения. Более интенсивное тепловыделение при увеличении количества топливной добавки (φ = 2 и 3.5), вызывает более активное выделение газа по сравнению с φ = 0.7, и это приводит к формированию большего количества крупных пор диаметром >10 нм (рис. 4, кривые 4 и 5).
Согласно литературным данным существует несколько причин повышения удельной поверхности при использовании SCS-метода. Основной является интенсивное выделение тепла при реакции азотнокислых солей с глицином, сопровождающееся бóльшим выходом газообразных продуктов, что ведет к формированию новых пор в керамическом носителе [33], ослаблению агломерации и диспергированию частиц активного компонента [18, 19, 38]. Другой причиной предполагают возникновение эффекта “нанореактора” при протекании экзотермической реакции в наноразмерных порах носителя [16]. Таким образом, в развитие удельной поверхности SCS-катализаторов вносят вклад процессы и порообразования, и диспергирования активного компонента. Влияние метода приготовления на дисперсность платины продемонстрировано на снимках ПЭМ и HAADF-STEM (рис. 2).
Локализация активных компонентов в катализаторе
Метод дифференцирующего растворения позволяет определить локализацию компонентов катализатора. Так, в мягких условиях растворения – в воде или в слабоконцентрированных кислотах – при температурах в интервале от комнатной до 50°С в раствор из катализатора переходят легкорастворимые соединения, расположенные на поверхности пор и в приповерхностных слоях. Ужесточение условий растворения (температура 50–90°С, сильные кислоты) приводит к растворению труднорастворимых соединений и труднодоступных компонентнов катализатора, расположенных в глубоких порах в объеме носителя. Кривые растворения всех компонентов Pt–MnOx-катализаторов, приготовленных пропиткой и методом SCS (для φ = 3.5), представлены на рис. 5.
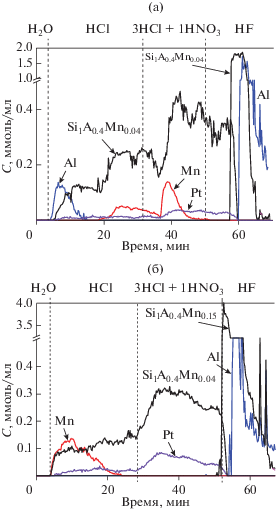
Основным отличием картин ДР исследованных образцов является кривая растворения Mn-содержащих соединений. В пропиточном образце Pt–Mn/AlSi (рис. 5а) 26% марганца растворяется вместе с компонентами носителя в стехиометрии Si1Al0.4Mn0.04. Поскольку эта стехиометрия сохраняется в течение длительного периода растворения, можно предположить существование фазы взаимодействия оксидов марганца с носителем. В пользу этого предположения свидетельствуют и данные ПЭМВР. Около 70% марганца растворяется без стехиометрии вместе с фазой Si1Al0.4Mn0.04 частично в HCl и большей частью в царской водке. Вероятнее всего, это часть MnOx находится в глубоких труднодоступных для растворителя порах носителя. И, наконец, 4% марганца растворяется в составе труднорастворимого соединения Si1Al0.4Mn0.04. Это соединение переходит в раствор только при использовании HF. Дифференцированное растворение Si1Al0.4Mn0.04 связано, вероятно, с неоднородностью морфологии носителя, содержащего и аморфные, и окристаллизованные фазы [32] с различной способностью к растворению. Платина растворяется только в царской водке и не образует соединений с другими компонентами катализатора. Следовое количество Pt в растворе HCl на начальных этапах растворения связано с уносом частиц платины при растворении носителя.
При растворении катализатора Pt–Mn/AlSi(3.5) (рис. 5б) до 10% уменьшается количество марганца, входящего в состав фазы Si1Al0.4Mn0.04, растворимой в HCl, и до 2% – растворимой в HF, что в 2–2.5 раза меньше по сравнению с пропиточным катализатором. 88% MnOx растворяется полностью в мягких условиях на начальных этапах растворения и, значит, эта фаза локализована в приповерхностных порах носителя. Картина растворения платины такая же, как и в пропиточном катализаторе.
Картину ДР соединений марганца дополняют данные EDX-исследований. На рис. 6 представлены изображения, полученные в режиме регистрации характеристического рентгеновского излучения K-линий Al (рис. 6а, 6д), Si (рис. 6б, 6е), Mn (рис. 6в, 6ж) от пропиточного (верхний ряд) и SCS (нижний ряд) образцов. На изображении для пропиточного катализатора Pt–Mn/AlSi, полученном с наложением всех сигналов (рис. 6г), видно, что Mn локализован на участках наложения сигналов К-линий Al и Si, но отсутствует на участках “чистого” алюминия (выделено белым контуром) и в очень малом количестве содержится в поровом пространстве между кристаллитами носителя. В катализаторе Pt–Mn/AlSi(3.5), приготовленном методом SCS (рис. 6з), Mn частично распределен на поверхности кристаллитов, содержащих одновременно Al и Si, и в большом количестве существует в виде отдельного соединения между кристаллитами носителя. Также как и в пропиточном образце, линии Mn не совпадают с линиями Al (выделено белым контуром).
Рис. 6.
ПЭМ-изображения, полученные в режиме регистрации характеристического рентгеновского излучения K-линий: а, д – Al; б, е – Si; в, ж – Mn; г, з – с наложением всех перечисленных сигналов. Верхний ряд изображений – для катализатора Pt–Mn/AlSi, нижний ряд – для Pt–Mn/AlSi(3.5). Белым контуром обведены участки изображения от излучения K-линий Al, свободные от наложения сигналов K-линий Mn.

Суммируя данные методов ДР и EDX-картирования, можно заключить, что независимо от способа получения катализаторов марганец распределяется на поверхности кристаллитов алюмосиликатной фазы и с высокой долей вероятности образует с ней фазу взаимодействия. SCS-метод обеспечивает локализацию марганца преимущественно на поверхности пор и создает условия, снижающие возможность образования фазы взаимодействия марганца с компонентами носителя. Подобный эффект наблюдался нами в работе [31], посвященной изучению влияния способа синтеза на распределение марганца в двухкомпонентных Mn–La- и Mn–Ce-катализаторах. В указанной работе для всех Mn-содержащих катализаторов, полученных методом SCS, было обнаружено обогащение поверхностных и приповерхностных слоев керамического носителя марганцем, особенно в случае образцов, приготовленных в условиях избытка топливной добавки. В пропиточных катализаторах преобладало взаимодействие катионов марганца с компонентами носителя. В [31] было установлено, что обогащение приповерхностных слоев SCS-катализаторов оксидом марганца по сравнению с пропиточными способствует повышению каталитической активности в окисления бутана благодаря большей доступности активных центров для реактантов.
Активность катализаторов в реакции окисления бутана и метана
Для выявления роли оксида марганца в активности смешанных Pt–Mn-систем в реакции окисления бутана и метана были испытаны и сопоставлены каталитические характеристики Mn- и Pt–Mn-образцов, приготовленных в одинаковых условиях. На рис. 7 и 8 приведены температурные зависимости конверсии бутана для пропиточных и SCS-катализаторов. В случае пропиточных образцов добавление Pt повышает активность Mn-катализатора (рис. 7, кривые 1 и 2). Однако характеристики полученных SCS-методом Pt–Mn-систем в окислении бутана (рис. 7, кривые 3–5) уступают таковым для Mn-катализаторов (рис. 8, кривые 2–4). Температуры 50%-ной конверсии бутана на Mn-образцах на 30–35°С ниже, чем на Pt–Mn-системах, приготовленных при таких же значениях φ. Несмотря на это, как для Mn-, так и для Pt–Mn-систем прослеживается тенденция к большей активности SCS-образцов по сравнению с пропиточными. Температуры 50%-ной конверсии бутана в присутствии катализаторов, полученных с помощью SCS и пропиточным методом, различаются на 23–40°С для Pt–Mn- и на 90–113°С для Mn-образцов. Отметим, что в обеих сериях SCS-катализаторов активность возрастала с увеличением значения φ, но в случае Pt–Mn/AlSi влияние φ было меньше, чем в случае Mn/AlSi.
Рис. 7.
Температурная зависимость конверсии бутана для пропиточных (1, 2) и SCS (3–5) катализаторов, различающихся составом и условиями синтеза: 1 – Mn/AlSi; 2 – Pt–Mn/AlSi; 3 – Pt–Mn/AlSi(0.7); 4 – Pt–Mn/AlSi(2); 5 – Pt–Mn/AlSi(3.5).
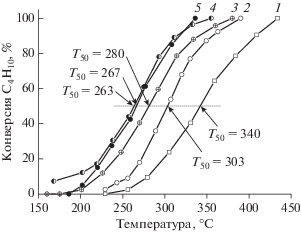
Рис. 8.
Температурная зависимость конверсии бутана для пропиточного (1) и SCS (2–4) Mn-катализаторов, различающихся условиями синтеза: 1 – Mn/AlSi; 2 – Mn/AlSi(0.7); 3 – Mn/AlSi(2); 4 – Mn/AlSi(3.5).
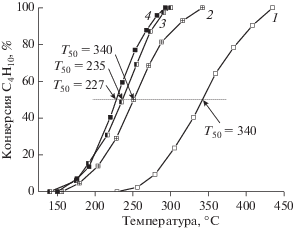
Похожие закономерности наблюдаются и в реакции окисления метана (рис. 9). Сравнение температурных зависимостей конверсии метана для пропиточных образцов показывает значительное увеличение активности Mn/AlSi после добавления в его состав Pt. Так, различие между Т50% на кривых конверсии метана для Mn/AlSi и Pt–Mn/AlSi (рис. 9, кривые 1 и 3) составляет 85°С. Практически такой же эффект (~95°С) проявляется при изменении способа синтеза Mn-катализаторов с пропитки на SCS-метод (рис. 9, кривые 1 и 2). Использование SCS для приготовления Pt–Mn-систем также приводит к повышению активности по сравнению с методом пропитки (рис. 9, кривые 3 и 4), хотя выигрыш в Т50% в этом случае не очень большой (всего 25°С) в отличие от Mn-образцов.
Рис. 9.
Температурная зависимость конверсии метана для пропиточных (1, 3) и SCS (2, 4) катализаторов, различающихся составом: 1 – Mn/AlSi; 2 – Mn/AlSi(2); 3 – Pt–Mn/AlSi; 4 – Pt–Mn/AlSi(2).
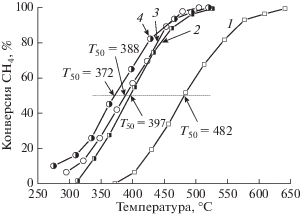
Необходимо отметить, что добавка Pt оказывала разное действие на каталитическое поведение SCS-катализаторов в реакциях окисления углеводородов, которое зависело от длины углеводородной цепи окисляемого алкана. В окислении метана активность SCS Pt–Mn-систем была несколько выше активности Mn-образцов (рис. 9, кривые 2 и 4), тогда как в окислении бутана SCS Mn-катализаторы превосходили Pt–Mn-системы (рис. 7, кривые 3–5, и рис. 8, кривые 2–4), синтезированные при одинаковых φ.
Сопоставление активности в окислении бутана и метана с физико-химическими свойствами катализатора
С учетом обсуждаемых выше данных о размере, составе и локализации каталитически активных частиц можно полагать, что наблюдаемые различия в каталитической активности пропиточных и SCS Pt–Mn-катализаторов в окислении бутана и метана в основном обусловлены составом оксидов марганца и их локализацией в порах носителя и в меньшей мере дисперсностью частиц платины в силу их высокодисперсного состояния во всех катализаторах. Размер частиц Pt в пропиточном и SCS Pt–Mn-катализаторах различался в 2–3 раза, не превышая, однако, 1–3 нм по снимкам ПЭМ для пропиточного катализатора. С одной стороны, такого различия может оказаться достаточно для улучшения эффективности каталитического действия в структурно-чувствительных реакциях окисления метана и бутана. Диспергирование частиц Pt в SCS-катализаторах давало некоторый выигрыш в окислении метана (ΔТ50% ≈ 16°С) и бутана (ΔТ50% ≈ 40°С) в сравнении с пропиточным катализатором, имевшим больший размер Pt-частиц. Размерные эффекты в катализе нанесенными металлами обсуждаются в ряде обзорных работ [64–67]. Согласно терминологии, введенной М. Будар (M. Boudart) с соавт. [68, 69], реакции окисления углеводородов на нанесенных платиновых катализаторах являются структурно-чувствительными, поскольку их скорость зависит от дисперсности активного компонента. По данным В.И. Бухтиярова с соавт. [70], при заданной температуре конверсия алканов С1–С6 на катализаторе 0.8% Pt/Al2O3 с разным диаметром частиц зависела от длины молекул углеводородов, вступающих в реакцию. К примеру, конверсия метана снижалась при увеличении размера частиц Pt от 1 до 3–4 нм, тогда как в случае н‑С4Н10 или н-С6Н14 конверсия углеводорода наоборот повышалась [70]. Подобные размерные эффекты авторы наблюдали и на носителях другой химической природы: Pt/TiO2 и Pt/SiO2 [71]. Наши данные по окислению метана хорошо коррелируют с результатами работ [70, 71], тогда как в окислении бутана противоречат им. Согласно [72] для реакции окисления метана зависимость каталитической активности от размера частиц Pt имела колоколообразный вид, а максимум удельной каталитической активности достигался в достаточно узком интервале значений – около 2 нм. При этом понижение активности частиц Pt размером менее 2 нм авторы связали с изменением природы платины от металлической на “неметаллическую” [72], а для частиц размером более 2 нм – с кажущимися энергиями активации процесса [70, 72]. И хотя скоростьопределяющей стадией реакции является взаимодействие адсорбированных кислорода и алкана [73], процесс лимитируется адсорбцией алкана на поверхности катализатора в силу высокой теплоты десорбции с нее хемосорбированного кислорода [74, 75]. Бриот (P. Briot) с соавт. наблюдали корреляцию между каталитической активностью катализаторов Pt/Al2O3 и теплотой адсорбции кислорода, составлявшей 250 кДж/моль для частиц размером 12 нм и 280 кДж/моль для частиц размером менее 2 нм [74]. Кислород, адсорбированный на 12 нм частицах, практически полностью снимался водородом уже при 195 К (–78°С), тогда как на 2 нм частицах платины кислород не реагировал с водородом вплоть до 276 К (3°С), удаляясь полностью лишь в интервале температур 3–20°С. Опубликованные в [74] факты указывают на ослабление связи Pt–O по мере увеличения размера частиц, что обеспечивает повышение скорости окислительных реакций. Естественно, что влияние десорбции хемосорбированного кислорода на скорость окисления алкана зависит от температурного интервала протекания реакции, и для бутана проявляется сильнее в силу меньших температур, необходимых для его окисления [70, 71].
С другой стороны, значимое влияние на каталитические характеристики в реакциях окисления CH4 и C4H10 может оказать изменение окислительно-восстановительных свойств Pt–Mn-катализаторов, приготовленных разными методами. В экспериментах ТПВ-Н2 (рис. 3а) температура максимума поглощения водорода смещалась в высокотемпературную сторону при переходе от пропиточного катализатора Pt–Mn/AlSi к SCS Pt–Mn-образцам (ΔТ = 20°С), а далее – при повышении φ от 0.7 до 3.5 (ΔТ еще 29°С), тогда как температурные зависимости конверсии метана и бутана, наоборот, сдвигались в низкотемпературную область (рис. 7 и 9). Обнаруженный эффект противоречит закономерностям, обсуждаемым в литературе для Pt-катализаторов [76, 77], в том числе, модифицированных оксидом марганца [42, 50], где зачастую утверждается повышение активности при увеличении способности катализатора к восстановлению. Аналогично предполагаемому в [42, 43], способность к восстановлению MnOx-компоненты Pt–Mn-систем определяется, по-видимому, контактами между частицами MnOx и PtOх/Pt. При их наличии восстановление MnOx в Pt–Mn-катализаторе облегчается в сравнении с Mn-образцом в силу переноса водорода с Pt на MnOx [42], что хорошо заметно в профилях ТПВ-Н2 пропиточных образцов (рис. 3а и 3б, кривые 1). По данным ДР (рис. 5а) в пропиточном катализаторе Pt–Mn/AlSi соединения Pt и Mn достаточно равномерно распределены по всему объему катализатора, что и обеспечивает большое число таких контактов и способность катализатора к восстановлению. В случае же SCS Pt–Mn-катализаторов (рис. 5б) бóльшая часть MnOx локализована в приповерхностных слоях катализатора, причем эта тенденция усиливается с увеличением φ, тогда как платина по-прежнему находится во всем его объеме. В результате снижения числа контактов между частицами MnOx и PtOх/Pt происходит ухудшение способности SCS Pt–Mn-катализаторов к восстановлению. Таким образом, отклонение поведения SCS Pt–Mn-катализаторов от известных закономерностей “восстанавливаемость системы–каталитическая активность” [42, 50, 76, 77] связаны с особенностями переноса водорода с частиц Pt на MnOx и их локализацией в катализаторе.
В целом SCS Pt–Mn-катализаторы имели несколько бóльшую активность в окислении метана и бутана, чем пропиточный катализатор Pt–Mn/AlSi, однако этот экспериментальный факт связан не с размерным или структурным эффектами частиц Pt, а скорее с высокой реакционной способностью приповерхностных частиц Mn3O4. Приповерхностное расположение частиц Mn3O4 в SCS-катализаторах (данные ДР, рис. 5б) обеспечивало бóльшую доступность оксидов марганца для реактантов и высокую эффективность окисления бутана и метана. Ключевую роль локализации оксидов марганца в активности SCS-катализаторов подтверждает повышенная активность в окислении бутана Mn-образцов (рис. 8) в сравнении с Pt–Mn-системами (рис. 7). Отметим, что для серии Mn-катализаторов каталитическая активность в окислении углеводорода растет (рис. 8 и 9) со способностью системы к восстановлению (рис. 3б), то есть общеизвестная тенденция [56, 78] выполняется, исключение составляет образец Mn/AlSi(3.5). Для него температура 50%-ного превращения С4Н10 была несколько ниже, чем для катализатора Mn/AlSi(2) (227 против 235°С, рис. 7), тогда как восстанавливался он труднее (максимум широкого пика приходится на 330–390°С против 280–380°С, рис. 3б). Это связано с тем, что в составе каталитически активного оксида присутствуют разные оксиды: Mn3O4 в Mn/AlSi(3.5) и Mn2O3/Mn3O4 в Mn/AlSi(2), как следует из соотношения H2/Mn в экспериментах ТПВ-Н2. Поэтому высокая реакционная способность катализатора Mn/AlSi(3.5) вполне закономерна в силу большей реакционной способности Mn3O4 в сравнении с другими оксидами марганца [50, 79].
Улучшение характеристик пропиточного Pt–Mn-катализатора в окислении бутана и метана в сравнении с Mn-образцом обусловлено вкладом в активность Pt-частиц, поскольку частицы Mn3O4 в обоих пропиточных катализаторах локализованы в труднодоступных порах, и часть катионов Mn(II,III) входила в состав фазы взаимодействия с компонентами носителя (Si1Al0.4Mn0.04, данные ДР), что ослабляло окислительно-восстановительные и каталитические свойства MnOx.
Таким образом, высокая активность и доступность приповерхностных частиц Mn3O4 в SCS-катализаторах полностью нивелирует роль высокодисперсных частиц платины в окислении бутана. Негативным фактором оказался слишком маленький размер частиц Pt0, сформированных в условиях SCS – менее 1 нм по данным ПЭМВР, в результате чего они имели сильное взаимодействие с носителем и легко изменяли свое состояние с металлического на оксидное. Следовательно, сильное диспергирование частиц Pt в условиях SCS-метода оказалось нежелательным для катализаторов глубокого окисления углеводородов. С другой стороны, SCS-метод обеспечивает активацию Mn-компоненты катализаторов. Для достижения синергетического эффекта между Mn3O4 и Pt, по-видимому, необходимо увеличение размера частиц Pt до размеров 2–4 нм.
ЗАКЛЮЧЕНИЕ
Исследования комплексом физико-химических методов и в реакции окисления бутана и метана катализаторов на основе Pt–MnOx, нанесенных на блочные керамические носители, позволили сделать следующие выводы.
В пропиточных катализаторах формирование активных компонентов происходит за счет процесса термического разложения азотнокислых солей марганца и платины. В результате образуются смешанные оксиды Mn3O4 и Mn5O8, последний обогащен катионами Mn(IV). Оксиды марганца взаимодействуют с компонентами носителя, образуя в приповерхностных слоях катализатора фазу взаимодействия. Платина окристаллизована в частицы размером около 2 нм и находится в металлическом состоянии.
Активные компоненты в SCS-катализаторах формируются в результате протекания совокупности процессов: разложения исходных солей и их комплексов с глицином, термической реакции между солями и глицином и реакции восстановления нитратов марганца и платины глицином. В результате этих процессов образуется Mn3O4, который преимущественно расположен в приповерхностных слоях носителя, и высокодисперсная Pt0 с размером частиц до 1 нм. Интенсивное выделение тепла при реагировании азотнокислых солей с глицином, сопровождающееся большим выходом газообразных продуктов, приводит к повышению удельной поверхности SCS-катализаторов за счет формирования новых пор в керамическом носителе и диспергирования частиц активного компонента.
Активность пропиточных Pt–Mn-систем в реакциях глубокого окисления метана значительно выше таковой для Mn-катализаторов, благодаря частицам Pt размером около 2 нм. В реакции окисления бутана эффект от введения Pt в пропиточный Mn-образец оказался слабее.
Активность SCS Pt–Mn-катализаторов определяется составом оксидов марганца и их локализацией в порах носителя, поскольку платина находится в высокодисперсном состоянии, преимущественно в виде частиц размером до 1 нм. SCS Pt–Mn-катализаторы хотя и имели несколько бóльшую активность в окислении бутана и метана, чем пропиточный Pt–Mn-катализатор, но этот экспериментальный факт был связан не с размерным или структурным эффектами частиц Pt, а с высокой реакционной способностью приповерхностных частиц Mn3O4. Таким образом, SCS-метод обеспечивает значительную активацию Mn-компоненты, в результате чего SCS Mn-катализаторы становятся более активными в окислении бутана, чем Pt–Mn-системы, синтезированные при аналогичном φ. Сильное диспергирование частиц Pt оказывается скорее сдерживающим фактором в развитии каталитических свойств Pt–Mn-катализаторов, однако этот эффект зависит от длины цепи окисляемого алкана.
Список литературы
Forzatti P., Ballardini D., Sighicelli L. // Catal. Today. 1998. V. 41. P. 87.
Yashnik S.A., Ismagilov Z.R., Porsin A.V., Denisov S.P., Danchenko N.M. // Top. Catal. 2007. V. 42–43. № 1–4. P. 465.
Merzhanov A.G. // J. Mater. Chem. 2004. V. 14. P. 1779.
Taniguchi K., Okinaka N., Akiyama T. // J. Alloys Compd. 2011. V. 509. № 10. P. 4084.
Mossino P. // Ceram Int. 2004. V. 30. № 3. P. 311.
Hirano T., Tosho T., Watanabe T., Akiyama T. // High Temp Mater P-us. 2012. V. 31. № 4–5. P. 513.
Kingsley J.J., Patil K.C. // Mater. Lett. 1988. V. 6. P. 427.
González-Cortés S.L., Xiao T.-C., Green M.L.H. // Stud. Surf. Sci. Catal. 2006. V. 162. P. 817.
González-Cortés S.L., Imbert F.E .// Appl. Catal. A: General. 2013. V. 452. P. 117.
Groven L.J., Pfeil T.L., Pourpoint T.L. // Int. J. Hydrogen. Energ. 2013. V. 38. № 15. P. 6377.
Reddy L.H., Reddy G.K., Devaiah D., Reddy B.M. // Appl. Catal. A: General. 2012. V. 445–446. P. 297.
Mukasyan A. S., Dinka P. // Intern. J. Self-Propagating High-Temperature Synthesis. 2007. V. 16. P. 23.
Ghose R., Hwang H.T., Varma A. // Appl. Catal. A: General. 2014. V. 472. P. 39.
Varma A., Mukasyan A.S., Rogachev A.S., Manukyan K.V. // Chem. Rev. 2016. V. 116. P. 14 493.
Alves A.K., Bergmann C.P., Berutti F.A. Novel synthesis and characterization of nanostructured materials. Ch. 2. Combustion synthesis. Berlin: Springer, 2013. 92 p.
Завьялова У.Ф., Третьяков В.Ф., Бурдейная Т.Н., Лунин В.В., Шитова Н.Б., Рыжова Н.Д., Шмаков А.Н., Низовский А.И., Цырульников П.Г. // Кинетика и катализ. 2005. Т. 46. С. 795.
Завьялова У.Ф., Барбашова П.С., Лермонтов А.С., Шитова Н.Б., Третьяков В.Ф., Бурдейная, Т.Н. Лунин В.В., Дроздов В.А., Яшник С.Я., Исмагилов З.Р., Цырульников П.Г. // Кинетика и катализ. 2007. Т. 48. № 1. С. 171.
Sharma S., Hegde M.S. // Catal. Lett. 2006. V. 112. P. 69.
Russo N., Mescia D., Fino D., Saracco G., Specchia V. // Ind. Eng. Chem. Res. 2007. V. 46. P. 4226.
Aldashukurova G., Mironenko A., Mansurov Z., Shikina N., Yashnik S., Ismagilov Z. // Chem. Eng. Trans. 2011. V. 25. P. 63.
Ismagilov Z.R., Mansurov Z.A., Shikina N.V., Yashnik S.A., Aldashukurova G.B., Mironenko V.A., Kuznetsov V.V., Ismagilov I.Z. // Nanosci. Nanotechnol. 2013. V. 3. № 1. P. 1. https://doi.org/10.5923/j.nn.20130301.01
Aldashukurova G.B., Mironenko A.V., Mansurov Z.A., Shikina N.V., Yashnik S.A., Kuznetsov V.V., Ismagilov Z.R. // J. Energy Chem. 2013. V. 22. P. 811.
Piumetti M., Fino D., Russo N. // Appl. Catal. B: Environ. 2015. V. 163. P. 277.
Manukyan K.V., Cross A., Roslyakov S., Rouvimov S., Rogachev A.S., Wolf E.E., Mukasyan A.S. // J. Phys. Chem. 2013. V. 117. P. 24 417.
Najjar H., Lamonier J.-F., Mentre O., Giraudon J.-M., Batis H. // Appl. Catal. B: Environ. 2011. V.106. P.149.
Aruna S.T., Mukasyan A.S. // Curr. Opin. Solid State Mater. Sci. 2008. V. 12. P. 44.
Nersisyan H.H., Lee J.H., Ding J.R., Kim K.-S., Manukyan K.V., Mukasyan A.S. // Prog. Energy Combust. Sci. 2017. V. 63. P. 79.
Twigg M.V. // Catal. Today. 2011. V. 163. P. 33.
Dinka P., Mukasyan A.S. // J. Phys. Chem. B. 2005. V. 109. P. 21627.
Manukyan K.V., Chen Y.S., Rouvimov S., Li P., Li X., Dong S., Liu X., Furdyna J.K., Orlov A., Bernstein G.N., Porod W., Roslyakov S., Mukasyan A.S. // J. Phys. Chem. C. 2014. V. 118. P. 16 264.
Shikina N.V., Yashnik S.A., Gavrilova A.A., Ishchenko A.V., Dovlitova L.S., Khairulin S.R., Ismagilov Z.R. // Catal. Lett. 2019. V. 149. № 9. P. 2535.
Шикина Н.В., Яшник С.А., Гаврилова А.А., Довлитова Л.С., Хайрулин С.Р., Козлова Г.С., Исмагилов З.Р. // Кинетика и катализ. 2018. Т. 59. № 4. С. 517.
Bera P., Patil K.C., Jayaram V., Subbanna G.N., Hegde M.S. // J. Catal. 2000. V. 196. P. 293.
Priolkar K.R., Bera P., Sarode P.R., Hegde M.S., Emura S., Kumashiro R., Lalla N.P. // Chem. Mater. 2002. V. 14. P. 2120.
Bera P., Malwadkar S., Gayen A., Satyanarayana C.V.V., Rao B.S., Hegde M.S. // Catal. Lett. 2004. V. 96. P. 213.
Bera P., Gayen A., Hegde M.S., Lalla N.P., Spadaro L., Frusteri F., Arena F. // J. Phys. Chem. B. 2003. V. 107. P. 6122.
Chourashiya M., Gyergyek S., Andersen S.M. // Mater. Chem. Phys. 2020. V. 242. P. 122 444.
Morfin F., Nguyen T.-S., Rousset J.-L., Piccolo L. // Appl. Catal. B: Environ. 2016. V. 197. P. 2.
Zavyalova U., Girgsdies F., Korup O., Horn R., Schlogl R. // J. Phys. Chem. C. 2009. V. 113. P. 17 493.
Royer S., Duprez D. // ChemCatChem. 2011. V. 3. P. 24.
Yu W., Porosoff M.D., Chen J.G. // Chem. Rev. 2012. V. 112. P. 5780.
Carno J., Ferrandon M., Bjornbom E., Jaras S. // Appl. Catal. A: General. 1997. V. 155. P. 265.
Ferrandon M., Carno J., Jaras S., Bjornbom E. // Appl. Catal. A: General. 1999. V. 180. P. 141.
Ferrandon M., Carno J., Jaras S., Bjornbom E. // Appl. Catal. A: General. 1999. V. 180. P. 153.
Yashnik S.A., Kuznetsov V.V., Ismagilov Z.R., Ushakov V.V., Danchenko N.M., Denisov S.P. // Top. Catal. 2004. V. 30/31. P. 293.
Yashnik S.A., Ismagilov Z.R., Kuznetsov V.V., Ushakov V.V., Rogov V.A., Ovsyannikova I.A. // Catal. Today. 2006. V. 117. P. 525.
Yashnik S.A., Ismagilov Z.R., Porsin A.V., Denisov S.P., Danchenko N.M. // Top. Catal. 2007. V. 42–43. № 1–4. P. 465.
An K., Alayoglu S., Musselwhite N., Plamthottam S., Melaet G., Lindeman A.E., Somorjai G.A. // J. Am. Chem. Soc. 2013. V. 135. P. 16 689.
Yashnik S.A., Ismagilov Z.R., Denisov S.P., Danchenko N.M. // Appl. Catal. B: Environ. 2016. V. 185. P. 322.
Yashnik S.A., Ishchenko A.V., Dovlitova L.S., Ismagilov Z.R. // Top. Catal. 2017. V. 60. P. 52.
Givera A., Pavese M., Saracco G., Specchia V. // Catal. Today. 2003. V. 83. P. 199.
Малахов В.В., Васильева И.Г. // Успехи химии. 2008. Т. 77. С. 370.
Wang X., Qin M., Fang F., Jia B., Wu H., Qu X., Volinsky A.A. // J. Alloys Compd. 2017. V. 719. P. 288.
El-Shobaky G.A., El-Shobaky H.G., Badawy A.A.A., Fahmy Y.M. // Appl. Catal. A: General. 2011. V. 409–410. P. 234.
Lu H., Zhou Y., Huang H., Zhang B., Chen Y. // J. Rare Earths. 2011. V. 29. P. 855.
Яшник С.А., Гаврилова А.А., Суровцова Т.А., Шикина Н.В. // Вестник Томского государственного университета. Химия. 2018. № 10. С. 45.
Stobbe E.R., de Boer B.A., Geus J.W. // Catal. Today. 1999. V. 47. P. 161.
Weimin W., Yongnian Y., Jiayu Z. // Appl. Catal. A: General. 1995. V. 133. P. 81.
Kapteljn F., Smgoredjo L., Andreml A., Moljin J.A. // Appl. Catal. B: Environ. 1994. V. 3. P. 173.
Carvalho L.S., Pieck C.L., Rangel M.C., Figoli N.S., Grau J.M., Reyes P., Parera J.M. // Appl. Catal. A: General. 2004. V. 269. P. 91.
Lieske H., Lietz G., Spindler H., Volter J. // J. Catal. 1983. V. 81. P. 8.
Lietz G., Lieske H., Spindler H., Hamker W., Volter J. // J. Catal. 1983. V. 81. P. 17.
Santos A.C.S.F., Damyanova S., Teixeira G.N.R., Mattos L.V., Noronha F.B., Passos F.B., Bueno J.M.C. // Appl. Catal. A: General. 2005. V. 290. P. 123.
Бухтияров В.И., Слинько М.Г. // Успехи химии. 2001. Т. 70. № 2. С. 167.
Хаджиев С.Н. // Нефтехимия. 2011. Т. 51. № 1. С.3.
Попов Ю.В., Мохов В.М., Небыков Д.Н., Будко И.И. // Известия ВГТУ. 2014. Т. 12. № 7. С. 5.
Chen J., Arandiyan H., Gao X., Li J. // Catal. Surv. Asia. 2015. V. 19. P. 140.
Boudart M., Aldag A., Benson J.E., Dougharty N.A., Girvin C. // J. Catal. 1966. V. 6. P. 92.
Boudart M. // Adv. Catal. 1969. V. 20. P. 153.
Гололобов А.М., Бекк И.Э., Брагина Г.О., Зайковский В.И., Аюпов А.Б., Телегина Н.С., Бухтияров В.И., Стахеев А.Ю. // Кинетика и катализ. 2009. Т. 50. № 6. С. 864.
Стахеев А.Ю., Гололобов А.М., Бекк И.Э., Брагина Г.О., Зайковский В.И., Аюпов А.Б., Телегина Н.С., Бухтияров В.И. // Изв. Наук. Серия химич. 2010. № 9. С. 1667.
Beck I.E., Bukhtiyarov V.I., Pakharukov I.Yu., Zaikovsky V.I., Kriventsov V.V., Parmon V.N. // J. Catal. 2009. V. 268. № 1. P. 60.
Otto K., Andino J.M., Parks C.L. // J. Catal. 1991. V. 131. P. 243.
Briot P., Auroux A., Jones D., Primet M. // Appl. Catal. 1990. V. 59. P. 141.
Burch R., Loader R.K. // Appl. Catal. A: General. 1995. V. 122. P. 169.
Meephoka C., Chaisuk C., Samparnpiboon P., Praserthdam P. // Catal. Comm. 2008. V. 9. P. 546.
Tiernan M.J., Finlayson O.E. // Appl. Catal. B: Environ. 1998. V. 19. P. 23.
Яшник С.А., Шикина Н.В., Гаврилова А.А., Суровцова Т.А., Исмагилов З.Р. // Химия в интересах устойчивого развития. 2019. Т. 27. С. 211.
Tsyrulnikov P.G., Tsybulya S.V., Kryukova G.N., Boronin A.I., Koscheev S.V., Starostina T.G., Bubnov A.V., Kudrya E.N. // J. Molec. Catal. A. 2002. V. 179. P. 213.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ