

Кинетика и катализ, 2020, T. 61, № 5, стр. 595-653
Окисление олефинов до карбонильных соединений: современный взгляд на классическую реакцию
О. Н. Темкин *
МИРЭА – Российский технологический университет – (Институт тонких химических технологий
имени М.В. Ломоносова)
119571 Москва, просп. Вернадского, 86, Россия
* E-mail: olegtemkin@mail.ru
Поступила в редакцию 18.11.2019
После доработки 27.03.2020
Принята к публикации 27.03.2020
Аннотация
Проанализированы результаты 60-летних исследований механизмов реакции окисления олефинов до карбонильных соедиений (Вакер-окисление), катализируемой комплексами палладия в растворах. Рассмотрены также представления о механизмах близких реакций окислительного карбоксилирования (реакция Моисеева) и алкоксилирования олефинов. В обзоре обсуждаются закономерности процессов в водно-органических растворителях, проблемы регио- и стереоселективности реакций, роль со-катализаторов, окислителей и лигандов, включая и асимметрический катализ. Детально рассмотрено влияние на механизм реакции окисления олефинов перехода от анионных или нейтральных хлоридных комплексов палладия к катионным в водных и в водно-органических средах.
СОДЕРЖАНИЕ
1. Введение
2. История открытия реакции окисления олефинов с образованием карбонильных соединений
3. О механизмах реакции окисления олефинов в растворах хлоридных комплексов палладия(II)
3.1. Реакции обмена нуклеофилами в винильных и аллильных соединениях и реакции окисления олефинов в уксусной кислоте и в метаноле
3.2. Исследование стереохимии процессов гидрокси- и алкоксипалладирования олефинов
3.3. Окисление и изомеризация аллиловых спиртов
3.4. Исследования реакций изомеризации и окисления хиральных аллиловых спиртов
3.5. Механизмы превращений σ-палладийорганических интермедиатов
3.6. Теоретические исследования механизма реакции Вакер-окисления
4. Реакции окисления олефинов в бинарных водно-органических растворителях с участием различных окислителей и лигандов
4.1. Различные окислители и со-катализаторы
4.2. Пероксид водорода и ROOH – стехиометрические окислители
4.3. Кислород – стехиометрический окислитель без со-катализаторов
5. Региоселективность реакций окисления олефинов
6. Окисление олефинов в растворах катионных комплексов палладия
6.1. Реакционная способность катионных комплексов непереходных и переходных металлов в реакциях с олефинами
6.2. Катионные комплексы палладия в окислении олефинов
6.3. Кинетика и механизм реакций окисления этилена и циклогексена п-бензохиноном в растворах комплексов ${\text{Pd}}{{({\text{AN}})}_{x}}({{{\text{H}}}_{2}}{\text{O}})_{{4 - x}}^{{2 + }}$ в системе AN–Н2О
7. Некоторые дополнительные аспекты химии и технологии процессов окисления олефинов
7.1. Синтезы 1,2-продуктов окисления олефинов
7.2. Реакции Вакер-окисления в гетерофазных системах
7.3. Другие переходные металлы в катализе окисления олефинов до карбонильных соединений
8. Заключение
1. ВВЕДЕНИЕ
В 2019 г. исполнилось 60 лет первой научной публикации, посвященной реакции окисления олефинов до карбонильных соединений, катализируемой комплексами палладия(II), важной для промышленного органического синтеза, для препаративной органической химии и сыгравшей значительную роль в становлении современного гомогенного металлокомплексного катализа. Эта реакция давно вошла в учебники по катализу, координационной и металлоорганической химии, обсуждается в монографиях [1–7], обзорах [8–22] и энциклопедиях [23–26] (здесь приведена лишь небольшая часть публикаций).
В результате исследований этой удивительной реакции был установлен ее механизм и найдены различные модификации каталитической системы, в том числе в водно-органических средах с разными окислителями олефинов. Была показана также возможность селективного анти-Марковниковского окисления терминальных олефинов до альдегидов [15, 19, 20] и найдены условия и механизмы окислительного алкоксилирования олефинов до кеталей и ацеталей и окислительного карбоксилирования олефинов c образованием винильных и аллильных продуктов [1, 3, 10].
Целью настоящего обзора является анализ основных достижений в области каталитической химии палладия в реакциях окисления органических соединений с терминальными и интернальными кратными связями С=С при наличии любых заместителей (для краткости – олефинов). История и результаты 60-летних исследований механизмов этих реакций представляют, по мнению автора, особый интерес для специалистов в области металлокомплексного катализа, занимающихся изучением кинетики, теории механизмов сложных реакций и реакционной способности π- и σ-металлоорганических соединений.
2. ИСТОРИЯ ОТКРЫТИЯ РЕАКЦИИ ОКИСЛЕНИЯ ОЛЕФИНОВ ДО КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
Каталитическая реакция окисления олефинов до карбонильных соединений была открыта практически одновременно и независимо двумя группами исследователей в конце 50-х гг. в Германии (ФРГ) и в СССР. История открытия этой реакции описана участниками работ д-ром Р. Ирой (R. Jira) [9, 27] и акад. И.И. Моисеевым [1, 28].
В 1956 г. в исследовательском центре компании “Wacker-Chemie” (Consortium für Electrochemische Industrie, CEI) в Мюнхене молодому химику В. Хафнеру было предложено заняться новыми путями использования этилена. Поскольку окисление этилена кислородом до этиленоксида на гетерогенном серебряном катализаторе уже было известно и использовалось в промышленности, а другие олефины на серебре не окислялись до эпоксидов, Хафнер решил проверить возможность эпоксидирования олефинов кислородом на катализаторе Pd/C, но с добавкой водорода. Вероятнее всего, он рассчитывал получить пероксид водорода in situ на палладии для эпоксидирования этилена (и других олефинов) в мягких условиях, что было весьма революционным решением в то время. В первых же опытах Хафнер заметил образование ацетальдегида (по запаху, поскольку в то время в лаборатории CEI еще не было хроматографа [27]). Этим наблюдением заинтересовался руководитель CEI Йюрген Смидт (J. Smidt) и сразу же в 1956 г. организовал из сотрудников CEI группу исследователей для разработки гетерогенного процесса синтеза ацетальдегида. Эта группа должна была протестировать другие металлы платиновой группы и другие олефины на возможность синтеза карбонильных соединений. Оказалось, что добавки к палладиевому катализатору со-окислителей CuCl2 или FeCl3 ускоряют реакцию синтеза ацетальдегида, но катализатор быстро теряет активность. Тем не менее в 1957 г. был получен патент на газофазный процесс синтеза ацетальдегида (DP 1049845) по реакции
Кроме того, вторая группа исследователей во главе с Р. Ирой занялась изучением гомогенного жидкофазного процесса. Как вспоминает Р. Ира [27], участники проекта нашли в литературе информацию о том, что еще в 1894 г. Ф. Филлипсом было показано [29], что хлорид палладия в водных растворах легко восстанавливается этиленом (и СО) до металла с образованием ацетальдегида. Также было известно, что давно описанная соль Цейзе (π-комплекс K[PtCl3(C2H4)]) при кипячении в воде распадается с образованием ацетальдегида и металлической платины [30]. Казалось вполне естественным использовать CuCl2 для окисления Pdмет и воздух (или кислород) для окисления CuCl.
Поскольку семейство Wacker владело только 50% собственности фирмы “Wacker- Chemie”, a остальное принадлежало фирме “Farbwerke Hoechst”, эта фирма, получив предварительную информацию, также начала работать над процессом синтеза ацетальдегида и запатентовала газофазный процесс, опередив третий патент фирмы “Wacker-Chemie” (по жидкофазному процессу). В результате сложных коммерческих отношений первая подробная публикация по перспективному жидкофазному процессу синтеза ацетальдегида появилась только осенью 1959 г. [31] с описанием возможных технологий, отработанных на пилотных установках (осень 1957 г. [27]), и реакций окисления разнообразных олефинов. Установка производительностью 1 т в день вступила в строй в марте 1958 г. В январе 1960 г. были запущены установки производительностью 15 000 т в год фирмами “Wacker-Chemie” и “Farbwerke Hoechst”.
Советские исследователи решали задачу создания жидкофазной каталитической реакции окисления олефинов на основании тех же наблюдений Филлипса [29] и Андерсона [30], но с самого начала полагали, что суть реакции восстановления палладия этиленом заключается в особой реакционной способности олефиновых π-комплексов Pd(II) (комплексов Караша [32]) по отношению к нуклеофилам (молекулам воды, спирта, карбоновой кислоты и др.) [1, 8, 10]. Идея об участии π-комплексов ацетилена с соединениями Hg(II) в реакции гидратации по Кучерову была убедительно обоснована в кандидатской диссертации Моисеева И.И. (М.: МИТХТ им. М.В. Ломоносова, 1958 г.) и получила экспериментальное подтверждение позднее (1963 г.) в работе с участием автора настоящего обзора [33]. Первые “капли” ацетальдегида из этилена, по словам Моисеева [28], появились в 1957 г., а основные результаты были получены Моисеевым и М.Н. Варгафтиком в 1959–1960 гг. на Кафедре физической химии МИТХТ, возглавляемой чл.-корр. АН СССР Я.К. Сыркиным. По ряду субъективных и внешних причин [28] первая статья этих авторов появилась только в 1960 г. [34]. В работе использовали в качестве окислителей олефинов кислород (с со-катализатором CuCl2), пероксид водорода и п-бензохинон. Интересно, что обе группы исследователей тем или иным путем оказались вовлечены в химию π-комплексов металлов. Хафнер В. и Ира Р. – ученики Нобелевского лауреата Э.О. Фишера (см. [35–37]), синтезировавшего в 1955 г. вместе с Хафнером комплекс (η6-С6Н6)2Cr. Я.К. Сыркин – крупнейший специалист по теории строения координационных соединений и квантовой химии. Обе исследовательские группы одновременно получили первые η3-аллильные комплексы Pd(II) при взаимодействии аллилового спирта с раствором хлорида палладия и описали их в 1959 г. в статьях Хафнера со Смидтом [38] и Моисеева с Э.А. Федоровской [39].
Глубокое понимание советскими авторами механизма реакций координационных соединений и, в частности, π-комплексов металлов (см. [1]) привело к открытию новых реакций окислительного алкоксилирования олефинов до ацеталей и кеталей (Пат. СССР, SU 137511A1, 1960) и окислительного винилирования карбоновых кислот до сложных виниловых эфиров (например, винилацетата из этилена, так называемой реакции окислительной этерификации (oxidative esterification).
В работе [40] впервые упоминается принципиально важная гипотеза об участии σ-палладийорганического соединения с фрагментом [Pd]CH2CH2OH в механизме реакции образования ацетальдегида. Реакцию окислительной этерификации олефинов справедливо называют реакцией Моисеева (см., например, [26] и [7, рр. 5, 187]). Компании “Bayer” (совместно с “Hoechst” и “Knapsack”) и “National Distillers Products” (США) разработали два гетерогенных катализатора синтеза винилацетата по реакции Моисеева, которые нашли промышленное применение (см. [23 (2012. V. 38. р. 111)]).
Вследствие успехов фирмы “Wacker-Chemie” в открытии реакции и создании технологии процесса окисления олефинов [31, 41] синтез карбонильных соединений из олефинов получил в литературе название Вакер-окисление (Wacker-oxidation, Wacker-type oxidation). Возникшее понимание важной роли реакционноспособных π-комплексных и σ-металлоорганических соединений палладия [1] и других металлов в катализе сразу продвинуло развитие гомогенного металлокомплексного катализа [1–5, 7, 8, 42] и поддержало принципиальную парадигму о преимущественно химической природе катализа.
3. О МЕХАНИЗМАХ РЕАКЦИИ ОКИСЛЕНИЯ ОЛЕФИНОВ В РАСТВОРАХ ХЛОРИДНЫХ КОМПЛЕКСОВ ПАЛЛАДИЯ(II)
Реакция окисления этилена протекает в воде или водно-органических растворах комплексов Pd(II) и Cu(II) при атмосферном давлении и температурах 25–95°С по реакции (I)
(I)
${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{4}} + \,\,{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}{{{\text{O}}}_{2}}\xrightarrow{{{\text{PdC}}{{{\text{l}}}_{2}} - {\text{CuC}}{{{\text{l}}}_{2}}}}{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHO}}.$(II)
$\begin{gathered} {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{4}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} + {\text{PdC}}{{{\text{l}}}_{2}} \to \\ \to {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHO}} + {\text{P}}{{{\text{d}}}^{0}} + 2{\text{HCl}}, \\ \end{gathered} $(III)
${\text{P}}{{{\text{d}}}^{0}} + 2{\text{CuC}}{{{\text{l}}}_{2}} \to {\text{PdC}}{{{\text{l}}}_{2}} + 2{\text{CuCl}},$(IV)
$2{\text{CuCl}} + \,\,{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}{{{\text{O}}}_{2}} + 2{\text{HCl}} \to 2{\text{CuC}}{{{\text{l}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}.$Очевидно, что главные события, приводящие к очень интересному превращению этилена в ацетальдегид с участием H2O, происходят в реакции (II). Для упрощения системы кинетические исследования проводили с окислителем п-бензохиноном (Q) по реакции (V):
(V)
${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{4}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} + {\text{Q}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}{\text{O}} + {\text{Q}}{{{\text{H}}}_{2}},$(1)
$R = \frac{{k\left[ {PdCl_{4}^{{2 - }}} \right][{{C}_{2}}{{H}_{4}}]}}{{[{{H}^{ + }}]{{{[C{{l}^{ - }}]}}^{2}}}}.$Из уравнения (1) следует, что процесс протекает с лимитирующей стадией и что в стадиях до лимитирующей при взаимодействии ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}}--}},$ H2O и C2H4 выделяются ион Н+ и два иона Cl–. Для выяснения вопроса о том, из какой частицы выделяется Н+, была проведена реакция π-комплекса Караша [PdCl2(C2H4)]2 [32] с D2О (99.7%) [41]. Оказалось, что ацетальдегид из C2H4 не содержит атома D. Авторы сообщили, что ранее (неопубликованные результаты д-ра Х. Крекелера из “Farbwerke Hoechst”) были получены близкие результаты с D2O (85%). Позднее П. Генри [48] провел реакцию PdCl2 с меченым этиленом C2D4 в H2O и получил только CD3CDO. Таким образом, убедительно показано, что источником Н+ может служить только молекула H2O. В работе [45] было получено то же уравнение (1) в закрытом и проточном реакторах полного смешения. Кинетическое уравнение двухмаршрутной реакции синтеза ацетальдегида со вторым маршрутом (второй порядок по ${\text{[PdCl}}_{{\text{4}}}^{{{\text{2}}--}}])$ справедливо в условиях бóльших концентраций PdCl2 (>0.02 М) [46, 47]. В работе [44] впервые был измерен кинетический изотопный эффект (КИЭ) в реакции (V) для пары H2O/D2O (kH/kD = 4.05 ± 0.15), подтвердивший с большой вероятностью существование стадии диссоциации молекулы воды до лимитирующей стадии.
Схема 1 механизма, соответствующая уравнению (1) и подтвержденная независимым исследованием равновесий в этой системе [1, 45], включает стадии
Схема 1 .
Гипотеза об участии в механизме реакции окисления σ-палладийорганического интермедиата (σ-палладоэтанола) [40, 44] оказалась весьма плодотворной [1, 3, 10, 45, 49]. Из факта отсутствия обмена атомов водорода в олефинах со средой следовало, что распад σ-палладийорганического интермедиата происходит в результате 1,2-сдвига гидрида, т.е. внутримолекулярного переноса атома водорода, вероятно, при содействии атома палладия [45] или даже с элиминированием гидрида палладия с последующим присоединением к π-комплексу винилового спирта с образованием ${\text{C}}{{{\text{l}}}_{{\text{2}}}}{\text{PdCH(OH)CH}}_{{\text{3}}}^{ - }$ [50].
Механизмы стадий образования σ-палладоэтанола и его превращения в ацетальдегид (здесь стадии (IX) и (X)), включая природу лимитирующей стадии цис (син)-гидроксипалладирования (стадия (IX) в представленном механизме), до сих пор являются предметом дискуссий. Вероятность образования промежуточного σ-палладоэтанола былa подтвержденa данными о продуктах взаимодействия PdCl2 c ClHgCH2CH2OH в диэтиловом эфире, приводящего к ацетальдегиду в условиях, когда процесс дезоксимеркурирования меркуроэтанола не протекает [51].
При анализе кинетических изотопных эффектов в реакциях окисления этилена и дейтероэтилена (C2H4/C2D4) было установлено, что полученные значения КИЭ 1.07 [45] и 1.11 (для первого маршрута) [46] характеризуют вторичные КИЭ, не связанные с разрывом связей С–Н, и объясняются разной устойчивостью π-комплексов и, вероятно, несколько различающейся электрофильностью координированных палладием C2H4 и C2D4.
В отличие от кинетики окисления линейных терминальных олефинов [1] скорость окисления стерически затрудненного циклогексена в тех же условиях не зависит от концентрации Н3О+ [52]. Предположение о возможности реализации другого варианта механизма гидроксипалладирования (ГП) в результате транс(анти)-присоединения нуклеофила Н2О к π-комплексу обсуждалось в работах [45, 52] и в монографиях Моисеева [1] и Генри [3].
Уже в первых работах по окислению монозамещенных олефинов [53] было показано, что в зависимости от условий региоселективность окисления может быть различной: возможно не только образование кетонов (по правилу Марковникова), но и образование альдегидов (анти-Марковниковское ГП олефинов, анти-М-ГП). Так, при окислении стирола в стандартной Вакер-системе (PdCl2–CuCl2) обнаружено образование фенилуксусного альдегида, помимо ацетофенона, т.е. продукта анти-М-ГП. Кинетические исследования окисления замещенных стиролов [54, 55] п-бензохиноном в водно-тетрагидрофурановых растворах PdCl2 выявили интересные особенности процесса, касающиеся и механизма гидроксипалладирования. Так, было установлено, что с ростом концентраций ионов Cl– и H+ не только падает скорость окисления в соответствии с уравнением (1), но и заметно растет отношение количеств продуктов альдегид/кетон (nа/к).
Анализ кинетики окисления стирола п-бензохиноном [56, 57] при постоянной ионной силе раствора, равной 3, в условиях [PdCl2]Σ/[C8H8]Σ $ \gg $ 1 привел к предположению, что при транс-присоединении молекулы воды к π-комплексу преимущественной оказывается анти-М-ориентация [57]. Более детальный кинетический анализ показал [58] и существование маршрута, не зависящего от [Н3О+]. Увеличение [Cl–] или [Н3О+] (при [Cl–] = 0.2 М), т.е. вклада предполагаемого транс-присоединения H2O, повышало отношение nа/к до ~10. Предполагается, что оба продукта (кетон и альдегид) образуются в ходе внутрисферного цис-ГП и внешнесферного транс-ГП стирола соответственно [58], но с преимущественным образованием альдегида по маршруту, не зависящему от концентрации Н3О+.
цис-Внедрение олефина по связи Pd–OH требует наличия H2O и OH– в цис-положении к олефину в плоском квадратном комплексе Pd(II). Очевидно, что сильный транс-эффект π-лиганда несомненно приводит к замещению транс-хлорида водой и к диссоциации транс-аква-π-комплекса с образованием транс-гидроксокомплекса. В этом случае есть два возможных объяснения протекания цис-внедрения:
1) существование небольшой равновесной концентрации цис-аква- и цис-гидроксикомплексов,
2) цис-внедрение в результате изомеризации транс-комплекса при атаке молекулы воды по пятой координате квадратного транс-гидроксикомплекса и образования тригональной бипирамиды [1, 48].
Этапы развития исследований стадий механизма реакций окисления олефинов в водных и неводных средах описаны в обзоре Кейта и Генри 2009 г. [49], название которого “Механизм Вакер-реакции – повесть о двух гидроксипалладированиях” точно отражает состояние проблемы. Более детальный анализ исследований, выполненных до 2009 г. и в течение последнего десятилетия, представленный в настоящем обзоре, интересен драматичностью столкновений альтернативных гипотез о механизме реакций окисления, выдвинутых различными исследовательскими группами. Кроме того, этот анализ весьма поучителен с точки зрения выбора стратегии корректной экспериментальной дискриминации гипотез о механизме сложных каталитических реакций. Рассмотрим сначала проблемы, связанные с образованием σ-палладийорганических интермедиатов, т.е. с механизмом образования связи С–О в палладоэтаноле и в других палладийорганических интермедиатах процессов окисления олефинов в спиртах и карбоновых кислотах.
Поскольку установление механизма присоединения нуклеофилов к π-комплексам является общей проблемой для многочисленных каталитических процессов в металлокомплексном катализе и касается не только молекулы воды [49, 59], были предприняты многочисленные кинетические, стереохимические и теоретические исследования реакций, механизмы которых предположительно включают стадии присоединения нуклеофилов (Cl–, RCOO–, ROH и др.), продолжавшиеся вплоть до 2015 г. В ходе этих исследований была получена полезная информация о возможных механизмах гидроксипалладирования, алкоксипалладирования и карбоксипалладирования олефинов. Поскольку разные направления исследований проводились часто параллельно, а кинетические и стереохимические исследования иногда выполнялись в одной работе, довольно трудно сохранить хронологический порядок изложения результатов.
3.1. Реакции обмена нуклеофилами в винильных и аллильных соединениях и реакции окисления олефинов в уксусной кислоте и метаноле
Начнем с работ Генри и соавт. 1971–1976 гг., посвященных исследованию стадий присоединения нуклеофилов к π-комплексам, аналогичных стадиям гидрокси-, алкокси- и карбоксипалладирования олефинов, но в неокислительных реакциях обмена нуклеофилами в виниловых и аллиловых сложных эфирах, изомеризации аллиловых эфиров [60–68], обмена хлора в алкенилхлоридах [65, 67]. Рассмотрим также исследования механизмов окисления олефинов в растворах хлоридных и ацетатных комплексов палладия в уксусной кислоте [69–71] и в растворах хлоридных комплексов в метаноле [72].
Реакции обмена нуклеофилами (XIII) и (XIV), катализируемые комплексами палладия, – это неокислительные процессы, позволяющие проследить стереохимические особенности присоединения Nu к π-комплексам палладия(II).
(XIII)
${\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} = {\kern 1pt} {\text{CHX}} + {{{\text{Y}}}^{--}} \rightleftarrows {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} = {\kern 1pt} {\text{CHY}} + {{{\text{X}}}^{--}},$(XIV)
$\begin{gathered} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} = {\kern 1pt} {\text{CHC}}{{{\text{H}}}_{{\text{2}}}}{\text{X}} + {{{\text{Y}}}^{--}} \rightleftarrows \\ \rightleftarrows {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} = {\kern 1pt} {\text{CHC}}{{{\text{H}}}_{{\text{2}}}}{\text{Y}} + {{{\text{X}}}^{--}}, \\ \end{gathered} $Первый пример обмена виниловых эфиров с АсОН исследовали еще в 1962 г. [34].
Исследование равновесий в системе PdCl2–LiCl–AcOH в условиях существования димера Li2Pd2Cl6 [60] позволило подойти к исследованию кинетики обмена в реакциях CH2=CHOCOCD3 [61] и CH2=CHOCOC2H5 [62] с LiOAc. В условиях отсутствия ацетатного комплекса палладия получили кинетическое уравнение (2), где S – олефин:
(2)
$R = \frac{{[L{{i}_{2}}P{{d}_{2}}C{{l}_{6}}][S]}}{{[LiCl]}}\left( {k_{1}^{/} + k_{1}^{{//}}[LiOAc]} \right).$Изучение обменов в реакциях замещенных аллиловых эфиров позволило провести дискриминацию возможных для аллиловых эфиров механизмов замещения – SN2, π-аллильного и механизма со стадией ацетоксипалладирования [63, 64] с подтверждением наибольшей вероятности стадии ацетоксипалладирования. Механизм превращений π-аллильных комплексов палладия в аллильные производные рассматривался в работе [73], где былa показана необходимость присутствия окислителя для превращений такого рода. Кинетика обменов с аллилпропионатом [63] и кротилпропионатом [64] описывается уравнением, аналогичным уравнению (2), но обмен происходит с изомеризацией
(XV)
$\begin{gathered} {\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{СН}}{\kern 1pt} = {\kern 1pt} {\text{СНС}}{{{\text{Н}}}_{{\text{2}}}}{\text{ОСО}}{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{5}} + {\text{Ас}}{{{\text{О}}}^{--}} \rightleftarrows \\ \rightleftarrows {\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{СН}}\left( {{\text{ОАс}}} \right){\text{СН}}{\kern 1pt} = {\kern 1pt} {\text{С}}{{{\text{Н}}}_{2}} + {{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{5}}}}{\text{СО}}{{{\text{О}}}^{--}}. \\ \end{gathered} $Обмен Cl на Cl* (радиоактивный изотоп) в винилхлориде и алкенилхлоридах [65] не дал ясных результатов по стереохимии присоединения этого нуклеофила. Было получено кинетическое уравнение
(3)
$R = {{k}_{3}}[{\text{L}}{{{\text{i}}}_{{\text{2}}}}{\text{P}}{{{\text{d}}}_{{\text{2}}}}{\text{C}}{{{\text{l}}}_{{\text{6}}}}{\text{][S}}]$В случае аллилтрифторацетата и кротилтрифторацетата также получено кинетическое уравнение (3) [66] с изомеризацией кротилтрифторацетата в процессе обмена с хлорид-ионом. Предполагается цис-хлоропалладирование с участием связи [Pd]–Cl*. Исследование стереохимии обмена Cl* на АсО в алкенилхлоридах [65, 68] показало, что цис-1-хлорпропен превращается в 85% цис- и 15% транс-1-ацетоксипропены, а транс-1-хлорпропен дает преимущественно транс-1-ацетоксипропен. Кинетическое уравнение (4) может указывать, во-первых, на координацию АсО– палладием и цис-ацетоксипалладирование и, во-вторых, на транс-ацетоксипалладирование и на необходимость свободной координационной вакансии для цис-дехлоропалладирования. Второй вариант принимается на основании предыдущих стереохимических результатов [68].
(4)
$R = \frac{{[L{{i}_{2}}P{{d}_{2}}C{{l}_{6}}][S]}}{{{{{[LiCl]}}^{2}}}}\left( {k_{5}^{/} + k_{5}^{{//}}[LiOAc]} \right).$Сравнение результатов изучения обменов RCOO на АсО, Cl на АсО и Cl на Cl* в замещенных олефинах на основе кинетики и стереохимии указывает на стереоспецифическое транс-ацетоксипалладирование олефина [68], что было подтверждено и в работах по окислению дейтерозамещенного циклогексена (d4) в системе PdCl2–CuCl2 в AcOH в присутствии АсО– [69, 70]. Предложенная схема механизма объясняет образование всех изомеров циклогексенилацетата и хлорацетатциклогексана и включает транс-ацетоксипалладирование с последующим перемещением Pd(II) по кольцу в результате цис-[Pd]–Н-элиминирования и присоединения [Pd]–Н к образующейся кратной связи. Окисление интермедиатов R[Pd]C хлоридом Cu(II) приводит к образованию изомеров хлорацетатциклогексана и диацетатциклогексана [70].
Подтверждение стадии транс-ацетоксипалладирования олефинов было получено и при детальном изучении кинетики и механизма реакции Моисеева (XVI) [33] в системе Pd3(OAc)6–AcOH–NaOAc [71].
(XVI)
${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{4}} + {\text{AcOH}} + {\text{Q}} = {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} = {\kern 1pt} {\text{CHOAc}} + {\text{Q}}{{{\text{H}}}_{2}}.$В результате исследования равновесий образования ацетатных комплексов Pd(II) в системе Pd3(OAc)6–AcOH–NaOAc [80] установлено, что при [NaOAc] = 0.2 М в растворе присутствует, в основном, димерный ацетат Na2Pd2(OAc)6. Константа равновесия K1 реакции (XVII) при 25°С равна 7.77 × 104 М–5, а K2 реакции (XVIII) равна 5.8 × 10–3 М–4.
(XVII)
$2{\text{P}}{{{\text{d}}}_{{\text{3}}}}{{\left( {{\text{OAc}}} \right)}_{6}} + 6{\text{NaOAc}} \rightleftarrows 3{\text{N}}{{{\text{a}}}_{{\text{2}}}}{\text{P}}{{{\text{d}}}_{{\text{2}}}}{{\left( {{\text{OAc}}} \right)}_{6}},$(XVIII)
${\text{P}}{{{\text{d}}}_{{\text{3}}}}{{\left( {{\text{OAc}}} \right)}_{6}} + 6{\text{NaOAc}} \rightleftarrows 3{\text{N}}{{{\text{a}}}_{{\text{2}}}}{\text{Pd}}{{\left( {{\text{OAc}}} \right)}_{4}}.$(5)
$R = \left( {{{k}_{1}}\left[ {{\text{P}}{{{\text{d}}}_{{\text{3}}}}{{{\left( {{\text{OAc}}} \right)}}_{6}}} \right] + {{k}_{2}}\left[ {{\text{N}}{{{\text{a}}}_{{\text{2}}}}{\text{P}}{{{\text{d}}}_{{\text{2}}}}{{{\left( {{\text{OAc}}} \right)}}_{{\text{6}}}}} \right]} \right)\left[ {\text{S}} \right].$Изучение кинетики окисления этилена и пропилена п-бензохиноном [71] с учетом описанной выше информации при ${{Р}_{{{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{4}}}}}}}$ = 1 атм = const привело к кинетическим уравнениям (6) и (7):
(6)
$R = {{k}_{{набл}}}[{{N{{a}_{2}}P{{d}_{2}}{{{(ОАс)}}_{6}}][{{C}_{2}}{{H}_{4}}]} \mathord{\left/ {\vphantom {{N{{a}_{2}}P{{d}_{2}}{{{(ОАс)}}_{6}}][{{C}_{2}}{{H}_{4}}]} {[NaOAc]}}} \right. \kern-0em} {[NaOAc]}},$(7)
$R = \frac{{{{k}_{{набл}}}{{{[N{{a}_{2}}P{{d}_{2}}{{{(OAc)}}_{6}}]}}_{\Sigma }}[{{C}_{2}}{{H}_{4}}]}}{{K[{{C}_{2}}{{H}_{4}}] + [NaOAc]}}.$Схема 2 .
Из кинетических результатов следует необходимость освобождения координационного места в стадии (XXI) для β-элиминирования [Pd]–Н в лимитирующей стадии (XXII). При допущении заметного вклада в материальный баланс по [Pd]Σ комплекса π1 ([Pd2]Σ = π1 + [Pd2]) получим уравнения (8) и (9):
(8)
$R = {{k}_{{22}}}[{{\sigma }_{2}}] = {{k}_{{22}}}{{K}_{{19}}}{{K}_{{20}}}{{K}_{{21}}}\frac{{[P{{d}_{2}}][{{C}_{2}}{{H}_{4}}]}}{{[NaOAc]}},$(9)
$R = \frac{{{{k}_{{22}}}{{K}_{{19}}}{{K}_{{20}}}{{K}_{{21}}}{{{[P{{d}_{2}}]}}_{\sum }}[{{C}_{2}}{{H}_{4}}]}}{{{{K}_{{19}}}[{{C}_{2}}{{H}_{4}}] + [NaOAc]}},$В работе [71] объясняется и влияние [NaOAc] на соотношение изомерных σ-палладоорганических интермедиатов, образующихся в стадии (XX) и распадающихся в стадиях (XXI) и (XXII). Так, предполагается, что при малых значениях [NaOAc] соотношение продуктов (М- и анти-М-) определяется кинетическим контролем, т.е. соотношением констант скорости стадий (XX) и (XXI), а при больших [NaOAc] процесс контролируется термодинамикой, т.е. определяется константами равновесия K20 и K21 для двух региоизомеров.
Кинетику окисления этилена п-бензохиноном до диметилацеталя ацетальдегида изучали в метаноле [72] как в растворителе, более близком по свойствам к воде, чем к АсОН, в каталитической системе PdCl2–LiCl при 25°С. Константы равновесия образования комплексов PdCl2(C2H4)(CH3OH), PdCl3(CН3OH)– и PdCl3(C2H4)– из ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}$ определяли спектрофотометрически в области длин волн 200–400 нм. Влияние концентрации Н+ на скорость окисления изучали в растворах кислот CH3SO3H, AcOH и ClCH2COOH. В интервалах концентраций [Н+] 10–2–10–5 М, [Cl–] = 0.05–0.5 M и [Pd]Σ = 0.006–0.1 M скорость реакции, выраженная через равновесные концентрации реагентов, описывается уравнением, аналогичным уравнению (1). Торможение ионом Н+ в этих условиях авторы [72] связывают с квазиравновесной стадией депротонирования комплекса PdCl2(C2H4)(CH3OH) до PdCl2(C2H4)(OCH3)–. Анализируя возможные стадии транс-присоединения метанола к этому π‑комплексу и цис-внедрение этилена по связи [Pd]–ОСН3 под действием МеОН, авторы, по ряду кинетических соображений и, в частности, вследствие отсутствия маршрута с участием π‑комплекса PdCl3(C2H4)– в стадии транс-присоединения метанола считают процесс цис-метоксипалладирования более вероятным. КИЭ в системе CH3OH/CH3OD равен 1.34, что существенно меньше КИЭ в водных системах, но также подтверждает наличие лимитирующей стадии превращения π → σ-интермедиатов. Появление максимума на зависимости R = f(pH) при рН 6 (падение скорости до рН 10), наблюдавшееся в работе [50], не имеет корректного объяснения.
Возможность образования продукта транс-метоксипалладирования этилена из π-комплекса Pd(II) была промоделирована с катионным комплексом палладия [82]
(XXIV)
$\begin{gathered} ({{\eta }^{{\text{5}}}}{\text{ - }}{{{\text{C}}}_{{\text{5}}}}{{{\text{H}}}_{{\text{5}}}}){\text{Pd}}({\text{PP}}{{{\text{h}}}_{{\text{3}}}}){{({{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}})}^{ + }}{\text{ClO}}_{{\text{4}}}^{ - } + \\ + \,\,{\text{Me}}{{{\text{O}}}^{--}} \to \\ \to \left( {{{\eta }^{{\text{5}}}}{\text{ - }}{{{\text{C}}}_{{\text{5}}}}{{{\text{H}}}_{{\text{5}}}}} \right){\text{Pd}}\left( {{\text{PP}}{{{\text{h}}}_{{\text{3}}}}} \right)\left( {{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OMe}}} \right) + \\ + \,\,{\text{ClO}}_{{\text{4}}}^{ - }. \\ \end{gathered} $Аналогичные соединения получены и с другими спиртами. Медленно распадающийся в бензоле и хлороформе при 25°С трео-конформер, полученный из цис-CHD=CHD, однозначно подтверждает транс-присоединение MeO– к π-комплексу. Нагревание σ-комплекса (η5-C5H5)Pd(PPh3)(CHDCHDOR) до 80°С приводит к образованию алкениловых эфиров в результате β-элиминирования [Pd]–Н(D) и циклопентадиена.
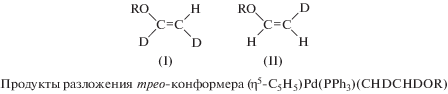
Соотношение изомеров I/II равно ~2, что близко к соотношению изомерных альдегидов, полученных из CHD=CHD в Вакер-процессе [48].
Таким образом, в описанной серии экспериментов установлены возможности транс-ацетоксипалладирования олефина в хлоридных и ацетатных π-комплексах и цис-алкоксипалладирования олефина в хлоридных π-комплексах. Убедительно показана возможность транс-алкоксипалладирования в случае π-комплексов палладия, не имеющих координационных вакансий. Полученная информация безусловно полезна и подтверждает принципиальную возможность реализации двух вариантов присоединения нуклеофилов к координированному в π-комплексе олефину. Авторы этих исследований, к сожалению, не изучили реакции обмена алкенилхлоридов и алкенилкарбоксилатов с Н2О (реакции гидролиза) в условиях катализа комплексами палладия, включающие стадии ГП, тем более что молекула Н2О отличается от других нуклеофилов не только величиной нуклеофильности, но и высокой степенью ассоциированности в жидкой фазе.
3.2. Исследование стереохимии процессов гидрокси- и алкоксипалладирования олефинов
Поскольку проследить стереохимию присоединения фрагмента ОН к π-комплексу в процессе Вакер-окисления олефинов невозможно, две группы исследователей начали изучать стереохимию присоединения ОН при образовании продуктов окислительного распада σ-комплексов [Pd]CH2CH2OH при взаимодействии с окислителями (CuCl2) или с другими нуклеофилами. При этом обе группы исходили из гипотезы (недоказанной), что карбонильные продукты и продукты 1,2-окисления олефинов получаются из одного и того же интермедиата. Аналогичные исследования были описаны выше на примере хлорацетоксилирования циклогексена-d4 [69, 70].
В серии работ Дж. Стилле и сотр. [83–87] была исследована стереохимия реакций окислительного метоксикарбонилирования олефинов и синтеза лактонов. В реакции цис-бутена-2 с СО в метанольном растворе PdCl2 (0.05 М) и CuCl2 (1.33 М) [84] получены трео-изомер СН3ОСН(СН3)–СН(СН3)СО2СН3 и d,l-мезо-дикарбоксилаты. Полученные продукты свидетельствуют о транс-метоксипалладировании и о цис-метоксикарбонилпалладировании бутена (с последующим карбонилированием и образованием сложных мезо-диэфиров):
(XXV)
${\mathbf{C}}{{{\mathbf{H}}}_{{\mathbf{3}}}}{\mathbf{O}}{\text{CH}}\left( {{\text{C}}{{{\text{H}}}_{{\text{3}}}}} \right){\text{CH}}\left( {{\text{C}}{{{\text{H}}}_{{\text{3}}}}} \right)[{\text{Pd}}]\xrightarrow[{ - \left[ {{\text{Pd}}} \right] - {\text{H}}}]{{{\text{CO,C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}}}}трео{\text{ - }}{\mathbf{C}}{{{\mathbf{H}}}_{{\mathbf{3}}}}{\mathbf{O}}{\text{CH}}\left( {{\text{C}}{{{\text{H}}}_{{\text{3}}}}} \right){\text{CH}}\left( {{\text{C}}{{{\text{H}}}_{{\text{3}}}}} \right){\text{C}}{{{\text{O}}}_{2}}{\text{C}}{{{\text{H}}}_{{\text{3}}}},$(XXVI)
${\mathbf{C}}{{{\mathbf{H}}}_{{\mathbf{3}}}}{{{\mathbf{O}}}_{{\mathbf{2}}}}{\mathbf{С}}{\text{CH}}\left( {{\text{C}}{{{\text{H}}}_{{\text{3}}}}} \right){\text{CH}}\left( {{\text{C}}{{{\text{H}}}_{{\text{3}}}}} \right)[{\text{Pd}}]\xrightarrow[{ - \left[ {{\text{Pd}}} \right] - {\text{H}}}]{{{\text{CO,C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}}}}мезо{\text{ - }}{\mathbf{C}}{{{\mathbf{H}}}_{{\mathbf{3}}}}{{{\mathbf{O}}}_{{\mathbf{2}}}}{\mathbf{С}}{\text{CH}}\left( {{\text{C}}{{{\text{H}}}_{{\text{3}}}}} \right){\text{CH}}\left( {{\text{C}}{{{\text{H}}}_{{\text{3}}}}} \right){\text{C}}{{{\text{O}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{3}}.$транс-Бутен-2 образует эритро-изомер метоксикарбоксилата и d,l-дикарбоксилат (при добавлении NaOAc для подавления изомеризации бутенов). Специальными экспериментами [85] с ртутьорганическими соединениями в присутствии NaOAc было установлено, что, например, в реакции (XXVII) сохраняется транс-структура продукта.
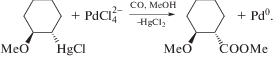
Таким образом, в процессе (XXVIII) (в присутствии NaOAc)
сохраняется конфигурация атома углерода как в стадии замещения HgX хлоридом Pd(II), так и при внедрении СО по связи C–Pd с последующим алкоголизом интермедиата, содержащего фрагмент C–C(O)Pd. Полученная конфигурация продукта стадии (XXVIII) возможна и при образовании интермедиата с фрагментом C–Pd(COOCH3) с последующим восстановительным элиминированием Pd(0).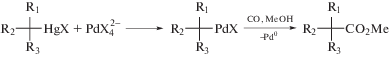
Подтверждение механизма транс-гидроксипалладирования олефинов молекулой воды было получено при карбонилировании циклооктадиена-1,5 (COD) до лактона [86] (XXIX):
Промежуточный продукт гидроксипалладирования COD был также получен с выходом 96% из π‑комплекса COD с PdCl2 при действии Н2О (в присутствии Na2СO3). Этот транс-изомер легко превращается при действии СО и Н2О в тот же лактон.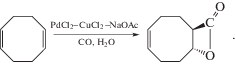
Еще более подробно изучен синтез β-пропиолактона из димерного π-комплекса Караша с этиленом или непосредственно из этилена в системе PdCl2–CuCl2–CH3CN–H2O в присутствии NaOAc при –20–(–25)°С и при отсутствии даже следов ацетальдегида (!) [87] (XXX). Стереохимию β-пропиолактона изучали по продукту окислительного карбонилирования π-комплекса цис-CHD=CHD в той же системе. В результате был получен транс-2,3-дидейтеропропиолактон, указывающий на транс-присоединение Н2О к π-комплексу в синтезе лактона.
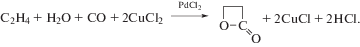
По мнению авторов [87], такая же стереохимия ГП реализуется и в Вакер-окислении этилена, механизм которого включает обратимые стадии присоединения Н2О к π-комплексу и депротонирования σ-металлоорганического интермедиата. В качестве лимитирующей стадии рассматривается стадия диссоциации интермедиата по связи [Pd]–Cl для освобождения координационного места, необходимого для β-элиминирования [Pd]–Н с образованием π-комплекса Н[Pd](СН2=СНОН).
Главным недостатком подходов, развитых в работах [84, 86, 87], является использование реакционных систем, далеких от условий Вакер-окисления, в которых присутствуют такие лиганды, как СО, СН3СN, OAc– и COD, при больших концентрациях Cl– и CuCl2. В самом деле, какое отношение к Вакер-процессу в системе CuCl2 (0.15–0.8 M)–LiCl (0.1–0.5 M)–H2O (>50 M)–HClO4 (<1.0 M) имеют, например, система CuCl2 (1.33 M)–NaOAc (1.3 M)–H2O (0.67 M)–СН3СN (19.0 М) [81] и комплексы Pd(II), образующиеся в ней?
Практически одновременно с рассмотренными выше работами стереохимию присоединения различных нуклеофилов к π-комплексам изучали Окермарк и Бэкквал и также на примере образования 1,2-продуктов окисления олефинов [88–95] в рамках той же парадигмы соответствия стереохимии продуктов 1,2-окисления стереохимии интермедиатов Вакер-процесса. В качестве нуклеофилов исследовали Н2О, ОАс– и амины. В работе Бэкквала [89] приведено следующее утверждение: “It is now reasonably clear that oxypalladation occurs with trans-stereochemistry”. При этом в его другой работе [91] было показано, что присоединение OAc– к π-аллильному комплексу Pd(II) в АсОН в присутствии п-бензохинона зависит от состава каталитической системы и протекает как транс-присоединение в присутствии Cl– и как цис-присоединение в отсутствие хлорид-иона. Рассмотрим кратко результаты этой научной группы, касающиеся стадии оксипалладирования в ходе превращений π-комплексов.
Ранее было показано, что в условиях промышленного Вакер-процесса образуется менее 1% этиленхлоргидрина (β-хлорэтилового спирта, ClCH2CH2OH) [41]. При увеличении концентраций CuCl2 до 3–4 М и LiCl до 2–4 М этиленхлоргидрин (ЭХГ) становится основным продуктом процесса [96]. Изучение конформации ЭХГ в реакциях цис- и транс-CHD=CHD в растворе PdCl2 (0.03 M)–CuCl2 (2.7 M)–LiCl (3.3 M) при 20°С показало [90, 93], что из транс-CHD=CHD получается трео-изомер ЭХГ, указывающий на транс-присоединение Н2О к π-комплексу. цис-CHD=CHD образует, соответственно, эритро-конформер. Для подтверждения этих выводов был изучен продукт реакции (XXXI).
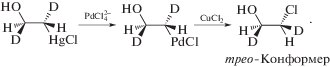
Поскольку обмен Hg(II) на Pd(II) происходит с сохранением конфигурации атома углерода [85], образование трео-конформера ЭХГ свидетельствует об инверсии конфигурации атома С при редокс-распаде палладоэтанола и нуклеофильном замещении Pd(II) хлорид-ионом с участием окислителя CuCl2.
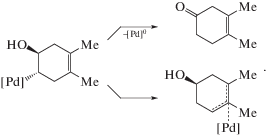
Рассмотрим еще работу 1987 г. [95], посвященную проблеме гидроксипалладирования. Как было установлено, 1,2-диметилциклогексадиен-1,4 превращается в растворе комплекса PdCl2(CH3CN)2 в ацетоне в присутствии KHCO3 и CuCl2 (при концентрации воды 4.6 М) в два продукта – транс-π-аллильный комплекс (77%) и 3,4-диметилциклогексен-1-он (18%). Предполагалось, что оба продукта образуются через один и тот же интермедиат – продукт транс-гидроксипалладирования диметилциклогексадиена – по реакциям (XXXII) и (XXXIII):
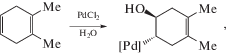
Общие проблемы механизмов присоединения различных “жестких” и “мягких” Nu к π-комплексам Pd(II) на этом этапе исследований рассматривались в экспериментальных работах Бэкквала [90, 93] и в квантово-химических исследованиях [97–99]. Теоретический анализ, выполненный в работе [99], показал, что прочность связей [Pd]–OH и [Pd]–F в π-комплексах в два раза выше прочности связей [Pd]–H и [Pd]–CH3 (с мягкими нуклеофилами).
Разность энергий низшей незаполненной и высшей заполненной молекулярных орбиталей (LUMO и HOMO соответственно) (например, этилена в π-комплексах) для связей [Pd]–Nu составляет 3.6 и 2.9 эВ для Н и СН3, 10.8 и 9.5 эВ для ОН и F (в соответствии с более ранним анализом [98]). На основании этих результатов был сделан вывод, что только нуклеофильные группы Н и СН3 способны к цис-внедрению в ходе орбитально-контролируемого процесса, тогда как жесткие нуклеофилы ОН и F мало реакционноспособны в таком процессе. Эти выводы, по мнению авторов [98], хорошо согласуются с изложенными выше наблюдениями о транс-присоединении Н2О, СН3ОН и АсО– к олефину в π-комплексе, а также с результатами исследований по присоединению Cl– и аминов (см. ссылки в работе [99]) к π-комплексам, полученными, как отмечалось выше, в условиях, сильно отличающихся от реальных условий процессов Вакер-окисления в воде, спиртах и уксусной кислоте. Кинетика окисления циклогексена [52] и стирола [58] п-бензохиноном показала, однако, что кроме маршрута, включающего диссоциацию молекулы воды в квазиравновесной стадии, существует и второй маршрут гидроксипалладирования – без стадии диссоциации молекулы воды, которая предшествует лимитирующей стадии.
Одновременная реализация обоих маршрутов была обнаружена и в случае окисления этилена [100]. Оказалось, что в растворах с относительно большой суммарной концентрацией Cl– (и со-окислителя FeCl3) при температурах 70–90°С маршрут с участием гидроксокомплексов Pd подавляется и скорость окисления этилена перестает зависеть от Н3О. В системе, использованной для изучения кинетики реакций окисления СО, гидратации и гидрохлорирования ацетилена, содержащей 0.02 М PdCl2, [HCl] + [LiCl] = 5 М, 1 М FeCl3 и 2 М HgCl2, зависимость скорости образования ацетальдегида (АА) RAA из этилена от [HCl] (рис. 1) в стационарных условиях в проточном по газу реакторе полного смешения описывается уравнением (10) [100]
(10)
${{R}_{{AA}}} = \frac{{{{k}_{3}}{{P}_{{{{C}_{2}}{{H}_{4}}}}}}}{{[HCl]}} + {{k}_{4}}{{P}_{{{{C}_{2}}{{H}_{4}}}}}.$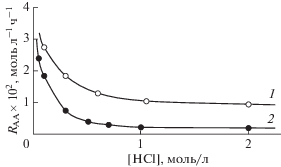
Оценка влияния температуры на отношение констант k3/k4 показывает, что энергия активации процесса с вероятным транс-присоединением молекулы воды к π-комплексу палладия (k4) на ~16 ккал/моль больше энергии активации процесса с цис-внедрением этилена (k3) по связи Pd–OH [100] (если диссоциация воды происходит в аква-π-комплексе палладия, стадия (VIII)). К сожалению, в этой серии экспериментов не были измерены КИЭ для системы H2O/D2O.
В связи с обсуждением стереохимии этиленхлоргидрина (ЭХГ) [90, 93] – продукта окислительного хлоргидроксилирования этилена – и роли хлорид-иона [41] при больших концентрациях CuCl2 следует отметить, что концентрация CuCl2 также может влиять на соотношение маршрутов, особенно если учитывать возможность образования гетерометаллических комплексов PdCl2(CuCl2)n.
В результате изучения кинетики и механизма окисления этилена в метанольных и этанольных растворах в системе PdX2–CuX2–LiX–HX–LiClO4 (X = Cl или Br) до диметилацеталя ацетальдегида (ДМА), диэтилацеталя ацетальдегида (ДЭА) и хлор(бром)этилметиловых эфиров (ХЭМЭ, БЭМЭ) [7 (Ch. 4), 102–104] был сделан вывод о существовании параллельных маршрутов образования продуктов 1,1- и 1,2-окисления с участием комплексов PdX2 и PdX2(CuX2)2 соответственно (см. подробности в [7 (Ch. 4, пример 4.1)]).
На основе изучения электронных спектров в указанных выше системах были определены константы равновесий реакций образования хлоридных комплексов меди(II) и установлено образование гетерометаллических комплексов [7 (Ch. 4)]. Интересно, что скорости образования всех трех продуктов (ДМА, ХЭМЭ и БЭМЭ) в условиях постоянной высокой ионной среды описываются кинетическим уравнением, предполагающим торможение реакции кислотой НХ (11):
(11)
${{R}_{i}} = \frac{{{{k}_{i}}\left[ {PdCl_{4}^{{2 - }}} \right]{{P}_{{{{C}_{2}}{{H}_{4}}}}}}}{{[HX]}}.$Из приведенного анализа экспериментальных работ по стереохимии продуктов 1,2-окисления олефинов и по кинетике реакций окисления этилена в системах PdCl2–FeCl3–LiCl–H2O и PdCl2–CuCl2–LiCl–ROH следуют два вывода.
1). Установлено существование двух маршрутов в механизме окисления этилена до ацетальдегида, различающихся порядками по [H+], и в механизме алкоксилирования этилена, различающихся порядками по [CuCl2], и, соответственно, составами каталитически активных комплексов. Только один маршрут обнаружен в механизме окисления этилена п-бензохиноном в метаноле (разд. 3.2).
2). Стереохимические исследования, проведенные вне связи с кинетикой двухмаршрутной реакции, не могут служить тестом на стереохимию стадии гидроксипалладирования в условиях Вакер-процесса.
3.3. Окисление и изомеризация аллиловых спиртов
Наблюдения, сделанные в работе [100], согласуются с результатами серии статей Генри и сотр. за период 1981–1988 гг., посвященных окислению аллиловых спиртов (АС) [105–108] и изомеризации d2-дейтерированного аллилового спирта [109]. Реакция изомеризации АС оказалась очень полезной для выяснения деталей механизма Вакер-окисления олефинов, поскольку катализируется комплексами палладия с вероятным образованием в качестве ключевого, близкого к механизму Вакер-окисления интермедиата. Уже в кратком сообщении [105] было показано, что реакция изомеризации (XXXIV)
(XXXIV)
${\text{C}}{{{\text{D}}}_{2}}{\kern 1pt} = {\kern 1pt} {\text{CHC}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}} \rightleftarrows {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} = {\kern 1pt} {\text{CHC}}{{{\text{D}}}_{{\text{2}}}}{\text{OH}}$(12)
${{R}_{{изо}}} = {{k}_{{изо}}}\frac{{\left[ {PdCl_{4}^{{2 - }}} \right][AC]}}{{[C{{l}^{ - }}]}}.$В работе [109] более детально изучали процессы изомеризации АС и обмена АС с ${\text{Н}}_{{\text{2}}}^{{{\text{18}}}}{\text{О}}{\text{.}}$ Константа скорости первого порядка по АС была определена как k1, изо $ \cong $ 1.5 × 10–3 c–1 (см. уравнение (12)) в интервалах [H+] = 0.2–1.0 М и [Cl–] = = 0.2–1.0 М. При этом константа скорости обмена АС с ${\text{Н}}_{{\text{2}}}^{{{\text{18}}}}{\text{О,}}$ проходящего также через стадию гидроксипалладирования, рассчитанная аналогично, имеет тот же порядок величины – (1.5–1.7) × 10–3 c–1. Исследовали также [106, 107] процесс стехиометрического окисления АС тетрахлорпалладат-анионом при [H+] = 0.2 М и [Cl–] = 0.2 М в условиях 98–99%-ного превращения аллиловых спиртов в продукты окисления с образованием следов π-аллильных комплексов палладия(II). Основными продуктами окисления стали гидроксипропаналь (40%), акролеин (30%) и ацетол (12–15%). Такие же дейтерозамещенные продукты были получены и из d2-AC, причем КИЭ = 1.9 ([HOCD2CH2CHO]/[HOCH2CHDCDO]). Для всех продуктов окисления АС был проведен анализ возможных путей их образования.
Поскольку скорость изомеризации, описываемая уравнением (12), является скоростью гидроксипалладирования, то при больших концентрациях Cl– скорость ГП должна быть больше скорости окисления, которое в этих условиях практически не протекает, вероятно, из-за отсутствия вакантных мест в координационной сфере атома палладия, необходимых для β-элиминирования [Pd]–H. При низких значениях [H+] и [Cl–] ситуация другая – происходит только окисление АС.
Итоги этих интересных исследований были подведены в работе [108].
1). Окисление АС п-бензохиноном привело к кинетическому уравнению вида (1) при I = 2, [Cl–] = 0.1–1.0 М, [H+] = 0.1–1.0 М и [Pd]Σ = = 0.002–0.05 M.
2). В этих условиях процесс ГП необратим, поскольку в ходе реакции окисления реакция изомеризации АС не идет.
3). На схеме 3 на примере образования гидроксипропаналя представлен общий механизм двух процессов. Видно, что при k–1 $ \gg $ k2 гидроксипалладирование обратимо и при малой скорости окисления получается равновесная смесь изомерных АС (до появления заметных количеств продуктов окисления). При k–1 $ \ll $ k2 гидроксипалладирование станет необратимым, и будет протекать только процесс окисления до гидроксипропаналя. Так, в условиях изомеризации за период полупревращения АС (t1/2) окисляется <3% АС.
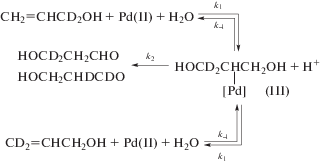
Схема 3 .
Из описанных выше результатов следует, что σ‑палладоорганический интермедиат (III) на схеме 3 при заполненной хлорид-ионом координационной сфере палладия в Cl3PdR2– не распадается окислительно предположительно из-за отсутствия вакантных мест для протекания β-элиминирования [Pd]–H (см. стабильный комплекс (η5-C5H5)Pd(PPh3)(CH2CH2OМe) в работе [82]).
В работе [110] при исследовании распада н-бутильных комплексов Pt(II) было показано, что комплекс (n-Bu)2Pt(PPh3)2 распадается с образованием комплекса Pt(0), бутена-1 и н-бутана только после диссоциации комплекса с выделением одного фосфинового лиганда и образованием комплекса (n-Bu)2Pt(PPh3)..
Процесс цис-гидроксипалладирования с участием связи [Pd]–ОН с большой вероятностью ведет к освобождению одного координационного места (если внедрение этилена по связи [Pd]–ОН в π-комплексе происходит без участия других лигандов, например молекулы воды).
Следует упомянуть, что такой продукт окисления АС, как ацетол, образуется также в результате гидроксипалладирования АС формально по правилу Марковникова, причем АС и d2-AC превращаются в ацетол, естественно, с КИЭ, равным 1.
(XXXV)
$\begin{gathered} {\text{Cl}}\left[ {{\text{Pd}}} \right]{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{CH}}\left( {{\text{OH}}} \right){\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}} \to \\ \to {\text{Cl}}\left[ {{\text{Pd}}} \right]{\text{H}} + {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{C}}\left( {\text{O}} \right){\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}}. \\ \end{gathered} $В работе Т. Хосокавы и сотр. [111] было убедительно показано, что процесс ГП может быть обратимым и в отсутствие кислот. В случае неактивированных олефинов процесс β-элиминирования [Pd]–H протекает быстрее β-элиминирования [Pd]–ОH, однако при наличии сильной электроноакцепторной группы у двойной связи происходит исключительно элиминирование [Pd]–ОH. Так, в результате алкоксилирования метил(α-гидроксиметил)акрилата в растворе PdCl2 в 1,2-диметоксиэтане в токе аргона при 50°С получается только метил(α-алкоксиметил)акрилат при R1 = H и R2 = Me :
(XXXVI)
$\begin{gathered} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} = {\kern 1pt} {\text{C}}\left( {{\text{C}}{{{\text{O}}}_{{\text{2}}}}{\text{Me}}} \right){\text{CH}}\left( {{\text{OH}}} \right){{{\text{R}}}^{1}} + \\ + \,\,{{{\text{R}}}^{{\text{2}}}}{\text{OН }} \to {\text{ }} \\ \to {{{\text{R}}}^{{\text{1}}}}{\text{CH}}{\kern 1pt} = {\kern 1pt} {\text{C}}\left( {{\text{C}}{{{\text{O}}}_{{\text{2}}}}{\text{Me}}} \right){\text{С}}{{{\text{Н}}}_{{\text{2}}}}{\text{О}}{{{\text{R}}}^{2}} + {{{\text{Н}}}_{{\text{2}}}}{\text{О}}. \\ \end{gathered} $Результаты, подтверждающие концепцию [108], были получены позднее [112] при исследовании арилирования α-фенилзамещенных аллиловых спиртов в АсОН в реакции с PhPd(tmeda)I и LiCl и в реакциях CH2=CHCH2Y (Y = OH, OEt, OAc) с PhHgOAc в присутствии Pd(OAc)2 и LiCl. Было показано, что повышение [LiCl] меняет направление распада σ-палладийорганического интермедиата, полученного внедрением АС по связи Ph–Pd, с образования карбонильного соединения (β-элиминирование [Pd]–H) на синтез фенилированных олефинов (реакция (XXXVII)). При [LiCl] = 0 получается 59% (IV) и 18% (V)), а при [LiCl] = 10 мМ (при [Pd]Σ = 1 мМ) – 13% (IV) и 64% (V). В случае CH2=CHCH2Y также происходит смена направлений элиминирования различных групп [Pd]–Х (Х = Н, Y), зависящая от концентраций [LiCl] и природы группы Y [112] (схема 4 ).
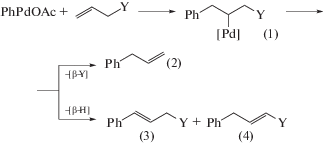
Схема 4 .
При Y = ОН продукт (4) превращается в альдегид PhСН2СН2СНО.
При [LiCl] = 0 получаются небольшие количества продуктов (3) и (4), так как элиминирование [Pd]–H делает процесс невозможным из-за исчезновения Pd(OAc)2. В условиях избытка Cl– по отношению к палладию (200 : 1) идет только β-элиминирование группы с гетероатомом [Pd]–Y и образованием продукта (2) и небольшого количества дифенилированного олефина PhCH=CHCH2Ph. Авторы [112] предполагают, что в этих условиях хлорид-ион, атакуя σ-PdR, стимулирует Е2-процесс элиминирования группы Y– (в растворах АсОН!), например, в переходном состоянии процесса (схема 5 ).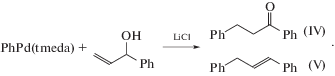
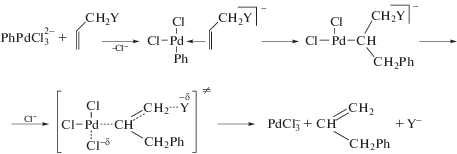
Схема 5 .
В результате описанных выше исследований реакций изомеризации, обмена групп ОН и окисления аллиловых спиртов было установлено, что скорость реакции окисления АС п-бензохиноном в условиях, близких к Вакер-окислению, описывается кинетическим уравнением типа (1), и был подтвержден также факт влияния концентраций Cl– на реализацию маршрутов цис- и транс-ГП олефинов.
3.4. Реакции изомеризации и окисления хиральных аллиловых спиртов
Описанные выше исследования внесли определенную ясность в “проблему гидроксипалладирования”, однако Генри с соавт. продолжили поиск дополнительных тестов [113–119], подтверждающих концепцию о влиянии концентрации хлорид-ионов на соотношение маршрутов с необратимой стадией цис-внедрения и с возможной обратимой стадией транс-присоединения молекулы воды (и других нуклеофилов). Эту концепцию на примере реакции окисления этилена в каталитической системе Вакер-процесса иллюстрирует схема 6 : соотношение маршрутов определяется концентрациями ионов Cl– и H+.
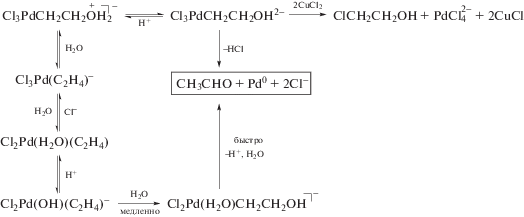
Схема 6 .
Вопрос о природе лимитирующей стадии маршрута транс-ГП в рамках схемы 6 остается невыясненным. Присоединение Н2О к π-комплексу Cl3Pd(C2H4)– может сопровождаться и синхронным депротонированием присоединяющейся молекулы воды. Заметим, что наличие стадии диссоциации Н2О должно всегда приводить к появлению КИЭ за исключением маловероятного случая, когда стадия присоединения молекулы воды к π-комплексу становится лимитирующей.
Для исследования стадий ГП в реакции изомеризации АС с обменом 16ОН на 18ОН был выбран аллиловый спирт (АС) с четырьмя заместителями, не окисляющийся в растворах ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}$ при 25°C (схема 7 ) [113] и позволяющий сопоставить кинетику изомеризации и обмена со стереохимией ГП. Опыты проводили в системе с [Cl–] = 0.1–1.0 M, [HClO4] = 0.05–0.4 M и с ионной силой, равной 2, которая поддерживалась добавками LiClO4. Поскольку вращение вокруг связи =СН–С в соединении (А) затруднено и происходит с высоким энергетическим барьером, исследовали хиральный спирт 2а[(−)-(R)] с геометрией Е.
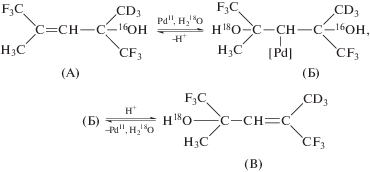
Схема 7 .
Использование хиральных спиртов позволяет определить, происходит ли геометрическая изомеризация и меняется ли конфигурация хирального центра. На схеме 8 (обозначения авторов) [113, 114] показано, что транс-ГП спирта 2а[(−)-(R)] дает продукт 3а, превращающийся в 2b[(R)-Z], а цис-ГП дает продукт 3b, превращающийся в 2b[(S)-E]. Предсказания строения интермедиатов и энантиомерных продуктов в рамках схемы 8 были сделаны при допущении, что исходный спирт (А) координируется палладием единственным способом (одной плоскостью олефина), представленным структурой (VI).
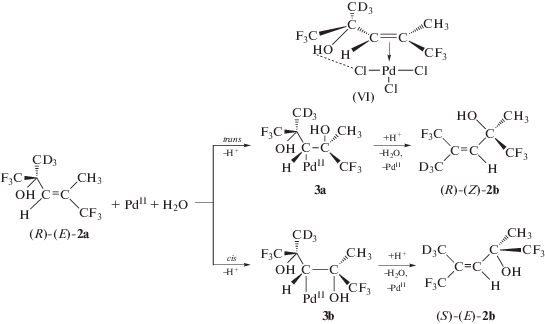
Схема 8 .
Согласно схеме 7 [113], в условиях низкой концентрации хлорид-иона получено уравнение (13), аналогичное уравнению (1), с равными константами скорости изомеризации и изотопного обмена (1.1 и 1.01 с–1 соответственно). В случае “аллильного” механизма изомеризации и обмена с образованием η3-алильного комплекса Pd(IV) скорость изомеризации должна быть равной половине скорости обмена.
Анализ полученных энантиомеров из хиральных 2-Е-спиртов показал, что из 2[R)-Е]-изомера при ~50%-ной степени изомеризации получается 50% 2[(S)-Е]-продукта, а из 2[(S)-Е]-спирта – 50% 2[R)-Е]-изомера, что указывает на инверсию конфигурации хирального атома углерода в условиях низкой концентрации хлорид-иона. В соответствии со схемой 8 этот результат указывает на цис-гидроксипалладирование в условиях изомеризации, которое, согласно кинетическому уравнению, свидетельствует о диссоциации молекулы воды в квазиравновесной стадии, предшествующей процессу ГП.
Таким образом, использование хиральных АС с инверсией хирального центра в ходе изомеризации сможет служить независимым тестом на цис-ГП (при указанных выше допущениях). Отсутствие рацемизации энантиомеров свидетельствует о том, что скорости оптической и геометрической изомеризаций равны скорости брутто-процесса изомеризации спиртов, рассчитанной по уравнению (13).
В работе [114] изучали кинетику и стереохимию процесса изомеризации тех же хиральных спиртов, но в условиях большей концентрации хлорид-иона (2–4 М). При этом было получено кинетическое уравнение типа уравнения (12) [98], не включающее концентрации протона. Показано также, что отсутствует инверсия конфигурации хирального центра и из 2[(R)-Е]-спирта образуется изомер 2[(R)-Z], свидетельствующий, по мнению авторов, только о транс-ГП при большой концентрации Cl– и о возможной стабилизации π-комплекса взаимодействием ОН-группы АС с анионом ${\text{PdCl}}_{{\text{3}}}^{ - }.$ Стабильность интермедиата ГП 3а (cхема 8), включающего фрагмент PdCl3, связана, возможно, и с неоднократно обсуждавшимся выше ингибированием β-элиминирования [Pd]–H хлорид-ионом, занимающим координационное место.
Для уточнения влияния концентрации LiCl на соотношение скоростей маршрутов процесса окисления исследовали кинетику реакций окисления этилена (с образованием ацетальдегида и этиленхлоргидрина (ЭХГ)) и изомеризации хиральных аллиловых спиртов в растворах пиридинового комплекса PdCl3(Py)–. При окислении этилена п‑бензохиноном [115] в интервале концентраций [LiCl] = 0.05–0.2 M, [H+] = 0–1.0 M и [PdCl3(Py)–] = = (3.5–14.0) × 10–3 M получено уравнение (14), аналогичное уравнению (1).
(14)
$R = \frac{{{{k}^{/}}{{K}^{/}}[PdC{{l}_{3}}{{{(Py)}}^{ - }}][{{C}_{2}}{{H}_{4}}]}}{{[{{H}^{ + }}]{{{[C{{l}^{ - }}]}}^{2}}}}.$В растворах PdCl3(Py)– исследовали также кинетику изомеризации тетра- и тризамещенных хиральных спиртов, например хирального (R)-(R)-АС F3CC*H(OH)CH=C(CF3)CH3 (метилпентенола, Г) [116]. Если в случае ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}$ изомеризация спирта А (схема 7 ) при малых концентрациях хлорид-иона протекает в соответствии с кинетическим уравнением Вакер-процесса (1), то при катализе комплексом PdCl3(Py)– процесс описывается уравнением (15)
полученным ранее для ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}$ при большой концентрации Cl– [114]. Таким образом, изомеризация, катализируемая пиридиновым комплексом при малых концентрациях хлорида, протекает как транс-ГП в отличие от цис-ГП в случае ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}.$ Стереохимия реакции с хиральным АС (А) в случае пиридината палладия также демонстрирует идентичность со стереохимией реакции, катализируемой ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}$ при больших концентрациях хлорида.Окисление метилпентенола (Г) происходит только при малых концентрациях хлорида, и при этом наблюдается 100%-ная инверсия хирального центра в случае ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}$ в процессе окисления до кетона. Для проведения процесса окисления вместо изомеризации в растворах пиридината палладия необходимо снижать концентрацию хлорида до 0.05 М. При этом стереохимическим результатом, в основном, является инверсия конфигурации, но содержание энантиомерного избытка ее намного ниже, чем в исходных реагентах, что говорит о протекании ГП одновременно по двум путям. Эти результаты прекрасно согласуются с данными, полученными при исследовании конформаций ЭХГ при больших концентрациях Cl– [89, 90, 93].
Таким образом, на этом этапе исследований было установлено, что стереохимия присоединения воды к π-комплексам олефинов различна при разных концентрациях хлорид-иона. Поскольку и кинетика, и стереохимия окисления Е- и Z-CHD=CHD при больших [Cl–] согласуются с транс-ГП [90, 93], в случае тетразамещенных АС также предполагается, что при больших [Cl–] происходит транс-ГП, что отражается на переносе хиральности в ходе изомеризации (R → R, S → S). Другими словами, допускается [116], что свойства и реакционная способность π-комплексов этилена и тетразамещенных АС с двумя трифторметильными и одной гидроксильной группами одинаковы.
Дальнейшие исследования дизамещенных хиральных аллиловых спиртов (АС) (пентенолов и гексенолов), способных изомеризоваться и окисляться [117–119] и создающих неизмеримо меньшие стерические препятствия при образовании π-комплексов, показали, однако, что наиболее стабильными и менее реакционноспособными π‑комплексами являются такие, в которых ОН-группы хирального центра взаимодействуют с PdCl2X– (X = Cl, OH, OMe, OAc). В таких комплексах с заместителями R1 и R2 в АС, находящимися в наиболее удаленном положении друг от друга, создаются условия повышения устойчивости π-комплексов. В результате изучения стереохимии продуктов окисления и изомеризации дизамещенных АС с различными нуклеофилами Х (кроме Ph) были установлены следующие факты.
1). При транс-присоединении Х к стабильным π-комплексам (R)-Z-АС происходит инверсия абсолютной конфигурация хирального центра реагента в продукте ГП и в продуктах изомеризации и окисления (кетон): R → S. При R1 = R2 = Ме кетон получается в форме рацемата. В случае тетра- и тризамещенных АС при транс-присоединении Н2О к π-комплексам не происходит инверсии конфигурации хирального центра, что, по мнению авторов, характерно для неустойчивых π‑комплексов. В таких π-комплексах плоскость олефина, к которой направляются Pd и Nu, будет зависеть от абсолютной конфигурации хирального центра в исходном АС. Это заключение проиллюстрировано на схеме 9 [117, 118] для очевидного цис-присоединения Ph−${\text{PdCl}}_{{\text{3}}}^{{{\text{2}} - }}$ к АС, где устойчивым является комплекс 2а с удаленными друг от друга метильными группами.
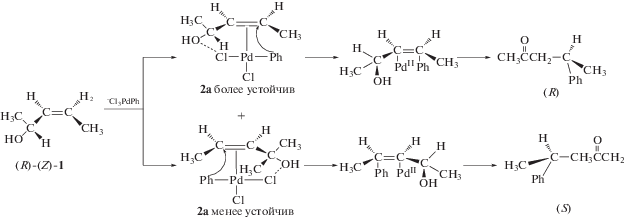
Схема 9 .
2). При высоких и низких концентрациях Cl– стереохимические результаты гидроксипалладирования АС противоположны.
3). Продукты окисления и изомеризации имеют одинаковую конфигурацию хирального центра.
4). Конфигурация хирального центра в реагентах и продуктах цис-фенилирования одинакова независимо от [Cl–].
5). В водных и метанольных растворах при низких значениях [Cl–] 4-гидрокси- и 4-метоксипентаноны-2 имеют (R)-конфигурацию хирального центра исходного АС$,$ свидетельствующую о цис-присоединении Nu (схема 10 [118]).
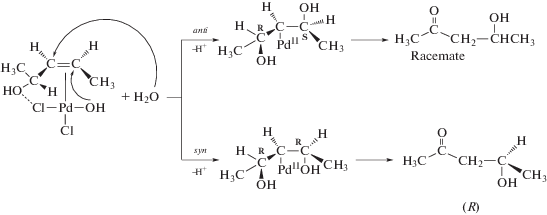
Схема 10 .
Из полученных результатов в этой серии экспериментов следует, что предложенный стереохимический тест по определению абсолютной конфигурации хирального центра продукта может быть полезным тестом на механизмы ГП или присоединения других Nu к π-комплексам только при наличии информации (или обоснованных гипотез) о составе и устойчивости π-комплексов с данным набором лигандов.
Что касается кинетических тестов применительно к Вакер-окислению, то они связаны, в первую очередь, с зависимостями скоростей реакции окисления от концентраций Cl– и Н+, т.е. с уравнениями (1), (10) и (15). Степень обратимости стадии ГП зависит от условий, определяющих реакционную способность σ-палладийорганического интермедиата. Возможность реакции, обратной ГП, т.е. дегидроксипалладирования, таких интермедиатов следует из исследований реакций изомеризации АС (см. разд. 3.3) и работ [111, 112].
Использование информации по механизму и условиям синтеза хлоргидринов (ХГ) позволило провести асимметрический синтез ХГ с моно- и биядерными комплексами палладия в качестве катализаторов [120–122]. Так, в водном тетрагидрофуране в системе ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}$–CuCl2(4 M)–LiCl(0.2 M) с использованием хирального сульфированного фосфинового лиганда (например, (R)-Tol-BINAP) из пропена, метилвинилкетона и аллилфенилового эфира получили хиральные (R)-энантиомеры ХГ с оптической чистотой 28–76% [120]. Выход 1-хлор-2-гидроксиизомера (ClCH2CH(OH)R) составил >95%. Димерные β-трикетонатные комплексы палладия с дифосфинами оказались наилучшими катализаторами синтеза хиральных ХГ с ее ≈ 95% [121, 122]. Отношение изомеров 1-Cl/2-Cl в случае пропена равно 3.5. Число оборотов катализатора при окислении олефинов кислородом достигает 300 с сохранением димерного катализатора.
3.5. Механизмы превращений σ-палладийорганических интермедиатов
После анализа проблем, связанных с ГП олефинов и с присоединением других Nu к π-комплексам, рассмотрим механизмы превращений σ-палладийорганических интермедиатов (например, [Pd]CH2CH2OR), полученных в результате гидрокси-, алкокси- и карбоксипалладирования, в продукты окисления олефинов.
Все экспериментальные результаты – КИЭ, кинетика, распределение изотопов в продуктах – однозначно указывают на перенос гидрида из β- в α-положение по отношению к [Pd]. Вопрос заключается в том, как этот так называемый “1,2-сдвиг гидрида” происходит? Эта проблема обсуждается в монографиях [1, 3] и в обзоре Генри [123], посвященном сравнению процессов окисления олефинов комплексами Pd(II) и Tl(III).
Первая гипотеза (А) была выдвинута в работах Моисеева с соавт. [44, 51, 124] и заключалась во внутримолекулярном переносе гидрид-иона из β‑положения к α-атому углерода, имеющему больший положительный эффективный заряд. Такое внутримолекулярное “нуклеофильное” замещение может приводить к высвобождению уходящей группы ClPd– (или Pd0) с парой электронов и к образованию иона карбения [1]. В переносе гидрида, вероятно, принимает участие атом палладия в переходном состоянии [1, 45, 125], т.е. образуется так называемая “агостическая” связь [126], и реакция (XXXVIII)
(XXXVIII)
$\begin{gathered} {\text{Cl}}\left[ {{\text{Pd}}} \right]{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}} \to \\ \to {\text{Cl}}{{\left[ {{\text{Pd}}} \right]}^{--}} + {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{C}}{{{\text{H}}}^{{\text{ + }}}}{\text{OH}} \\ \end{gathered} $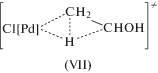
(XXXIX)
${\text{Pd}}_{{({\text{aq}})}}^{{{\text{2}} + }} + {\text{ }}{{{\text{Н}}}_{{{\text{2}}\left( {\text{г}} \right)}}} \to {\text{Pd}}_{{({\text{aq}})}}^{0} + {\text{2}}{{{\text{Н}}}^{ + }}$(XL)
${\text{Pd}}_{{({\text{aq}})}}^{{{\text{2}} + }} + {{{\text{Н}}}_{{{\text{2}}\left( {\text{г}} \right)}}} \to {\text{Pd}}_{{\left( {{\text{тв}}} \right)}}^{0} + {\text{2}}{{{\text{Н}}}^{ + }}.$Термодинамика стадии гетеролиза может стать более благоприятной при распаде σ-палладийорганического интермедиата с участием Nu (Н2О, ROH, AcO–) [1]. Нуклеофил может атаковать β‑атом углерода в переходном состоянии вследствие частичного освобождения орбитали в результате переноса, например, гидрид-иона при участии молекулы воды в реакции (XLII)
(XLII)
$\begin{gathered} {\text{Cl}}[{\text{Pd}}]{\text{C}}{{{\text{Н}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \to \\ \to {\text{ClPd}}_{{({\text{aq}})}}^{ - } + {\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{СН}}\left( {{{{\text{О}}}^{{\text{ + }}}}{{{\text{Н}}}_{{\text{2}}}}} \right){\text{ОН}}, \\ \end{gathered} $Термодинамические ограничения для стадии редокс-превращения σ-палладийорганического интермедиата естественным образом снимаются при участии окислителя (Ох) в стадии переноса гидрид-иона с образованием органического продукта. Следует отметить, что реакции нуклеофильного замещения (типа SN2) в случае металлоорганических соединений Pd(II), Pt(II) и Hg(II) также не известны при отсутствии окислителей и стабилизирующих лигандов, а в соединениях Pt(IV), например, c уходящей группой Pt(II) такие процессы хорошо изучены [127]. Скорость окисления олефинов в хлоридных растворах комплексов палладия в каталитических условиях в определенном интервале концентраций не зависит от концентрации Ох (CuCl2, Q, H2O2), что обычно объясняют участием окислителя в стадиях окисления ${\text{ХPd}}_{{({\text{aq}})}}^{ - }$ или ${\text{Pd}}_{{({\text{aq}})}}^{0}$ после превращения RPdX. В случае лимитирующей стадии ГП участие Ох в превращениях RPdX также может привести к нулевому порядку по окислителю при большой скорости окисления RPdX. Так, вероятно, и происходит в классическом процессе Вакер-окисления при низких концентрациях CuCl2 и относительно больших – Cl–. В реакции окисления этилена в спиртах скорость образования ацеталя не зависит от [CuCl2] при [LiCl] = 0 [7 (Ch. 4), 102].
К сожалению, кинетические уравнения Вакер-процесса, включающие [CuCl2] и [LiCl], получены в условиях с неустановленными функциями закомплексованности CuCl2 и PdCl2 и с неизвестными концентрациями свободного иона Cl– [1, 128–130]. Окислителем, образующим комплексы с RPdX может быть CuCl2, п-хинон (Q) и даже сам исходный комплекс ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}$ или PdCl3(Н2О)–. Так, при больших значениях [Pd]Σ появляется второй маршрут [1, 46, 47] со вторым порядком по $\left[ {{\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}} \right].$ Скорость изомеризации бутенов также зависит от [Pd]Σ во второй степени [131, 132]. В этих случаях перенос гидрид-иона и двух электронов приводит к образованию хлоридного комплекса Pd(I), например, ${\text{P}}{{{\text{d}}}_{{\text{2}}}}{\text{Cl}}_{n}^{{{\text{2}} - n}}$ (n = 4, 6). Интересно, что этот комплекс Pd(I), вероятнее всего, и катализирует реакцию изомеризации бутенов, которая прекращается при добавлении п-бензохинона в раствор в результате окисления комплекса Pd(I) до комплекса Pd(II) [1, 131–133].
Таким же образом Hg(OAc)2 помогает окислительным превращениям ртутьорганических соединений. Так, σ-бутенильное производное ртути CH3CH=CHCH2Hg(OAc) – интермедиат аллильного ацетоксилирования бутена-2 – распадается в присутствии Hg(OAc)2 при 25°С до бутенилацетата с образованием Hg2(OAc)2 в 275 раз быстрее, чем без добавок ацетата ртути [120, 134].
(XLIII)
$\begin{gathered} {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CH}}{\kern 1pt} = {\kern 1pt} {\text{CHC}}{{{\text{H}}}_{{\text{2}}}}{\text{Hg}}\left( {{\text{OAc}}} \right) + {\text{Hg}}{{\left( {{\text{OAc}}} \right)}_{2}} \to \\ \to {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CH(ОАс)CH}}{\kern 1pt} = {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}} + {\text{H}}{{{\text{g}}}_{{\text{2}}}}{{\left( {{\text{OAc}}} \right)}_{{\text{2}}}}. \\ \end{gathered} $Комплексы PdCl2(CuCl2)n участвуют в синтезе β-хлорэфиров из этилена [7 (Ch. 4), 104] и в реакциях окисления диенов в спиртах и гликолях без дополнительных окислителей [7 (Ch. 4), 135–137]. Так, например, в системе PdCl2–CuCl2–LiCl (2 М) из С4Н6 в этиленгликоле образуется 2-винил-1,4-диоксан [135], а в этаноле – изомеры эфиров бутендиола [138]. При этом скорость окисления бутадиена при 80°С описывается уравнением (16)
(16)
${{R}_{\Sigma }} = k{{P}_{{{{{\text{C}}}_{{\text{4}}}}{{{\text{H}}}_{{\text{6}}}}}}}{{[{\text{PdC}}{{{\text{l}}}_{{\text{2}}}}]}_{\Sigma }}[{\text{CuC}}{{{\text{l}}}_{{\text{2}}}}]_{\Sigma }^{{{\text{4}}.{\text{3}}}}.$Многоядерные комплексы Cu(II) обнаружены и в других неводных системах. Вероятно, появление гетерометаллических комплексов в реакции окисления диенов происходит еще до стадии образования η3-бутенильных интермедиатов или в реакции η3-аллильных комплексов с (CuCl2)n, если стадия образования таких комплексов обратима. Интересно, что FeCl3, неспособный к образованию гетерометаллических комплексов, не окисляет диены в этих условиях – процесс останавливается на стадии синтеза η3-бутенильных комплексов палладия. Роль гетерометаллических комплексов палладия и меди в окислении олефинов обсуждается в разд. 3.
Синтез диацетатов диолов из 1,3-диенов описан Бэкквалом [139–143] в системе Pd(OAc)2–Q–AcOH–LiOAc–Ox (Оx = MnO2), в которой наблюдается зависимость скорости окисления от [Q] (см. аналогичные зависимости в других процессах [7 (Ch. 4)]). В присутствии п-хинона процесс протекает с высокой стереоселективностью. Предполагается [93, 140, 141], что основная роль п-хинона заключается в стабилизации “плохой” уходящей группы в результате образования π-комплекса п-хинона с Pd0 (реакция (XLIV)), где πХ – π-комплекс палладия с диеном, а πXQ – η3-бутенилацетатный комплекс с п-хиноном:
(XLIV)
$\begin{gathered} {{{\text{C}}}_{{\text{4}}}}{{{\text{H}}}_{6}}\overset {{\text{Pd}}{{{\text{X}}}_{2}}} \leftrightarrows {{\pi }_{{\text{X}}}}\overset {{\text{OA}}{{{\text{c}}}^{ - }},{\text{Q}}} \leftrightarrows \\ \rightleftarrows {{\pi }_{{\text{X}}}}{\text{Q}}\xrightarrow{{{\text{LiOAc}}}}{\text{P}} + {\text{Pd}}{{{\text{Q}}}^{0}}, \\ {\text{Pd}}{{{\text{Q}}}^{0}} + 2{\text{HX}} \to {\text{Pd}}{{{\text{X}}}_{2}} + {\text{Q}}{{{\text{H}}}_{2}}. \\ \end{gathered} $Хотя окисление Pd0 в комплексе PdQ0 в присутствии кислоты хорошо известно [144, 145], такая схема механизма (XLIV) не согласуется с зависимостью скорости окисления η3-аллильных комплексов п-бензохиноном от [H+] в условиях первого порядка реакции по [Q] [73, 146]. В случае η3-С3H5PdCl [73] и η3-бутенильных комплексов палладия [146] установлено, что в реакции η3-аллильных комплексов с нуклеофилами непосредственно участвует протонированный п-хинон в η3-аллилхиноновом комплексе, например, когда нуклеофилом является Cl–:
(XLV)
$\begin{gathered} {{\pi }_{{\text{A}}}}{\text{Q}} + {{{\text{H}}}_{{\text{3}}}}{{{\text{O}}}^{ + }} \rightleftarrows {{\pi }_{{\text{A}}}}{\text{Q}}{{{\text{H}}}^{ + }} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}, \\ {{\pi }_{{\text{A}}}}{\text{Q}}{{{\text{H}}}^{ + }} + {\text{C}}{{{\text{l}}}^{--}} \to {\text{P}} + {\text{ClPdQH}}, \\ \end{gathered} $В качестве стехиометрических окислителей в реакциях окисления диенов используется и кислород, но с дополнительными катализаторами его активации – фталоцианинатами и порфиринатами Co(II) и Fe(II) [139, 143]. В водно-ацетонитрильных растворах катионных комплексов палладия в процессе окисления этилена п-бензохиноном гидрохинон окисляется кислородом при 30°С в присутствии нерастворимых и растворимых фталоцианинатов Fe(II) [147]. Кислородные комплексы фталоцианинатов переходных металлов РсМ(О2) способны в результате окислять QH2 до хинона, превращая процесс окисления в каталитический по Pd(II) и по Q, а также окислять η3-аллильные комплексы без п-хинонов при их взаимодействии с супероксидным комплексом LCo(O2) (L = замещенный порфиринат). Имеются и другие примеры участия п-хинонов в окислительном деметаллировании σ-палладийорганических соединений [7 (Ch. 4)]. Например, в реакции окислительного карбонилирования спиртов в системе PdCl2–LiCl–PhCOOH–PhCOONa–MeOH при увеличении [Q] понижается скорость образования диметилкарбоната и повышается скорость образования диметилоксалата в результате превращения предполагаемого интермедиата Pd(Q)(COOMe)2 [148, 149].
Вторая гипотеза (Б) о механизме превращения интермедиатов [Pd]CH2CH2X (X = OH, OR, OAc) рассматривает β-элиминирование [Pd]−H с образованием винильного продукта в π-комплексе H[Pd](CH2=CHX) [50, 90, 124]. Возможность реализации этой стадии при наличии свободного или легко освобождаемого координационного места в комплексе палладия не вызывает сомнений. Прекрасным подтверждением существования такой стадии является реакция Моисеева окислительной “эстерификации” олефинов [1, 10, 40]. В определенных условиях получаются также простые виниловые эфиры [150]. Для получения карбонильных продуктов (в условиях отсутствия обменов Н/D со средой) необходимо предположить, что “виниловый” спирт, не выходя из координационной сферы палладия в π-комплексе, сразу же после поворота вокруг оси связи палладия с виниловым спиртом Н[Pd]–(CH2=CHОН) присоединяется к [Pd]–H. Таким образом, вместо одной необратимой элементарной стадии превращения интермедиата [Pd]CH2CH2ОН в продукты (без участия Ох или с его участием) эта гипотеза требует трех дополнительных стадий (схема 11 , где π–π-комплекс).
Вторым “слабым” местом гипотезы (Б) является отсутствие вытеснения олефином винилового спирта из координационной сферы палладия в раствор из интермедиатов в стадиях (2) и (3) (схема 11 ) по аналогии с синтезами винилацетата и простого винилового эфира.
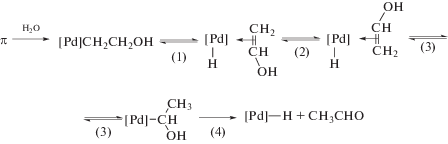
Схема 11 .
Если в случае этилена (вследствие его низкой концентрации в растворе) этот результат легко понять, то для высших олефинов, особенно в водно-органических растворителях, его трудно объяснить. Например, при окислении d2-аллилового спирта получаются карбонильные соединения с неизменным изотопным составом [106, 107].
“Слабые” места гипотезы (Б) можно, вероятно, смягчить предположением о быстром превращении π-комплекса винилового спирта в σ-комплекс H[Pd]CH2CHO при отщеплении протона (или НХ) с последующим восстановительным элиминированием [Pd0] и СН3СНО. При этом допущении сохраняется согласие со всеми экспериментальными результатами. Быстрое равновесие реакции
Кроме того, со второй гипотезой (Б) трудно согласовать образование ацеталей и кеталей в спиртах или этилидендиацетата в уксусной кислоте. Вопрос о механизмах образования 1,1-диэфиров (ацетали, кетали, диацетаты) детально обсуждался в работах [1, 3, 10, 123] (см. разд. 6). В рамках гипотезы (Б) необходимо предположить, что интермедиат Cl[Pd]CH(OR)CH3, полученный из этилена в соответствии со схемой 11 , должен быть атакован молекулой ROH с образованием Cl[Pd]– и Н+ (или Cl[Pd]H) и CH3CH(OR)2. В таком процессе возможно участие окислителя, содействующего восстановительному элиминированию продукта, например, по реакции (XLVI):
(XLVI)
$\begin{gathered} RO[Pd]CH(OR)C{{H}_{3}} + Ох \to \\ \to [Pd(II)] + C{{H}_{3}}CH{{(OR)}_{2}} + Red. \\ \end{gathered} $В рамках первой гипотезы (А) для объяснения механизма образования таких продуктов можно полагать, что 1,2-сдвиг гидрида происходит по аналогии с реакцией (XLII) синхронно с атакой нуклеофилом β-атома углерода [1, 10]:
В случае $R$ = Н получим 1,1-этилиденгликоль.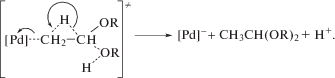
(XLVIII)
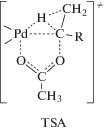
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CH}}{{\left( {{\text{OH}}} \right)}_{2}} \to {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHO}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}{\text{.}}$Близкая гипотеза о влиянии атаки олефина в π-комплексе ацетатной группой, связанной с палладием, на синхронный внутримолекулярный перенос гидрида обсуждается и при анализе механизмов образования 1,1-диацетатов [79]. Так, предполагается возможность образования, например, интермедиата Cl2PdC(CH3)(OAc)(R)– в результате переноса групп Н и АсО в рамках переходного состояния (TSA).
Полезная информация о механизмах редокс-превращений σ-металлоорганических соединений палладия(II) получена при изучении реакций трансметаллирования и мехинизмов катализа в полифункциональных каталитических системах (ПФКС).
Использование и исследование ПФКС, в которых разные катализаторы выполняют различные кинетические функции [7 (Ch. 4)], началось особенно активно после открытия процессa Вакер-окисления в системе PdCl2–CuCl2. В таких системах, как PdCl2–HgCl2, HgCl2–CuCl2, TlCl3–PdCl2 и CuCl–CuCl2, роль PdCl2 и CuCl2 заключается в окислении металлоорганических соединений непереходных металлов или катализе деметаллирования ртуть(II)- или таллий(III)-органических интермедиатов в присутствии других окислителей [7 (Ch. 4), 154–157]. Реакции трансметаллирования было предложено использовать в качестве основы для дизайна каталитических реакций [157]. Эта реакция в случае ClHgCH2CH2OH и PdCl2, как уже отмечалось, сыграла важную роль в доказательствах образования интермедиата [Pd]CH2CH2ОН в Вакер-окислении [1, 51]. Так, при 20°С в эфире выход ацетальдегида составил 93.7%. Из AcOHgCH2CH2OR в эфире в присутствии СаО образуются винилэтиловый эфир при R = Et и винилацетат при R = СН3СО. В уксусной кислоте с добавкой CH3COONa из AcOHgCH2CH2ОАс и PdCl2 образуются винилацетат и этилидендиацетат. На основании реакции транспалладирования в 1971 г. был разработан препаративный метод синтеза метилкетонов [158].
Реакции в системах RMXn–PdCl2 выявили интересные особенности реакционной способности RPdCl и самой реакции обмена металлами. Как уже отмечалось [80], перенос органического фрагмента с атома ртути на палладий происходит с сохранением конфигурации атома углерода, связанного с атомом ртути. Этот вывод был подтвержден в работе [159] при обмене трео- и эритро-CH(NMe2)2CH(Me)HgCl c PdCl2(PhCN)2 в тетрагидрофуране (ТHF). Изучение обмена неопентильного производного Hg(II) с Pd(OAc)2 [160] показало, что кроме 1,2-сдвига гидрида возможен и 1,2-сдвиг метильной группы. Реакция (СН3)3ССН2HgCl с CuCl2 (LiCl–AсОН) протекает очень медленно с образованием (CH3)3CCH2Cl. При добавлении каталитических количеств Pd(OAc)2 получаются метилбутены (СН3)2С=СНСН3 и СН2=С(СН3)С2Н5, а также хлор- или ацетатметилбутенил (СН3)2СХ(С2Н5). Предполагалось образование иона карбения (СН3)2С+(С2Н5) с протеканием последующих изомеризаций, однако оказался возможным и 1,2-сдвиг метила в палладийсодержащем интермедиате. Интересный результат был получен и при изучении реакции PhCH(OH)CH2HgCl c PdCl2 в ацетоне и хлороформе [161]. Продуктами реакции оказались фенилуксусный альдегид, ацетофенон и стирол (в результате разложения непрореагировавшего исходного реагента соляной кислотой). Авторы [161] объяснили это обратимостью стадии ГП (XLIХ), однако в соответствии с работой [160] можно предположить и перенос Ph-группы в переходном состоянии стадии редокс-превращения ClPdCН2CH(OH)Ph.
(XLIХ)
$\begin{gathered} {\text{ClPdC}}{{{\text{Н}}}_{{\text{2}}}}{\text{CH(OH)Ph}} \rightleftarrows \pi {\text{ - комплекс}} \rightleftarrows \\ \rightleftarrows {\text{ClPdCН(Ph)C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}}. \\ \end{gathered} $Карбонильные соединения, кетали и ацетали получаются и в реакции RTl(OAc)2 (R = = PhCH(OМе)CH2) c PdCl2 в метаноле в зависимости от условий обработки фильтратов после отделения Pdмет [162]. Так, в присутствии AcONa в метаноле при экстракции бензолом в качестве основного продукта образуется PhC(OМе)2CH3 (80%). При обработке же фильтрата водой до экстракции основным продуктом является PhCOCH3. Интересно, что в отсутствие AcONa и в случае, когда PhCH(OМе)CH2Tl(OAc)2 превращается без добавления PdCl2 [163], образуется ~40% PhCH2СН(OМе)2 (плюс 48% PhCOCH3 в реакции с PdCl2 [162]).
При обсуждении роли CuCl2 в ПФКС в водных растворах HgCl2 или PdCl2 возникает также вопрос о вероятности трансметаллирования с образованием RCuCl (RCuX). Так, например, в реакции гидрохлорирования ацетилена, протекающей с образованием промежуточного β-хлорвинилмеркурхлорида в системе HgCl2–HCl–Н2O, добавки CuCl2 приводят к демеркурированию этого интермедиата и образованию транс-1,2-дихлорэтилена с уменьшением скорости синтеза винилхлорида [164]. При этом транс-изомер ClHgCH=CHCl в реакции с CuCl2 превращается в транс-1,2-дихлорэтилен. Эти факты позволяют предположить образование промежуточного соединения транс-ClCuCH=CHCl, реакция которого со второй молекулой CuCl2 приводит к его деметаллированию и образованию 1,2-дихлорэтилена.
В системе CuCl–CuCl2–HCl–Н2O из ацетилена образуются винилхлорид, транс-1,2-дихлорэтилен и винилиденхлорид СН2=СCl2 [165]. Исследование кинетики реакции транс-ClHgCH=CHCl с CuCl2 в нестационарных и стационарных (FeCl3) условиях показало [166], что изменение первой производной скорости реакции по времени (при t → 0) описывается уравнением второго порядка по [CuCl2]. Этот факт подтверждает участие второй молекулы CuCl2 в деметаллировании медьорганического интермедиата. Моно- или бимолекулярный распад RCuCl так же, как и в случае RPdCl, термодиномически невероятен.
(L)
${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{{\text{2}}\left( {\text{г}} \right)}}} + {\text{Cu}}_{{({\text{aq}})}}^{{{\text{2}} + }} + {\text{2C}}{{{\text{l}}}^{ - }} = {\text{Cu}}_{{\left( {\text{г}} \right)}}^{0} + {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{l}}}_{{{\text{2}}\left( {\text{г}} \right)}}}.$Возможность образования очень реакционноспособных σ-медьорганических соединений RCuIIL и RCuIIIL следует из работ Д. Мейерштейна [167] и других исследователей. Стабильные σ-металлоорганические соединения Cu(II) и даже Cu(III) были синтезированы в 1993–2011-х гг. (см., например, [168–171]).
В механизме реакции синтеза хлоркарбонатов окислительным карбонилированием спиртов в системе PdCl2–CuCl2 предполагается стадия трансметаллирования с образованием алкоксикарбонильного комплекса Cu(II) [172]:
(LI)
$\begin{gathered} {{{\text{L}}}_{{\text{2}}}}{\text{Pd}}\left( {{\text{COOR}}} \right){\text{Cl}} + {\text{CuC}}{{{\text{l}}}_{2}} \to \\ \to {\text{ClCu}}\left( {{\text{COOR}}} \right){\text{Cl}} + {{{\text{L}}}_{{\text{2}}}}{\text{PdC}}{{{\text{l}}}_{2}}, \\ {\text{ClCu}}\left( {{\text{COOR}}} \right){\text{Cl}} + {\text{CuC}}{{{\text{l}}}_{2}} \to \\ \to 2{\text{CuCl}} + {\text{ClCOOR}}. \\ \end{gathered} $Реакция трансметаллирования в случае винильных производных Hg(II) и PdCl2, давно известная в органическом синтезе [173], приводит к синтезу диенов с высокой селективностью (до 100%). В реакции транс-ClHgCH=CHCl с PdCl2 [174] образуются Pdтв и дихлорполиены Cl(CH=CH)nCl (n = 2–6) c преимущественным образованием транс, транс-1,4-дихлорбутадиена. В присутствии FeCl3 процесс становится каталитическим по катализатору PdCl2 с числом оборотов 124–1960. В заключение этой части анализа исследований механизма Вакер-окисления и близких реакций следует отметить, что в каталитических процессах в зависимости от механизма редокс-распада σ-палладийорганических интермедиатов реализуются следующие пути регенерации исходных комплекcов Pd(II).
1). Если процесс образования продуктов доходит до появления ${\text{Pd}}_{{{\text{aq}}}}^{0},$ ${\text{PdX}}_{{{\text{aq}}}}^{ - }$ или комплексов Pd(I), окислители Q, CuCl2, FeCl3 и др. в определенных условиях легко справляются с задачей регенерации Pd(II). При наличии стабилизирующих лигандов молекулярный кислород также может окислять восстановленные частицы без дополнительных катализаторов [14]:
(LII)
${{{\text{L}}}_{n}}{\text{Pd(}}{{{\text{O}}}_{2}}) + 2{\text{HX}} \to {{{\text{L}}}_{n}}{\text{Pd}}{{{\text{X}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}.$2). Если в процессе окисления образуются гидридные комплексы [Pd]–H, те же окислители (Q, CuCl2 и О2) прекрасно справляются с их oкислением [175, 176], например,
(LIII)
$\begin{gathered} {\text{HPdX}} + {{{\text{O}}}_{{\text{2}}}} \to {\text{HOOPdX}}\xrightarrow{{HX}} \\ \to {\text{Pd}}{{{\text{X}}}_{{\text{2}}}} + {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}}. \\ \end{gathered} $3). При участии окислителя в стадиях превращения σ-палладийорганических интермедиатов катализатор Pd(II) не меняет степени окисления в ходе процесса, приводя к окислению органического фрагмента.
3.6. Теоретические исследования механизма реакции Вакер-окисления
Механизм этой важной для промышленной и препаративной органической химии реакции [4, 5, 9, 13, 16, 22, 23] не мог не привлечь внимания специалистов в области квантовой химии. Результаты ранних исследований 1977–1985 гг. [97–99, 177] и более поздние работы с применением методов теории функционала плотности (DFT) (с учетом влияния сольватации интермедиатов, переходных состояний и образования ионных пар [178–185]), а также методов молекулярной динамики [186–188] привели к заключению, что цис-гидроксипалладирование в газовой фазе или в растворах – энергетически запрещенный процесс (!). При этом по мнению ряда исследователей [49, 182], авторы модельных расчетов [обзор 59, 181, 186, 188] игнорируют огромное количество экспериментальных данных, рассмотренных в разд. 3. Квантово-химические расчеты [183, 184] выявили существенные недостатки современного состояния теоретических подходов к оценке энергетики процессов переноса протонов в водную или водно-органическую среду даже с учетом присутствия противоионов. Ошибка в расчетах ионных пар может составлять 15 ккал/моль [49]. В связи с этим обстоятельством квантово-химическое моделирование сложных механизмов с заряженными частицами в полярных средах вряд ли можно рассматривать как доказательство существования тех или иных интермедиатов и лимитирующих стадий. Вместе с тем, концепции и идеи, возникающие в ходе квантово-химического моделирования элементарных стадий, представляют несомненный интерес для планирования экспериментальной дискриминации гипотез о механизме и, конечно, для развития теории механизмов каталитических реакций. Поэтому такие исследования полезно рассмотреть и прокомментировать.
В работе П. Зигбана [178] была проанализирована энергетика (∆Н) реакций присоединения гидроксил-иона к π-комплексу PdCl2(C2H4) по двум направлениям: (а) к этилену с образованием Cl2PdCH2CH2OH– и (б) к атому Pd с образованием Cl2Pd(OH)(C2H4)–. Как было показано, положительный противоион Н3О+, образующий водородные связи с двумя хлоридными лигандами, определенно необходим даже для такой простой стадии нуклеофильного присоединения, не имеющей, кстати, никакого отношения к механизму Вакер-окисления в соответствии с кинетическими данными [1, 44, 45]. При добавлении катиона Н3О+ относительная стабильность продукта направления (а) по отношению к продукту направления (б) меняется с 40 ккал/моль в пользу направления (а) до 11 ккал/моль в пользу направления (б). Заметим, что модель с внешней (транс) атакой ОН– на π-комплекс была использована и в работе [179], в которой для квантово-химического моделирования была взята каталитическая система Pd(OAc)2–AcOH–H2О, ранее не исследованная экспериментально в реакции окисления олефинов до карбонильных соединений. В работе 1996 г. Зигбаном [180] была предложена интересная модель участия ассоциатов (Н2О)n в транс-атаке олефина в π-комплексе PdCl2(Н2О)(C2H4) и в стадии β-элиминирования Pd–Н из σ-палладоэтанольного интермедиата и установлен оптимальный размер ассоциата, включающий 3 молекулы воды. Расчеты с большим базисом и с учетом эффектов сольватации показали, что при значительном разделении зарядов при образовании интермедиатов очень важно описать и учесть дальнодействующую поляризацию растворителя. Такое разделение зарядов с дипольным моментом 9.3 D наблюдается при образовании $\sigma {\text{ - Pd}}({\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}}){\text{Cl}}_{{\text{2}}}^{ - }$ из интермедиата с тремя молекулами воды (VIII) через переходное состояние (IX) для транс-гидроксипалладирования с одновременным отрывом протона в виде ${{{\text{Н}}}_{{\text{5}}}}{\text{О}}_{{\text{2}}}^{{\text{ + }}}{\text{.}}$ Только учет этого эффекта делает нуклеофильное присоединение экзотермичным. Именно этот эффект и определяет, по мнению автора [180], преимущество транс-ГП по сравнению с цис-ГП: энергетический барьер для этой стадии составляет 5.7 ккал/моль (298 К) при ∆Н = – 4.5 ккал/моль. Для второй стадии β-элиминирования Pd–H величина барьера равна 26.0 ккал/моль, что слишком велико, чтобы соответствовать экспериментально установленному отсутствию КИЭ для пары С2Н4/С2D4 (КИЭ ≈ 1). При включении в расчет очень большого базисного набора этот барьер снижается до 13.3 ккал/моль, но и с такой величиной барьера эта стадия не может быть лимитирующей. По мнению автора [180], для согласования расчетов с экспериментом нужна другая лимитирующая стадия с более высоким барьером. Предполагается, что такой стадией может быть последняя стадия (4) с разрывом О–Н-связи и образованием ацетальдегида в рамках второй гипотезы (Б) (схема 11 ).
В работе [181] также использовали модель с ассоциатом (Н2О)3, и было установлено, что прямая атака ОН-группы, координированной палладием, на этилен (цис-ГП) приводит к высокой величине энергии активации (Еа ≈ 47 ккал/моль), а использование транс-присоединения воды из ассоциата снижает энергию активации до 13 ккал/моль.
При этом преимущество отдается транс-присоединению группы ОН из ассоциата, но к гидроксокомплексу [Cl2Pd(OH)(C2H4)–](H3O)+. В качестве лимитирующей принимают реакцию (LIV). Расчет наблюдаемой энтальпии активации $\Delta Н_{{{\text{298}}}}^{ \ne }$ для рассматриваемой кинетической модели в рамках уравнения (1) (23.8 ккал/моль) показал удовлетворительное соответствие с экспериментом (19.8 ккал/моль по данным Генри [45]).
Следует отметить, что в механизме синтеза ацеталя ацетальдегида и эфиров хлор(бром)гидринов в связи с торможением обоих процессов кислотой НХ со вторым порядком по спирту и транс-стереохимией алкоксипалладирования при образовании эфиров галогенгидринов также рассматривали транс-атаку спиртом π-комплекса с алкоголятным лигандом [Pd](C2H4)(OR)– [7 (Ch. 4)].
Критический анализ работы [181] приведен в статье Кейта и др. [184], по мнению которых в работе [181] не были учтены многие экспериментальные результаты по реакции Вакер-окисления и близким процессам. В частности, транс-ГП гидроксокомплекса палладия с участием ассоциата (Н2О)3, по мнению Кейта, не играет роли в механизме Вакер-окисления, поскольку при низких концентрациях Cl– и CuCl2 процесс протекает как цис-внедрение этилена по связи Pd–OH.
Варианты превращений σ-палладоэтанольного интермедиата и, в частности, в рамках гипотез (А) и (Б) анализировали в работе [183]. Так, для β → α-сдвига гидрида с переходным состоянием TS1 (X) получено очень высокое значение энтальпии активации (42.8 ккал/моль, 298 К).
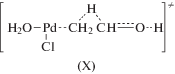
Анализ стадии β-элиминирования Pd–Н в результате разрыва связи О–Н (схема 11 ) в конечном интермедиате CH3CH(OH)PdCl(H2O) (XI) показал, что величина энергетического барьера $\left( {\Delta Н_{{{\text{298}}}}^{ \ne }} \right)$ переходного состояния TS2 (XII) при разных вариантах расчета составляет 25–30 ккал/моль.
И только распад этого же интермедиата по реакции (LV) с переходным состоянием TS3 (XIII) с отщеплением HCl и восстановительным элиминированием альдегида и Pd0 привел к значениям энтальпии активации 17–21 ккал/моль.
(LV)
$\begin{gathered} {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CH}}\left( {{\text{OH}}} \right){\text{PdCl}}\left( {{{{\text{H}}}_{{\text{2}}}}{\text{O}}} \right) \to \\ \to {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHO}} + {\text{P}}{{{\text{d}}}^{0}} + {\text{HCl}}{\text{.}} \\ \end{gathered} $
Поскольку цис-ГП при участии молекулы воды по результатам расчетов в работе [184] приводит к $\Delta G_{{{\text{298}}}}^{ \ne }$ = 33.4 ккал/моль, были рассмотрены другие пути образования продуктов ГП и, в частности, существование стадии синхронного с депротонированием аква-π-комплекса цис-внедрения этилена по связи Pd–OH (LVI) с величиной барьера $\Delta G_{{{\text{298}}}}^{ \ne }$ = 18 ккал/моль.
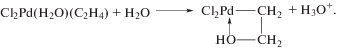
Такая стадия, несмотря на приемлемую высоту барьера, к сожалению, не может быть лимитирующей (скоростьопределяющей стадией), поскольку на основании кинетических данных она должна быть быстрой и квазиравновесной и, конечно, неэндотермической с $\Delta H_{{{\text{298}}}}^{ \ne }$ = 10.1 ккал/моль. Авторы работы [184] предложили в качестве лимитирующей еще один вариант изомеризации псевдометаллациклического интермедиата в реакции (LVI) в комплекс с агостической связью Pd…H…C (XIV) c $\Delta G_{{{\text{298}}}}^{ \ne }$ = 23.2 ккал/моль.
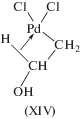
Участие ассоциата (Н2О)3 в механизме транс-ГП исследовалось и в экспериментально-теоретической работе [185]. Изучая кинетику реакции окисления децена-1 кислородом, катализируемое комплексом PdCl2[(–)-sparteine] в растворителе DMA–Н2О (DMA = диметилацетамид), авторы обнаружили третий порядок реакции по концентрации воды ([Н2О]∑), которую изменяли в интервале от 1 до 8 М. Этот результат рассматривают как подтверждение участия в реакции ассоциата (Н2О)3 в стадии транс-ГП. Теоретические оценки энтальпии активации для этой стадии с привлечением континуальной модели сольватации водой или DMA привели к значениям 5.2 и 11.9 ккал/моль соответственно. Причем авторы рассматривали интермедиат (XV), образующийся в обратимой стадии, который более похож на переходное состояние, чем на промежуточное соединение.
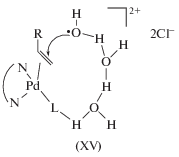
Отметим, что наблюдаемый порядок по [Н2О]∑ вряд ли отражает реальное количество участвующих в процессе молекул воды. Во-первых, концентрация свободных молекул воды или ассоциатов типа (Н2О)3 не пропорциональна [Н2О]∑ (см. обсуждение в разд. 6.3). Во-вторых, при варьировании [Н2О]∑ происходит весьма значительное изменение свойств среды и, соответственно, всех коэффициентов активности участников реакции и, особенно, заряженных частиц, поскольку комплекс палладия в этой системе является дикатионом. И в-третьих, при изучении кинетики не учитывалась возможность гидролиза дикатионного комплекса и соответствующее изменение концентрации Н+.
В следующем цикле работ использовали метод ab initio молекулярной динамики (AIMD) для построения энергетических профилей механизмов Вакер-окисления [186–188]. Так, в работе [186] метод AIMD был использован вместе с помещением исходного комплекса PdCl2(C2H4)(H2O) и интермедиатов в полость из 26 молекул воды. Эти молекулы воды, регулирующие кислотно-основные свойства воды, участвуют и в транс-присоединении молекулы воды к π-комплексу и в процессах замещения хлорид-иона и депротонирования координированных молекул воды. Авторы варьировали геометрию интермедиатов и способы цис- и транс-ГП, используя следующее требование (парадигму): “последовательные i-ые стадии механизма, вносящие вклад в общее кинетическое уравнение процесса, должны иметь величину свободной энергии активации $\left( {\Delta G_{i}^{ \ne }} \right),$ близкую (или немного большую) к наблюдаемой экспериментально в соответствии с уравнением (1): $\Delta G_{{{\text{300}}}}^{ \ne }$ = 22.4 ккал/моль.” В результате моделирования механизма был сделан вывод, что следует рассматривать только внешнесферную транс-атаку воды на этилен в π-комплексе и предложена схема механизма (схема 12 ) в соответствии с заданной парадигмой.
Схема 12 .
В связи с этими результатами и принятой парадигмой следует заметить:
– весь экспериментальный материал при малых концентрациях Cl– и CuCl2 свидетельствует о цис-ГП с участием связи Pd–OH,
– стадия депротонирования присоединяюшейся молекулы воды должна быть быстрой квазиравновесной стадией (кинетика), что вряд ли возможно при величине барьера 19 ккал/моль,
– не оценен барьер стадии, которую авторы считают лимитирующей,
– авторы не учитывали роль экзотермичности стадий, предшествующих лимитирующей, хотя при не очень больших значениях $\Delta H_{i}^{0}$ величины барьеров элементарных стадий могут быть выше наблюдаемого значения ∆G≠, поскольку скорость процесса (TOF) определяется величинами наибольшей “ямы” и наивысшего барьера на поверхности потенциальной или свободной энергий [189 (разд. 2.1.5)],
– необратимая лимитирующая стадия транс-ГП с одновременным переносом Н+ в среду вполне вероятна для процессов с большой концентрацией хлорид-иона в условиях независимости скорости реакции от концентраций Н+.
В статье [187] с участием группы авторов работы [186] в рамках того же подхода моделировали состояние всех комплексов палладия, предшествующих образованию двух важных для механизма Вакер-окисления изомеров – транс- и цис- PdCl2(C2H4)(H2O). В результате был сделан вывод, что величины барьеров $\left( {\Delta G_{{{\text{300}}}}^{ \ne }} \right)$ существенно выше для образования цис-изомера и поэтому его роль в процессе невелика. Этот результат расчетов вызывает удивление, поскольку процессы замещения лигандов в системе ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}$–C2H4–Cl––H2O являются быстрыми и квазиравновесными (в соответствии с экспериментальными результатами по изучению равновесий и с кинетикой реакции окисления [1, 45]) и не могут иметь барьеры по 20–25 ккал/моль. Если авторы допускают цепочку равновесных превращений
В работе [188] тем же методом AIMD были проанализированы два маршрута реакции окисления с участием транс- и цис-изомерных π-комплексов (маршруты I и II). При этом процесс изомеризации транс → цис-π-комплексов рассматривали через стадию образования цис-диаквакомплекса PdCl(C2H4)(H2O)2+ с барьером $\Delta G_{{{\text{3}}00}}^{ \ne }$ = 26.3 ккал/моль! В случае маршрута I транс-ГП (∆G≠ = 18 ккал/моль) не рассматривают в качестве лимитирующей стадии (схема 13 ), которой считают процесс β-элиминирования Pd–Н с той же величиной барьера 18 ккал/моль (с предшествующей стадией изомеризации транс → → цис с вытеснением Н2О в ходе изомеризации).
Схема 13 .
Если судить по значениям констант скорости стадии ГП и следующей стадии отщепления молекулы воды, стадия ГП не является и квазиравновесной. Максимальная величина свободной энергии переходного состояния на энергетическом профиле маршрута I составляет 21.5 ккал/моль (300 К). Для маршрута II, идущего через цис-изомер π-комплекса, максимальная величина свободной энергии переходного состояния равна 32.3 ккал/моль, причем в этом маршруте стадия ГП квазиравновесна, а лимитирующим становится процесс β-элиминирования Pd–Н с отщеплением цис-молекулы воды и образованием π-комплекса винилового спирта с барьером 12 ккал/моль. Эти результаты противоречат экспериментальным значениям КИЭ для пары С2Н4/С2D4. Последней стадией в обоих маршрутах является стадия восстановительного элиминирования альдегида и палладия(0) из комплекса Cl2Pd[CH(CH3)OH](H2O), образующегося из гидридного π-комплекса винилового спирта, с отщеплением Н3О+ и Cl–.
При отсутствии квазиравновесной стадии транс-ГП (с выделением протона) нельзя, однако, объяснить ни минус первый порядок по концентрации Н+ в системах с низкой концентрацией хлорид-иона, ни наличие квазиравновесной стадии ГП в реакции изомеризации дейтерированных аллиловых спиртов.
В связи с этим обстоятельством было предположено [188], что механизмы окисления этилена и аллилового спирта различны, хотя и отмечается, что эти реакции имеют близкие энергетические характеристики по результатам квантово-химического моделирования [182]. Кроме этих расчетов, отметим еще и одинаковые кинетические уравнения для обоих субстратов. В целом, результаты работы [188] вряд ли можно считать удовлетворительным решением проблемы механизма Вакер-окисления. Несмотря на огромный экспериментальный материал, убедительно свидетельствующий в пользу цис-ГП в механизме Вакер-окисления при низких концентрациях хлорид-иона, Коцьковский и Бэкквал в весьма интересном обзоре 2015 г. [59] продолжают настаивать на гипотезе о транс-ГП при любых концентрациях Cl– и CuCl2 и на идентичности стереохимии продуктов ГП и 1,2-окисления. Для подтверждения своих взглядов они привлекают результаты теоретических работ с использованием метода AIMD [186–188] о преимуществах транс-ГП, которые, как было видно выше, вызывают массу вопросов. Авторы обзора замечают, что не видят больших различий в системах с [Cl–] = 1 M и [Cl–] = 3 M. С этим можно согласиться, но при [LiCl–] = 1 M скорость Вакер-процесса близка к нулю. Если сравнить системы с [Cl–] = 0.2 M и с [Cl–] = 3 M, то скорость окисления при увеличении концентрации хлорид-иона уменьшается в 225 раз (!). При этом, естественно, остается только вариант транс-ГП со стереохимией этиленхлоргидрина, блестяще установленной Бэкквалом и соавт. [88–93].
В связи с изложенными результатами теоретических исследований последних 20 лет в области механизмов Вакер-окисления возникает вопрос, в какой мере результаты моделирования механизмов таких сложных реакций, как окисление олефинов, на современном уровне методологии теоретических расчетов металлокомплексов в растворах могут служить методом дискриминации гипотез о механизмах реакций? Как следует из приведенного выше анализа, исследователи приходят к значениям барьеров, близким (и ниже) к наблюдаемым экспериментально, при использовании различных допущений и подходов. Так, например, применяют: 1) различные наборы стадий, 2) отличающиеся по природе интермедиаты, 3) разные методы расчетов (DFT, AIMD), 4) различные способы учета сольватации и наличия ионных пар и, наконец, 5) заметно различающиеся количества участвующих в стадиях процесса молекул воды (1, 3 и 26)!
4. РЕАКЦИИ ОКИСЛЕНИЯ ОЛЕФИНОВ В БИНАРНЫХ ВОДНО-ОРГАНИЧЕСКИХ РАСТВОРИТЕЛЯХ С УЧАСТИЕМ РАЗЛИЧНЫХ ОКИСЛИТЕЛЕЙ И ЛИГАНДОВ
Поскольку высшие алифатические олефины и циклоолефины, а также олефины с функциональными группами практически не растворимы в водных растворах хлоридных анионных комплексов палладия, исследователи процессов Вакер-окисления еще на ранних этапах изучения этой реакции в 1964 г. [190] начали применять бинарные водно-органические растворители с лучшими результатами в системе DМF–Н2О. Приведем лишь часть исследований, посвященных использованию бинарных водно-органических растворителей в хлоридных каталитических системах Вакер-окисления. В результате изучения различных органических компонентов бинарных растворителей (DМF [191–198], AN [188, 190, 197], N-метилпирролидон (NMP) [191, 195, 197, 199], DМSО [190, 193, 195, 200, 201], DМА [197–199, 202], ROH [197, 200, 203–205], сульфолан [191], этиленкарбонат [199]) установлено, что ряды активности, обусловленные природой органических компонентов растворителя, в таких системах определяются составом каталитической системы (комплекс палладия, со-катализатор, мольная доля органического растворителя), природой олефина и условиями процесса. В растворах хлоридных комплексов палладия в первичных и вторичных спиртах даже при небольшом содержании воды (до 5 мас. %) с высокой скоростью и селективностью происходит каталитическое окисление олефинов до альдегидов и кетонов хлоридом меди(II) при незначительном образовании ацеталей и кеталей [1, 203]. Использовались и другие комбинации полярных растворителей с водой (ТHF, ацетон, диоксан, диглим, 1,2-диметоксиэтан (1,2-DME) [193]), а также АсОН, СН3NO2 и даже неполярные или малополярные растворители дихлорэтан и дихлорметан (с применением комплексов палладия с различными лигандами). Вероятно, первое кинетическое исследование реакции окисления октена-1 п-бензохиноном в бинарных растворителях было выполнено с катализатором PdCl2(PhCN)2 [193]. Особенно важную роль органические растворители сыграли в создании каталитических систем на основе катионных комплексов палладия в реакциях окисления высших и циклоолефинов, выполняя роль лигандов, стабилизирующих комплексы Pd(0) и Pd(I) (см. разд. 6).
Рассмотрим на примере бинарного растворителя DMF–H2O, какие функции выполняет растворитель в стандартной каталитической системе PdCl2–CuCl2 в отсутствие О2 и в его присутствии. В работе [206] описаны реакции комплексов PdCl2(DMF)2 с α‑олефинами в DMF с образованием η3-аллильных мономерных и димерных комплексов Pd(η3-C3H4R)(DMF)Cl и [Pd(η3-C3H4R)(DMF)Cl]2. В ходе этих реакций образуется и комплекс ${{\left[ {({\text{DMF}}){{{\text{H}}}^{ + }}} \right]}_{2}}\left[ {{\text{P}}{{{\text{d}}}_{{\text{2}}}}{\text{Cl}}_{{\text{6}}}^{{{\text{2}} - }}} \right].$ Аналогично ведет себя и диметилацетамид (DМА). Способность DMF взаимодействовать карбонильным кислородом с протоном кислоты приводит (например, в случае НCl) к образованию трех комплексов состава (DMF)2HCl, (DMF)HCl и [DMFH+]${\text{[HCl}}_{{\text{2}}}^{ - }]$ [207 ] . Структура комплекса (DMF)HCl была установлена методом рентгеноструктурного анализа (РСА) [208]. Особенно интересны биметаллические комплексы, содержащие PdCl2 и CuCl2, образующиеся в присутствии циклического амида – пирролидона – в качестве лиганда (L) в СН2Cl2 [209]. Реакция комплекса PdCl2(AN)2 с CuCl2 в избытке L приводит к образованию полимерного комплекса состава [CuCl2(L)4(PdCl2)2]n (A), содержащего, по данным РСА, октаэдрические комплексы CuL4Cl2, связанные хлоридными мостиками с димером Pd2Cl4. Как было показано, комплекс (А) катализирует в растворителе МеОН–DCE (DCE = 1,2-дихлорэтан) при 50°С окисление циклогексена кислородом до циклогексанона и циклогексенона. В амидных растворителях изучали также поведение каталитической системы PdCl2–CuCl, использованной, по-видимому, впервые в работах [193, 194]. Оказалось, что PdCl2(AN)2 и CuCl в 1,2-дихлорэтане в присутствии небольших количеств гексаметилфосфортриамида (НМРА = = L) и О2 превращаются в два комплекса – комплекс типа (А) [CuCl2(L)2(PdCl2)2] и комплекс типа (Б) Pd6Cu4Cl12O4L4 [210]. Предполагается, что в реакции CuCl с О2 и лигандом L образуется μ-пероксокомплекс Cu(I) (LVII)
(LVII)
$4{\text{CuCl}} + {{{\text{O}}}_{2}} + 2{\text{L}} \to 2{\text{CuC}}{{{\text{l}}}_{2}} + {\text{LCuOOCuL}}.$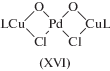
который содержит 2 частицы Cu=O и PdCl2, т.е. 2 атома Cu(II) и атом Pd(II) или 2 атома Cu(I) и атом Pd(IV). Из двух комплексов (XVI) и 4 молекул PdCl2 и получается комплекс (Б) (данные РСА) со структурой, содержащей 4 атома О, связанных с атомом Cu и тремя атомами Pd (μ4-О). Каждый из 4 таких атомов кислорода легко и количественно переносится на децен-1 при 50°С в аргоне с образованием кетона с выходом >99%. Аналогичная картина наблюдается и в DMF [196]. Полученный μ4-оксокомплекс типа (Б) также окисляет децен-1 с выходом кетона ~90%. Диметилформамидный комплекс типа (А) катализирует окисление децена-1 кислородом в DMF с небольшим содержанием воды. По мнению авторов [196], этот результат свидетельствует о процессе окисления с участием гетерометаллического комплекса без изменения степени окисления палладия в ходе процесса. Равновесия (LVIII)
зависят от природы Х, растворителя, лигандов и температуры [211], а степень окисления меди в этих соединениях меняется при переходе от ${\text{Cu}}_{{\text{2}}}^{{{\text{II}}}}$ (μ-O2) и ${\text{Cu}}_{{\text{2}}}^{{{\text{II}}}}$ (μ-η2, η2-O2) к ${\text{Cu}}_{{\text{2}}}^{{{\text{III}}}}$ (μ-O)2. В случае сильнооснóвных лигандов (например, аниона ${\text{Bu}}_{{\text{2}}}^{t}{\text{P}}({\text{NSiM}}{{{\text{e}}}_{{\text{3}}}})_{{\text{2}}}^{ - }$) этиленовый π-комплекс Cu(I) окисляется О2 с образованием устойчивого комплекса L2Cu2 (μ-O)2 с расстоянием Cu–Cu, равным 2.9 Å [212].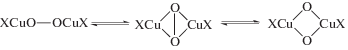
Система PdCl2–CuCl с кислородом в качестве окислителя в бинарных растворителях весьма эффективно применялась в работах Дз. Цудзи, и оказалась настолько удобной и селективно окисляющей огромное число органических соединений, содержащих двойную связь [195 и др.], что реакции окисления олефинов стали называть Wacker–Tsuji-oxidation [19, 21, 22, 202, 213, 214].
Помимо классического Вакер-окисления с хлоридными комплексами палладия, используют Pd(ОАс)2 в бинарных растворителях (с различными добавками) [14, 204, 215–223] и катионные и дикатионные комплексы палладия (см. разд. 6).
4.1. Различные окислители и со-катализаторы
По мере развития исследований каталитических систем для реакций окисления олефинов использовали различные со-катализаторы с конечным окислителем О2, способные, как и CuCl2, окислять олефины в присутствии палладия и без кислорода, или уже упоминавшийся выше со-катализатор CuCl, активный только в присутствии кислорода [192, 194, 196, 203, 210]. Ацетат меди также применяли в качестве со-катализатора в хлоридных [183] и в ацетатных системах [216, 224] и как окислитель [218] в системе Pd(OAc)2–Cu(OAc)2–DMSO–H2O. Различные неорганические соединения эффективно работают как окислители в различных бинарных растворителях с PdCl2 (например, Fe2(SO4)3 [198] и CrO3 [225] в системе AN–H2O), а также KBrO3 в системе THF–H2O [226], но с твердым катализатором Pd/С.
Весьма интересным компонентом (окислителем и лигандом) хлоридных палладиевых систем оказался нитрит-ион ${\text{NO}}_{{\text{2}}}^{ - }{\text{.}}$ Еще в работе Смидта и Иры 1960 г. [227] было показано, что в водном растворе PdCl2 и NaNO2 олефины окисляются до кетонов с образованием нерастворимого олигомера [Pd(NO)Cl]n. Позднее в серии работ М. Эндрюса и сотр. [228, 231, 233–235], А. Хоймана с сотр. [229, 230] и других исследователей [232] была обнаружена способность нитритных комплексов палладия катализировать окисление олефинов кислородом до кетонов и эпоксидов. Было показано, что комплекс Pd(AN)2Cl(NO2) в толуоле является катализатором окисления, а образующийся олигомер [Pd(NO)Cl]n окисляется кислородом до исходного нитритного комплекса. В работе [228] было предположено (по данным ИК- и ЯМР-спектров) образование металлагетероциклического интермедиата – гетеропалладациклопентана (XVII), который был позднее выделен и исследован [231]. Способность нитритных комплексов катализировать эпоксидирование кислородом напряженных циклических олефинов установлена и обсуждается в публикациях [229–232].
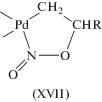
Влияние условий и природы олефина на процессы окисления, а также варианты механизмов, включая изотопные исследования, обсуждаются в работах [233–235]. Нитритные комплексы палладия оказались активными в ряде систем, катализирующих анти-М-окисление до альдегидов [15, см. разд. 5].
Другими окислителями, которые в определенных условиях могут стать и со-катализаторами процесса окисления, являются гетерополикислоты (ГПК), содержащие атомы V, Mo или W [236]. Начало использованию фосфомолибдованадатов в окислении олефинов в водных растворах катионных комплексов палладия было положено в работах [237, 238], которые будут проанализированы в разд. 5. Здесь же рассмотрим применение ГПК в хлоридных системах [239, 240]. Еще в первой работе 1977 г. [237] было установлено, что ГПK состава H6[PMo6V6O40] (Кеггин-тип полиоксометаллатов) окисляет восстановленные формы Pd(II) (например, Pd(0)) в результате перехода ванадия(V) в ванадий(IV), однако скорость окисления была невысока и понижалась со временем в связи частичным образованием палладийсодержащих наночастиц.
Использование добавок хлорид-ионов в процессе окисления этилена фирмы “Catalytica Inc.” [239] ([Cl–]/[Pd] = 50–250) привело к созданию устойчивой каталитической системы ${\text{PdCl}}_{n}^{{{\text{2}} - n}}$–ГПК (Na3H3[PMo9V3O40]). Процесс проводят в двух реакторах. В первом окисляется этилен гетерополикислотой со скоростью 8.1 ммоль С2Н4 л–1 с–1 и TOF = 81 c–1. В условиях, близких к условиям Вакер-процесса, скорость не зависит от концентрации ванадия(V) или ГПК и от количества атомов ванадия (от 1 до 4) в ГПК. Во втором реакторе скорость окисления ванадия(IV) кислородом лимитируется диффузией кислорода в реакционный раствор.
В работе [240] была изучена кинетика окисления этилена и бутена-1 кислородом, т.е. в условиях катализа хлоридными комплексами палладия и ГПК в стационарных условиях проточного по газу реактора с интенсивным перемешиванием фаз, исключающим диффузионное торможение процесса. Была использована каталитическая система, близкая по составу к системе фирмы “Catalytica Inc.”: [Cl–]/[Pd] = 50, рН ~ 1.3 и ГПК состава Na4H2[PMo9V3O40]. При 20–50°С установлен первый порядок по [Pd(II)] и нулевой по [ГПК] (при [Pd(II)] = const). Число оборотов палладиевого катализатора составляет 750–1500, а ГПК (по ванадию) – 1–2. Скорость поглощения олефинов примерно в 2 раза выше скорости поглощения кислорода во всех опытах в стационарных условиях, т.е. выполняется стехиометрия реакции окисления, хотя в случае этилена и бутена-1 скорость поглощения кислорода незначительно выше стехиометрической за счет побочных реакций окисления. Реакционная способность олефинов меняется в ряду С2Н4 ≤ 1-С4Н8 < С3Н6. Заметим, что нулевой порядок по концентрации ГПК в стационарных условиях одностадийного и в случае двухстадийного процессов может объясняться большой скоростью окисления паллладийорганических интермедиатов (см. разд. 2). Отметим также, что реокисление ГПК в соседнем реакторе (или в соседнем городе) не делает этот стехиометрический окислитель катализатором процесса, хотя такой термин часто встречается в публикациях.
К другим стехиометрическим окислителям в процессах окисления олефинов относятся “гипервалентные” соединения иода (HIO4 [241], органические периодинаны [223]), Н2О2 [153, 215, 242–245], ROOH [242–246] и, конечно, кислород с со-катализаторами и без них [14, 16–18, 22]. Окисление тетрадецена-1 в системе Pd(OAc)2–AN–H2O при 50°С периодинаном Десса–Мартина (DMP, степень окисления иода +III)
приводит к образованию α-кетона с выходом 95% (исследовано 62 олефина с различными заместителями [223]).
4.2. Пероксид водорода и ROOH – стехиометрические окислители
Пероксид водорода в качестве окислителя в процессах окисления олефинов впервые был применен Моисеевым и сотр. [1, 34]. В работе Цудзи [242] было показано, что интернальные олефины с функциональными группами, стиролы и α-олефины хорошо окисляются пероксидом водорода и трет-бутилгидропероксидом в системах АсОН, iPrOH, NMP, Н2О. В случае Н2О2 скорость окисления выше, чем в системе PdCl2–CuCl, но в реакциях α-олефинов наблюдается позиционная изомеризация олефина. Замещенные интернальные олефины окисляются tBuOOH с выходами 80–96% без изомеризации. Как было установлено, кетоны образуются не через эпоксиды. Значительный вклад в развитие процессов окисления олефинов пероксидами (Н2О2, ROOH), катализируемых комплексами Pd(II), внесли работы А. Мимуна и сотр. [215, 222–224]. Синтез и исследование строения и реакционной способности тетрамерного комплекса [RCO2PdOOtBu]4 [243] показали, что этот комплекс окисляет олефины до кетонов [215, 243] с образованием псевдопалладациклического интермедиата, распад которого приводит к образованию RCO2Pd(OtBu) и α‑метилкетона с переносом атома Н (или D) из группы СН (или СD) в метильную группу кетона. Регенерация алкилпероксидного комплекса происходит в реакции RCO2Pd(OtBu) с tBuООН. Детали механизма обсуждаются в работах [244, 245]. Работа [215] посвящена окислению олефинов пероксидом водорода. При 25–80°С в tBuОН или в АсОН с Pd(OAc)2 терминальные олефины окисляются селективно в избытке Н2О2 с частичным разложением пероксида, катализируемым палладием. Окисление в tBuОН с ${\text{Н}}_{{\text{2}}}^{{{\text{16}}}}{{{\text{О}}}_{{\text{2}}}}$ в избытке ${\text{Н}}_{{\text{2}}}^{{{\text{18}}}}{\text{О}}$ привело к образованию кетона, содержащего только 16О, т.е. к реализации “невакеровского” механизма окисления, и показало, что роль пероксида в этой системе заключается не в регенерации Pd(II). Предположено, что интермедиат аналогичен таковому в механизме с участием tBuООН. Образование промежуточного гидропероксидного комплекса [Pd]OOН происходит и в реакции L2PdO2 с CH3SO3H, причем L2Pd(OOН)+ окисляет олефин до кетона с выходом 50% на [Pd]∑ [244] (см. также обзор [14]). В случае стирола обмен Hg(II) в соответствующем ртутьорганическом псевдометаллацикле с ${\text{PdCl}}_{{\text{4}}}^{{{\text{2}} - }}$ приводит к образованию ацетофенона и HgCl2 [243, 244]. Окисление терпенов пероксидом водорода в системе Pd(OAc)2–АсОН было детально изучено в работах Гусевской и сотр. [153].
В еще одной важной работе, связанной с использованием tBuООН в качестве стехиометрического окислителя [246], в качестве лиганда применяли (-)sparteine и было показано, что комплекс Pd[(-)sparteine]Cl2 в метаноле при 35°С катализирует окисление виниларенов до кетонов с выходом >90% и децена-1 в DMA или NMP с Н2О при 70°С до деканона-2 с выходом 90–95% без изомеризации децена-1.
4.3. Кислород – стехиометрический окислитель без со-катализаторов
Использование О2 без со-катализаторов в системах с Pd(II) или Pd(0) давно считается важнейшей задачей в развитии процессов окисления олефинов (см. обзоры [14, 16–18, 22]). Первые работы по использованию О2 как единственного окислителя были, по-видимому, проведены при открытии и изучении реакций окислительной внутримолекулярной циклизации аллилфенолов с образованием гетероциклов Хосокавой и сотр. [216, 247–253] и другими исследователями [254]. Эти реакции (например, (LVIII)), протекают по “вакеровскому” типу механизмов:
Изомерные бензофураны получаются в системе Pd(OAc)2–МеОН–Н2О при атмосферном давлении кислорода и 25°С по реакции (LIX) [252] (один из изомеров):
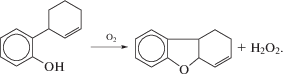
Внутримолекулярное фенокси- и карбоксиаминопалладирование субстрата, содержащего связь С=С, позволяет получать труднодоступные гетероциклы простым методом окислительной циклизации [195, 218, 219, 255–258]. Так, в работе [255] было установлено, что кислород окисляет 2-аллилбензойную кислоту и циклопентен-3-уксусную кислоту до лактонов в растворах Pd(OAc)2 в DMSO c выходом 86%. Окислительную циклизацию проводят также с участием со-катализаторов. Так, например, при проведении реакции синтеза фурановых производных (LX) [216] в системе Pd(OAc)2–Cu(OAc)2–MeOH(THF) O2 c хиральным пиненом
(или с комплексом палладия с η3-пиненовым лигандом L*) впервые получили хиральный винилдигидробензофуран по реакции окислительной циклизации и показали также, что Cu(OAc)2 участвует в процессе в составе гетерометаллического комплекса с палладием на этапе окисления гидридного комплекса палладия (XVIII).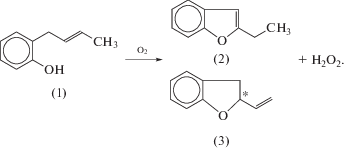
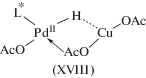
Прямое окисление олефинов до кетонов кислородом удалось успешно реализовать при использовании водорастворимых бидентатных фенантролиновых и дипиридильных лигандов в двухфазных системах (олефин–вода) при 100°С и 30 бар О2 [221, 222] в присутствии Pd(OAc)2 и NaOAc. Лучшие показатели в указанных условиях получены с батофенантролиндисульфонатом в реакции окисления α-олефинов и циклоолефинов. При использовании PdCl2 со спартеином [246] кислород также окисляет децен-1 при 70°С в системе DMA–H2O, но при высоком давлении. В работе [197] окисление децена-1 эффективно происходит в растворе PdCl2 в DMA–H2O при 80°С с выходом кетона 84%. Исследовано 13 различных олефинов с выходами, по данным газо-жидкостной хроматографии, 74–95%. В этиленкарбонате (PdCl2–NaOAc, 60°С и 10 бар О2) децен-1 окисляется до метилоктилкетона с выходом 74% и селективностью 95%. Другие α- и арилолефины селективно окисляются в этих же условиях, но с невысокой скоростью [199]. Стабилизация Pd(0) в форме палладиевых наночастиц этиленкарбонатом позволяет успешно проводить стадию регенерации катализатора кислородом. Удачным растворителем оказался и DMSO [201]. Использование Pd(OAc)2 в DMSO в присутствии трифторуксусной кислоты, т.е., по существу, Pd(OСОСF3)2 при 70°С, позволяет окислять стирол кислородом с выходом ацетофенона 86%. Для 14 изученных производных стирола и других функционально замещенных олефинов было установлено, что в случае гомоаллильных производных аренов и ряда других олефинов окисление до α‑кетонов сопровождается окислительным дегидрированием с образованием α,β-ненасыщенных кетонов с выходом 50–87%. Очень интересно, что комплексы палладия(II) в DMSO и в DMF [259, 260] окисляют альдегиды, кетоны, амиды и эфиры стехиометрически до α,β-ненасыщенных карбонильных соединений в присутствии оснований в очень мягких условиях (не выше 80°С), а также катализируют реакции окислительного дегидрирования этих же соединений с участием окислителей (tBuOH, O2, ГПК + O2).
Окисление кислородом олефинов может быть организовано и как сопряженный процесс окисления спирта и олефина в системе ROH–толуол–Pd(OAc)2 с добавлением пиридина (комплекс [Pd(OAc)2Py2]) при 60°С и высоком давлении О2 [204]. Суть процесса заключается в образовании интермедиата [Pd]OOH в результате окисления спирта (изопропанол → ацетон) и участии этого интермедиата в окислении олефина по типичному для пероксидных окислителей механизму (см. выше). Таким образом, в изопропаноле процесс протекает по уравнению (LXI)
(LXI)
$\begin{gathered} {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{4}} + {{{\text{(C}}{{{\text{H}}}_{{\text{3}}}}{\text{)}}}_{{\text{2}}}}{\text{CHOH}} + {{{\text{O}}}_{2}} \to \\ \to {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHO}} + {{({\text{C}}{{{\text{H}}}_{3}})}_{2}}{\text{CO}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}. \\ \end{gathered} $По аналогии с механизмами ферментативных реакций можно рассмотреть различные варианты механизмов окисления олефинов кислородом без участия со-катализаторов (М).
М1. Молекулярный кислород не участвует в переносе атома О на субстрат. Роль О2 заключается в окислении восстановленных форм катализатора-окислителя. В случае Вакер-окисления О2 является акцептором двух электронов и двух протонов с образованием Н2О2 (или Н2О) при окислении Pd(0) или [Pd2(I)]. Такая функция О2 реализуется в оксидазных процессах [14].
М2. Атом кислорода переносится на субстрат из молекулы О2. В этом случае можно рассмотреть два типа механизмов – “монооксигеназный” и “пероксидазный”.
Монооксигеназный механизм представлен на схеме 14 [13, 244, 245, 261]. Если субстратом (S2) является C2H4, итоговое уравнение маршрута, вытекающее из механизма, опишется стехиометрическим уравнением реакции
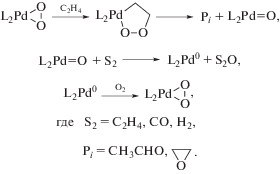
Схема 14 .
Пероксидазные механизмы (схема 15 а–15в). Основной чертой этих механизмов является участие гидропероксидного комплекса катализатора (палладия) с переносом атома кислорода на субстрат в стадиях образования продукта реакции Pi (например, СН3СНО).
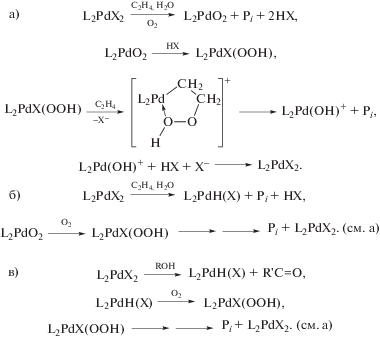
Схема 15 .
Вариант механизма (а) отличается от механизмов (б) и (в) способом образования пероксидного интермедиата, обоснованного в работах [175, 176]. В механизме (б) интермедиат [Pd]OOH образуется в рамках гипотезы (Б) о механизме превращений σ-палладоэтанола (разд. 3.5), например, в синтезе винилацетата. Если интермедиат L2PdX(OOH) вступит в реакцию с НХ, получится Н2О2 и реализуется оксидазный механизм окисления (М1). Механизм (в) реакции (LX) – сопряженного окисления кислородом спирта и олефина – несомненно относится к пероксидазным процессам, но в отличие от механизма (б) образование L2PdX(OOH) происходит за счет дополнительного восстановителя – спирта. Таким восстановителем в сопряженных процессах может служить и молекула СО [262, 263] и молекула Н2 [13].
5. РЕГИОСЕЛЕКТИВНОСТЬ РЕАКЦИЙ ОКИСЛЕНИЯ ОЛЕФИНОВ
Уже первые исследования региоселективности (РС) реакции окисления олефинов тетрахлоропалладат-анионом в 1962 г. [53] показали, что при стехиометрическом окислении в водном растворе, содержащем 1 М HClO4, при 70°С, наряду с кетонами (по Марковникову, М), образуются и альдегиды (анти-М). Выходы альдегидов из С3Н6, пентена-1 и стирола составляют 15, 20 и 75% соответственно. Увеличение концентраций кислоты и Cl– повышает выход пропионового альдегида с 7 до 20% (при [HCl] до 1 М) и с 4 до 17.5% (при [HClO4] до 1.85 М).
Более детально было исследовано каталитическое окисление стирола и замещенных стиролов п-бензохиноном в THF–Н2О [54, 55] и в воде [56–58] (разд. 3). Как было установлено [55–58], образование арилуксусных альдегидов (анти-М) происходит с выходом 75–90%, причем отношения количеств альдегида и кетона и скоростей Ra/Rк увеличиваются с ростом [Cl–] и [H3O+], т.е. РС определяется кинетикой реакций и механизмом гидроксипалладирования, если ГП является лимитирующей стадией.
В дальнейшем было показано, что высокая РС реакции образования альдегидов наблюдается и при окислении неактивированных α-олефинов в водно-органических средах. Обнаружено также влияние природы окислителя и механизма реакции на РС окисления в таких средах. Данные о РС процессов окисления, полученные на первом этапе исследований, проанализированы в монографии Моисеева [1].
После установления возможности окисления олефинов нитритными комплексами Pd(II) [227] в работах [228–235] было изучено окисление олефинов до кетонов и циклоолефинов до кетонов и эпоксидов кислородом в растворах Pd(CH3CN)2Cl(NO2). При этом использование катионного нитритного комплекса увеличивает скорость окисления октена-1 (по Марковникову) в 10–20 раз [230]. Добавление к нитритному комплексу Pd(II) трет-бутанола (tBuOH) и CuCl2 позволило при окислении децена-1 кислородом повысить отношение альдегид/кетон с 60/40 до 70/30 [264]. При окислении стирола выход альдегида составил 100%. В отсутствие CuCl2 или при использовании изопропилового спирта вместо tBuOH образуются кетоны с выходом 100%. В этой и в последующих работах [265–267] авторы придерживались гипотезы Эндрюса [231, 234, 235] об образовании циклического интермедиата с участием нитритного лиганда в качестве О-содержащего нуклеофила, считая, что анти-М-региоселективность определяется особыми свойствами tBuOH как лиганда, а роль CuCl2 заключается в образовании гетерометаллического комплекса с PdCl2. По мнению авторов работы [266], образование комплекса Pd(NO2)Cl c β‑гидроксиамидом Me2C(OH)CH2C(O)NMe2 (XIX), активного в окислении олефинов, подтверждает роль трет-бутанола в качестве лиганда.
Было показано [267], что нитритный комплекс палладия в спиртах (трет-бутаноле и изопропаноле) катализирует в присутствии кислорода образование алкоксифуранов из гомоаллиловых спиртов. В дальнейшем участие нитро- (Pd–NO2) и нитритных (Pd–ONO) лигандов в стадиях оксипалладирования и N-палладирования олефинов было подробно исследовано на примере реакций 1,1- и 1,2-окисления олефинов [152, 153].
Исследование Т. Венцелем [264–267] упомянутой каталитической системы показало [268], однако, что лиганд-окислитель ${\text{NO}}_{{\text{2}}}^{ - }$ может быть исключен из системы и что активной в процессе анти-М-окисления является система Pd(CH3CN)2Cl2–CuCl–LiCl–tBuOH. При 60°С из октена-1 в результате окисления кислородом образуется 66% альдегидов, а из аллилацетата – 86%. Отмечена ключевая роль tBuOH в появлении анти-М-продуктов. Предполагаемое образование алкенил-трет-бутилового эфира и его гидролиз согласуются с составом продуктов окисления аллилового спирта [264] (70% tBuOCH2CH2CHO и 30% tBuOCH=CHCH2OH) и образованием циклических трет-бутоксиацеталей [267] при окислении гомоаллиловых спиртов в растворах tBuOH с выходом до 100%. Окисление аллиловых эфиров, вероятно, происходит в результате анти-М-окисления субстрата до трет-бутилвинилового эфира, превращающегося в циклический алкоксиацеталь. Механизм образования альдегидов из аллиловых эфиров приведен на схеме 16 [268].
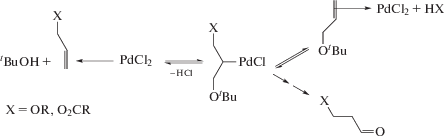
Схема 16 .
Предполагаемая схема превращений олефинов в растворах tBuOH на примере аллилацетата в системе PdCl2–CuCl–RCN–tBuOH–O2 [268] приведена и в обзоре [15]. Отношение альдегид/кетон в растворе MeCN равно 6. Атака трет-бутиловым спиртом π-комплекса Pd(II) по терминальному атому углерода (анти-М) может объясняться большей мягкостью этого нуклеофила по сравнению с H2O и стерическими затруднениями для цис- и транс-алкоксипалладирования по Марковникову.
При изучении окисления β-замещенных стиролов [269, 270] и замещенных в фенильной группе стиролов [150] было установлено, что заместители в кольце сильно влияют на РС окисления. Был сделан вывод, что образование η4-комплекса стирола с Pd(II) (X) и, возможно, η3-аллильного интермедиата (XI) способствует анти-М-региоселективности [271] реакций окисления стиролов (схема 17 ).
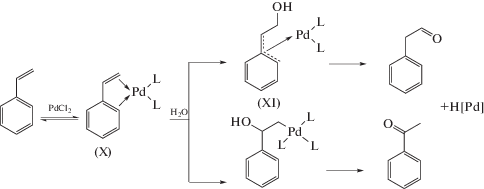
Схема 17 .
В работе [271] было сделано очень интересное наблюдение о влиянии природы окислителя на РС. При изучении окисления п-метоксистирола кислородом в системе PdCl2–CuCl–DМF–H2O в условиях стехиометрического окисления стирола хлоридом палладия (в отсутствие CuCl и O2) отношение альдегид/кетон nа/к= 9.8, а в присутствии CuCl и O2 – nа/к = 0.12.
При замене PdCl2 на Pd(OAc)2 образуется только кетон с выходом 72%. В этой же работе нашла убедительное подтверждение возможность участия окислителя (или со-окислителя) в стадиях, определяющих РС реакции, в водно-органических средах. Так, замена системы CuCl–O2 в качестве окислителя стирола на п-бензохинон приводит к nа/к = 0.09, а окисление гетерополикислотой H4PMo11VO40 дает nа/к = 6.4. Таким образом, окислитель в этих средах взаимодействует с активной формой комплекса палладия и может, по существу, изменить механизм реакции и РС. Влияние окислителя (CuCl2, CuCl–O2, п-хиноны, ГПК и O2) на РС означает, что в стадиях образования карбонильных групп в реакциях окисления, помимо нуклеофилов НО– и Н2О и стадий гидрокси(окси)палладирования (син-, анти-), могут участвовать и другие источники атомов кислорода (–PdOOH и –CuOPd–). В результате появляется возможность реализации неклассических механизмов окисления (“невакеровские” механизмы) с включением окислителя в активные металлосодержащие интермедиаты без изменения степени окисления металла в ходе процесса (см. обзор [15]).
Анализ реакций синтеза альдегидов путем окисления замещенных олефинов (которые содержат гетероатомы O и N в β- и γ-позициях по отношению к винильной группе или двойные связи, способные дополнительно координировать Pd(II), образующий π-комплекс с винильной группой за счет эффекта хелатирования) позволил сформулировать [15] вывод о том, что такое хелатирование способствует гидрокси- или оксипалладированию с анти-М-ориентацией. Так, например, при стехиометрическом окислении аллилового спирта комплексом Li2PdCl4 [107, 108] среди продуктов окисления были обнаружены HOCH2CH2CHO (40%) и CH3C(O)CH2OH (12–15%).
Таким образом, региоселективность гидроксипалладирования третичных аллиловых и гомоаллиловых спиртов и их эфиров зависит от природы заместителей при атоме углерода С3 (субстрата), от состава каталитической системы (катализатора) и от условий, меняющих механизм реакции [15]. Многочисленные примеры приведены в обзорах [15, 272]. Интересным примером являются реакции окисления несопряженных диенов. Так, из 4‑винилциклогексена-1 и нециклических 1,5-диенов, в которых возможна дополнительная π-координация С=С-связи, образуются альдегиды [205, 273]. Примером является реакция (LXII) [273]. Образование альдегидов в реакциях 1,5-диенов и других замещенных α-олефинов связывают с цис-гидроксипалладированием кратной связи [15]. Полагают, что в случае транс(анти)-присоединения нуклеофила существенно повышается вероятность образования продукта окисления по Марковникову.
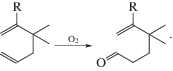
Вместе с тем, в реакции окисления стирола в системе PdCl2–BQ, как было отмечено выше [57, 58], наблюдается увеличение отношения альдегид/кетон в условиях, способствующих транс-присоединению воды. Поэтому пока не существует экспериментальных оснований связывать региоселективность со способом присоединения Nu (NuH). При окислении α-олефинов кислородом под давлением в отсутствие со-окислителей всегда происходит образование кетонов [274]. Это наблюдалось в системах LPd(OAc)2–H2O (L = производные фенантралина) [221], PdCl2–DМА–H2O [197] и LPdCl2–DМА–H2O (L = (–)-спартеин) [246].
В обзоре [15] содержится анализ региоселективности реакций окисления олефинов в спиртовых растворах или внутримолекулярного окислительного алкоксилирования. Направление присоединения нуклеофила OR с образованием ацеталей или циклических ацеталей (анти-М), как и в случае воды, определяется α-, β- и γ-гетероатомами, входящими в состав заместителей. Влияние природы лигандов, металла и окислителя на РС процесса обнаружено и в реакциях окислительного аминирования на стадиях амино(амидо)палладирования [15].
Важная роль и заместителей, и каталитических систем в управлении РС продемонстрирована в работе Б.Л. Феринги и соавт. [275] на примере изучения РС реакции окисления кислородом замещенных аллиламинов CH2=CНСН(R1)NR2R3, включая замещение и фтальимидом (ФИ) в положении С3. Использовали три каталитические системы: А – Pd(AN)2Cl(NO2)–CuCl2–tBuOH, B – PdCl2–CuCl–DMF–H2O, C – PdCl2–CuCl2–DMF–H2O. Приведем несколько примеров.
1). R1 = Ph, R2 = Ts, R3 = H. Система A, nа/к $ \cong $ 1; система B, nа/к = 3/97.
2). R1 = Ме, амин = ФИ. Для трех каталитических систем выходы альдегида близки и составляют >99% (B), 96% (А) и 94% (С).
3). Аллилфтальимид с различными заместителями R1 (7 соединений). Для систем А и В nа/к = 99/1.
Исследованная реакция с ФИ и с асимметрическим центром С3 позволяет, по данным авторов, получать хиральные аминокислоты через хиральные альдегиды.
Дальнейшие усовершенствования каталитических систем были предложены Р. Граббсом с соавт. [213, 276–278]. Разрабатывая полифункциональную каталитическую систему (ПФКС) для синтеза спиртов нормального строения (анти-М-гидратация олефинов) [279], авторы установили, что каталитическая система Pd(AN)2Cl2–CuCl2 в смеси tBuOH–Н2О–iPrOH с добавками BQ позволяет получать анти-М-интермедиат RCH=CHOtBu, который гидролизуется с образованием соответствующего альдегида. Восстановление альдегида добавленным изопропиловым спиртом в присутствии рутениевых комплексов приводит к н-спиртам с высокой селективностью. Исследования этой каталитической системы для улучшения анти-М-РС процессов окисления олефинов [276] показали, что в системе Pd(AN)2Cl2–BQ–tBuOH–Н2О стирол и его производные с электронодонорными и электроноакцепторными заместителями окисляются с селективностью 96–99% до альдегидов.
Для создания систем, способных катализировать окисление кислородом α-олефинов без функциональных групп или имеющих удаленные от винильной группы функциональные группы (“unbiased”) [277], авторы вернулись к нитритным системам, т.е. к системе Pd(AN)2Cl2–CuCl2–tBuOH в нитрометане с добавками различных источников нитритных окислителей. Оказалось, что использование tBuONO в качестве со-окислителя додецена-1 не приводит к существенному повышению РС по альдегиду (но не более 60%). Лучшие результаты получаются при использовании таких источников ${\text{NO}}_{{\text{2}}}^{ - }{\text{,}}$ как AgNO2 и NaNO2, когда селективность по альдегиду возрастает до 80%.
Использование AgNO2 оказалось удачным и для окисления олефинов типа СН2= СН(СН2)m–X. Для X, равных NO2 (m = 3), CO2Me (m = 8), CO2H (m = 3), Br (m = 6) и Ph (m = 2), РС по альдегиду достигает 70–80%. И только для Х = ФИ и m = 2 селективность по альдегиду составляет 90%. В случае же системы PdCl2–CuCl–DMF–H2O селективность по альдегиду при Х = ФИ и m = 2 достигает всего 15%. Применение разработанной системы [277] в реакции окисления β-замещенных олефинов с различными функциональными группами [213] также привело к еще более высоким результатам. Селективность по альдегиду в этом случае равняется 90–94%, поскольку в контроле РС участвует не только “катализатор”, но и заместители в субстрате, дополнительно предоставляя пару электронов субстрату и тем самым превращая олефин в бидентатный лиганд. В этих работах было детально изучено и влияние стерических факторов, обусловленных группой Х. Пространственно затрудненные α- и β-замещенные олефины в реакциях образования альдегидов исследовались и в работе [278].
Рассмотренные выше методы управления региоселективностью окисления олефинов, катализируемого каталитической системой, содержащей комплексы палладия, проанализированы в обзоре Феринги с соавт. [280]. Найдены и другие пути усовершенствования и упрощения каталитических систем. В работе Я.Б. Канга и соавт. [214] было обнаружено, что tBuONO при концентрации 20 мол. % в tBuOH окисляет аллилфениловый эфир с выходом альдегида 10% и в отсутствие Pd(II). В очень простой системе Pd(PhCN)2Cl2–tBuONO–tBuOH при комнатной температуре и атмосферном давлении кислорода различные β- и γ-замещенные олефины с О- и N-гетероатомами окисляются с селективностью по альдегидам 61–81% (без добавления CuCl2, AgNO2, NaNO2 и BQ!). Начальная скорость реакции в этой системе зависит в первой степени от [Pd]∑ и [tBuOH]0 и не зависит от концентраций tBuONO (в узком интервале 6.5–9.5 М) и субстрата (также в узком интервале концентраций 0.05–0.07 М). Применение радикального ингибитора 2,4,6-три(трет-бутил)фенола привело к торможению процесса окисления, снижению выхода альдегида и к образованию эфира RONO из ингибитора и NO, источником которого, по мнению авторов, является tBuONO. Окисление NO кислородом до NO2 и создает со-окислитель для регенерации Pd(II). Авторы этой работы полагают, что предложенный в работах Эндрюса [228] и Феринги [264, 265] механизм окисления олефинов в нитритных системах с внедрением олефина по связи Pd–ONO, согласующийся с квантово-химическими расчетами моделей с участием гетерометаллических комплексов Pd(II)–Cu(II) [281], должен быть исключен из рассмотрения.
6. ОКИСЛЕНИЕ ОЛЕФИНОВ В РАСТВОРАХ КАТИОННЫХ КОМПЛЕКСОВ ПАЛЛАДИЯ
При использовании металлокомплексных катализаторов давно замечено, что переход к катионным комплексам металлов весьма значительно повышает скорость реакций. Так, в реакциях, включающих стадию цис-внедрения олефина по связи М–Н (например, в реакции гидрирования олефинов), переход от нейтральных к катионным комплексам Rh(I) [282] и Ir(I) [283] повышает частоту оборотов катализатора (TOF) в 20–100 раз. Ускорение полимеризации олефинов катионными комплексами Ti(IV), впервые обнаруженное в 1959 г. в работах А.Е. Шилова и сотр. [284, 285], наблюдалось и в случае катионных дииминных комплексов Pd(II) и Ni(II) [286]. Полимеризация 1,3-бутадиена, катализируемая π-аллильным катионным комплексом Ni(II), ускоряется в 105 раз [287] по сравнению с нейтральным комплексом. Важную роль катионных комплексов, ионных пар и природы анионов в процессах полимеризации этилена в растворах комплексов Ti(III) и Zr(IV) подтверждают также результаты квантово-химических исследований [288–291]. Даже метатезисная полимеризация с раскрытием цикла на катионных комплексах Ru(IV) ускоряется в 140 раз [292], хотя в данном случае происходит присоединение олефина к карбеновому комплексу рутения как 2 + 2-циклообразование, при котором нуклеофил RСН2 остается связанным с металлом.
6.1. Реакционная способность катионных комплексов непереходных и переходных металлов в реакциях с олефинами
Эффективная электрофильная активация алкенов такими сильными электрофилами, как ионы Hg2+ [293, 294] и ${\text{Tl}}\left( {{\text{OAc}}} \right)_{{\text{2}}}^{ + }$ [297], была обнаружена довольно давно. При взаимодействии циклогексена с нитратом ртути(II) в ацетонитриле наблюдается образование симметричного π-комплекса (“иона меркуриния”) (XX), сольватированного ацетонитрилом ртутькарбениевого иона (XXI) и продуктов амидомеркурирования (XXII) [293, 294, 296].
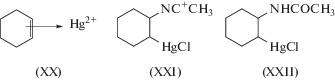
Комплекс (XX) был зафиксирован методом протонного магнитного резонанса (ПМР) в ацетонитриле [298] и в жидком SO2 при –60°C [299]. В случае катионных комплексов палладия(II) также получается продукт, аналогичный (XXII) (наши неопубликованные результаты, полученные Д.С. Захаровой). Так, комплекс Pd(AN)4(NO3)2 в реакции с циклогексеном, водой и хлорной кислотой в ацетонитриле (после исчезновения полосы поглощения при 380 нм) превращается в довольно устойчивый даже при 65°С интермедиат, который при взаимодействии с NaBH4 в растворе образует C6H11NHCOCH3 и Pd(0).
Предполагалось [295, 300], что в реакциях с участием несимметричных алкенов ион меркуриния больше соответствует металлкарбениевому иону (XXIII, МКИ)
Появление таких частиц приводит к депротонированию МКИ и появлению винилзамещенных ртутьорганических продуктов Ph2C=CHHg+. Обратимое взаимодействие перхлората ртути(II) с этиленом в водных растворах HClO4
(LXIII)
${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}} + {\text{H}}{{{\text{g}}}^{{{\text{2}} + }}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}{{ \rightleftarrows }^{ + }}{\text{HgC}}{{{\text{H}}}_{{\text{2}}}}{\text{CH}}_{{\text{2}}}^{ + }{\text{O}}{{{\text{H}}}_{{\text{2}}}}$(LXIV)
$\begin{gathered} {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{2}}}} + {\text{ArH}} \to {\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{ = CHAr}}\xrightarrow{{ArH}} \\ \to {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{CHA}}{{{\text{r}}}_{{\text{2}}}} \\ \end{gathered} $Катионные комплексы ${\text{Tl}}\left( {{\text{OAc}}} \right)_{{\text{2}}}^{ + }$ [297, 307] и Tl3+ [123, 308, 309] прекрасно окисляют этилен и другие олефины до карбонильных соединений и гликолей.
(LXV)
$\begin{gathered} {\text{Tl}}\left( {{\text{III}}} \right) + {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}\xrightarrow{{{{H}_{2}}O}}{\text{Tl}}\left( {\text{I}} \right) + {\text{C}}{{{\text{Н}}}_{{\text{3}}}}{\text{CHO}} + \\ + \,\,{\text{HOC}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}}. \\ \end{gathered} $Исследование кинетики (и изотопных эффектов) этой реакции в перхлоратных системах и в водных растворах уксусной кислоты показало близость системы к процессу Вакер-окисления. Так, изотопный состав альдегида в случае С2D4 в воде содержит более 95% дейтерия для обоих металлов, а КИЭ для С2Н4/С2D4 в случае таллия равен 0.8. Значение КИЭ, меньшее 1, объясняют большей электроотрицательностью протия по сравнению с дейтерием. КИЭ для образования альдегидов при распаде HOCHDCHDTl2+ для переносов H и D оказался равным 1.92, а для Pd(II) – 1.7 [309]. Эти результаты подтверждают существование лимитирующей стадии гидроксиметаллирования для обоих металлов [123, 309]. Отметим, что, в отличие от палладия, в случае таллия нет термодинамических ограничений при переходе Tl(III) в Tl(I) в ходе превращений σ-таллийорганических интермедиатов.
Дикатионные комплексы палладия вследствие суперэлектофильности и высокой окислительной способности вызвали большой интерес у исследователей. В работах А. Сена и Т.В. Лая [310–312] и Л. Хегедюша и соавт. [313] исследовали реакционную способность комплекса Pd(AN)4(BF4)2 (XXIV). В публикациях [310, 311] описана полимеризация олефинов и ацетиленовых соединений в ацетонитриле. Так, этилен и другие α-олефины при давлении 80 атм и 40°С превращаются в С4-, С6-, С8- и С10-олефины с величиной TON = 200, а стирол при комнатной температуре образует полистирол со средней мол. массой 7000 Дa. Суспензия (XXIV) в хлороформе катализирует изомеризацию бутенов до равновесной смеси изомеров. Замещение одной молекулы ацетонитрила (AN) в комплексе (XXIV) на PR3 в тех же условиях приводит к образованию только С4Н8 из этилена. В работе [310] было предположено прямое присоединение С2Н4 к π-комплексу
 (XXV)
(XXV)
который рассматривали как “зарождающийся” (“скрытый”) сольватированный ацетонитрилом МКИ (схема 18 ).
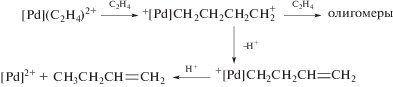
Схема 18 .
Поскольку возможность протолиза связи C–[Pd]+ маловероятна, превращения МКИ в бутен-1 можно представить как результат гидридных сдвигов по цепочке С–С-связей (LXVI):
(LXVI)
$\begin{gathered} ^{ + }\left[ {{\text{Pd}}} \right]{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{CH}}_{{\text{2}}}^{ + } \to \\ \to {{\,}^{ + }}\left[ {{\text{Pd}}} \right]{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}^{ + }}{\text{C}}{{{\text{H}}}_{{\text{3}}}} \to \\ \to {{\,}^{ + }}\left[ {{\text{Pd}}} \right]{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}^{ + }}{\text{C}}{{{\text{H}}}_{{{\text{2 }}}}}{\text{C}}{{{\text{H}}}_{{\text{3}}}} \to \\ \to {\text{ }}{{[{\text{Pd}}]}^{{{\text{2}} + }}} + {\text{1 - }}{{{\text{C}}}_{{\text{4}}}}{{{\text{H}}}_{{\text{8}}}}. \\ \end{gathered} $В работе [312] было показано, что в растворах комплексов PdLn(AN)4– n(BF4)2 при 25°С и P∑ < < 20 атм в CHCl3 и CH2Cl2 (L = PPh3) происходит сополимеризация С2Н4 и СО. Содержание СО и этилена в полимере с чередующимися мономерами составляет ~1 : 1. В спиртах образуются поликетоэфиры. Рост полимерной цепи подчиняется уравнению Шульца–Флори, описывающему цепные процессы со стадиями роста и обрыва кинетической цепи. Возможный механизм зарождения цепи в случае катионных комплексов состоит в образовании винильного производного, способного внедрять и СО и этилен, при депротонировании интермедиата (XXV):
(LXVII)
$\left[ {{\text{Pd}}} \right]{{\left( {{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}} \right)}^{{2 + }}} \to {{\,}^{ + }}\left[ {{\text{Pd}}} \right]{\text{CH}}{\kern 1pt} = {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}} + {{{\text{H}}}^{ + }}.$Как было установлено [313], катионный комплекс (XXV) алкилирует нитрилы с образованием промежуточных солей нитрилия (R–N=C–R/)Х– аналогично реакции Графа–Риттера. В качестве нуклеофилов, взаимодействующих с этими солями, выступают арены, спирты и амины с образованием различных гетероциклов и амидов (см. выше синтезы циклогексиламидов в растворах комплексов Hg2+ и Pd2+). Например, в случае аллилового спирта реакция приводит к σ-палладийорганическому интермедиату с различными вариантами образования конечных продуктов окислительно-восстановительных превращений (схема 19 ), включая оксазолиновые соединения.
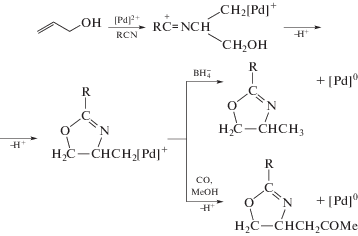
Схема 19 .
Более сложный вариант образования и превращения солей нитрилия включает участие в реакции атомов водорода ароматических соединений (например, в случае алкилирования нитрилов аллилиндолом (схема 20 )).
Схема 20 .
В статьях [314–319], посвященных синтезу и изучению реакционной способности дикатионных π‑комплексов Pd(II) [315] и Pt(II) [316, 319] с пинцерными лигандами 2,6-бис(дифенилфосфинометил)пиридином (PNP, XXVI) и трифосом (PPP, XXVII) [317], описаны реакции этих соединений с нуклеофилами типа NuH (H2O, ROH и RNH2) и охарактеризованы все основные продукты присоединения NuH, например, $({\text{PNP}}){\text{PdC}}{{{\text{H}}}_{{\text{2}}}}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}}_{{\text{2}}}^{{{\text{2}} + }},$ (PNP)PdCH2CH2OH+, (PNP)PtCH2CH2OR+, (PNP)PtCH2CH2NH2Ph2+.
В случае замещенных α-олефинов образуются продукты присоединения по Марковникову. Природа продукта присоединения метанола к стирольному π-комплексу определяется кинетикой реакции. Образующийся в виде суперкислоты катион [(PNP)PtCH2CH(Ph)OMe]+${\text{H}}_{{{\text{sol}}}}^{ + }$ через 2 ч полностью превращается в том же растворе в стирольное производное (PNP)PtCH=CHPh+ [316]. Реакция HBF4 с комплексом (PNP)PtCH2CH2OH+ приводит к образованию исходного π-комплекса, а не этанола.
При действии сухого хлористого водорода на продукты присоединения NuH к дикатионным комплексам Pt(II) выделяется алкен, а в случае монокатионного комплекса в результате протолиза образуется продукт присоединения NuH к олефину (например, CH3CH2NH2Ph+). В растворах дикатионного комплекса (PNP)Pd2+ в метиленхлориде осуществляется внутримолекулярное гидроамидирование (по Марковникову) с обратимой стадией цис-амидопалладирования и внутримолекулярным протолизом связи Pd–C [320].
При изучении реакций с участием дикатионных π-комплексов Pt(II), содержащих лиганд PNP, была обнаружена возможность присоединения к олефину в составе π-комплекса богатых электронами три- и тетразамещенных олефинов с образованием продуктов гидровинилирования (ГВ), т.е. продуктов димеризации олефинов, где R = H или Me [318, 319]:
При этом происходит миграция атома водорода и группы R. С участием аналогичных комплексов Pd(II), помимо образования продуктов ГВ, протекает реакция циклопропанирования (ЦП) [317]:
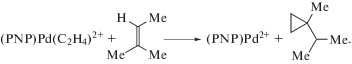
Комплекс (PNP)Pt(C2H4)2+, взаимодействуя с 2-метилбутеном-2, образует с выходом 95% продукт ГВ. Из комплекса (PNP)Pd(C2H4)2+ в зависимости от условий образуются в различных соотношениях продукты ГВ и ЦП, а комплекс (PPP)Pt(C2H4)2+ полностью превращается в продукт ЦП. Предложен новый механизм димеризации олефинов (ГВ) без участия гидридных интермедиатов и с промежуточным образованием МКИ [318], аналогичный механизму димеризации и олигомеризации алкенов с переносом гидрид-иона (LXVI). Показано, что все четыре атома D из C2D4 присутствуют в CD2CD2-группе циклопропанового кольца. Возможность циклопропанирования существенно расширяет перспективы создания новых каталитических процессов на катионных комплексах Pd(II) и Pt(II).
6.2. Катионные комплексы палладия в окислении олефинов
Интересные результаты были получены и при использовании катионных комплексов палладия в окислении олефинов. Окисление этилена сульфатом или нитратом палладия в водных кислых средах наблюдали Смидт и сотр. еще в 1962 г. [41], однако нехлоридные водные системы с хинонами (Q) и гетерополикислотами (ГПК) в качестве окислителей, по-видимому, впервые были предложены и исследованы К.И. Матвеевым и сотр. [237, 238, 321–326]. Это системы PdSO4–H2SO4(HClO4)–H2O с бензохиноном (ВQ), активные в сильно кислых средах (0.1–3.0 М HClO4) при 20–30°С в реакциях окисления этилена и пропилена, и PdSO4–ГПК [237, 238], активные в окислении этилена и бутиленов. В работах [323–326] был предложен ряд интересных гипотез.
1. Основное состояние палладия в каталитической системе Pd2+–H+–BQ–H2O – это восстановленные формы палладия Pd(0,I), стабилизированные хиноном и олефинами. На 1 моль палладия в комплексе может приходиться до 2 молей олефина (этилена, пропилена). Выделен комплекс Pd2SO4(BQ) · 3H2O [327], строение которого, однако, не было установлено.
2. Рост скорости реакции окисления олефинов хинонами по мере увеличения концентрации иона водорода объясняют медленной стадией процесса, которой, по мнению авторов [323–326], в отличие от хлоридных систем, является стадия окисления π-комплекса Pd0(BQ)(CnH2n)2 или π‑комплекса Pd(I) хиноном с участием протона с образованием π-комплекса Pd(II), например,
Некоторые результаты работ [323–326] были подтверждены при изучении водно-органических нехлоридных систем в реакциях окисления высших олефинов п-хинонами или ГПК в водно-органических растворах, содержащих катионные комплексы палладия и кислоты [328–330]. Такие системы (PdSO4, Pd(OAc)2 и Pd(NO3)2 c BQ или ГПК) в водно-органических гомофазных [220, 328–332] или двухфазных [333] средах оказались особенно эффективными в случае высших олефинов и циклоолефинов. Так, при переходе от аниона Cl– к аниону ${\text{ClO}}_{{\text{4}}}^{ - }$ в кислой среде производительность процесса окисления α-децена (моль л–1 ч–1) возрастает в ~24 раза при [H+] = 0.1 М и в ~60 раз при [H+] = 0.3 М [329]. Кроме того, в катионных системах наблюдается резкое изменение реакционной способности низших олефинов. Так, в водной сульфатной системе в одинаковых условиях скорость окисления пропилена превышает таковую для этилена в ~40 раз [326], тогда как в хлоридной системе отношение этих скоростей составляет 0.56 (Cl/Pd ≈ 20, [HClO4] = 1.0 М, 298 К) [334]. Окисление циклогексена кислородом в системе Pd(NO3)2–CuSO4–H3PMo12O40–AN–H2O [332] при 80°С и 50 бар О2 протекает с конверсией 80% (1 ч), селективностью более 99% и TOF = 260 ч–1. При окислении пероксидом водорода в тех же условиях достигается конверсия 95% и также без побочных продуктов.
Интересно отметить, что при изучении электрохимической регенерации п-бензохинона в системах Pd(OAc)2–Sol (Sol = DMF, AN, DМSО) [328] оказалось, что в присутствии п-бензохинона (но без электролиза) реакция окисления вообще не протекает. Этот факт объясняется отсутствием кислоты, образующейся в ходе электролиза и участвующей в переводе Pd(OAc)2 в катионный комплекс ${\text{Pd}}{{({\text{AN}})}_{x}}({{{\text{H}}}_{{\text{2}}}}{\text{O}})_{{{\text{4}} - x}}^{{{\text{2}} + }}.$ Ускорение реакций окисления сильными кислотами в нехлоридных системах, связанное с образованием катионных комплексов палладия, наблюдалось и в реакциях окисления аренов до бис-арилов в уксусной кислоте [335–337]. В работе [330] гидрохинон окисляли кислородом при катализе фталоцианинатным комплексом железа (PcFe) или комплексами кобальта с макроциклическими лигандами при 25°С и атмосферном давлении.
В дальнейшем катионные комплексы в виде синтезированных комплексов Pd(AN)4(BF4)2, ${\text{PdL}}({\text{AN}})_{{\text{2}}}^{{{\text{2}} + }}$ или образующихся in situ в системах Pd3(OAc)6–HA–Sol (HA = сильная кислота) или (PdCl2AN)2–Ag+–Sol активно использовали для синтеза сложных органических соединений окислением олефинов, включая асимметрический синтез [18, 21, 185, 338–343], с участием О2, Н2О2, ROOH и PhI(OAc)2 в качестве окислителей.
Кинетику и механизмы реакций окисления олефинов в различных водно-органических системах, содержащих катионные комплексы палладия с бидентатными азотными лигандами, исследовали весьма детально [185, 343]. Работа [185], в которой кинетику реакции изучали в растворах комплекса палладия со спартеином в качестве лиганда, обсуждалась выше в разд. 3.5. Следует отметить полезную практически систему с катионными комплексами [21] для синтеза кетонов из олефинов с различными функциональными группами. Окисление олефинов весьма эффективно происходит при комнатной температуре в системе Pd(OAc)2–HBF4 в тройном растворителе DMA–AN–H2O при соотношении 3.5 : 3.5 : 1 (v/v/v). В присутствии PcFe окислителем может служить кислород. В работе [343] изучали механизм окисления стиролов пероксидом водорода в присутствии воздуха в ацетонитрильном растворе катионных комплексов LPd(AN)2(OTf)2, где L = = PBO = 2-(пиридин-2-ил)бензоксазол. В результате было установлено образование нетипичного для Вакер-окисления “енолятного” интермедиата, обнаруженного ранее только в работах [152, 153]. Кинетические исследования позволили установить следующие факты.
1). Начальная скорость (R0) окисления стирола (при 10–15%-ной конверсии стирола) линейно зависит от концентрации стирола и от [Pd]∑ (при [H2O2] = 1.275 М, [H2O] = 2.53 М и объеме AN, равном 4.3 мл).
2). Зависимость R0 от [H2O2] имеет вид кривой насыщения, а зависимость R0 от [H2O] – слабый максимум в интервале 3–4 М.
в) Выход ацетофенона (АФ) в присутствии О2 и без H2O2 достигает 24% при 27°С за 24 ч. К сожалению, не было выяснено влияние ионов Н+ на R0 и роль Н+ в стадиях процесса, хотя дикатионный комплекс палладия ${\text{LPd}}({\text{AN}})_{{\text{2}}}^{{{\text{2}} + }}$ может гидролизоваться в этих условиях.
Изотопные исследования показали [343], что в системе AN–D2O2–D2O из стирола получаются с выходом 67% α-d1-PhCOCH2D и 33% PhCOCH3. КИЭ для H2O2/D2O2 равен 1.92. В тех же условиях дейтерированный ацетофенон не образуется. Анализ проб раствора методом ESI-MS обнаружил присутствие ионов LPd(OOH)+ и LPd(OOD)+ уже через 1 мин после начала реакции и в ходе процесса, но главное – иона LPdCH2COPh+, названного авторами “енолятным” интермедиатом (см. разд. 3, [152, 153]). Существование этого комплекса в растворе было подтверждено также методами масс-спектрометрии (CID-MS, DESI-MS) и ЯМР. Было установлено, что карбонильный кислород в этом комплексе на 74% происходит из пероксида водорода. В случае использования кислорода (без H2O2) КИЭ для системы H2O/D2O равен 0.97. При этом стирол в D2O превращается в недейтерированный ацетофенон, а α-d1-стирол в воде – в β-d1-ацетофеон с выходом 82%. Из PhCD=CH2 в системе AN–Н2О2–H2O образуется также PhCOCH2D с выходом 30%, а в системе AN–D2O2–D2O – с выходом 92%.
При использовании в качестве окислителя трет-бутилгидропероксида окисление α-d1-стирола в растворителе СН2Cl2–H2O привело к образованию PhCOCH2D, а методом ESI-MS было обнаружено присутствие в растворе LPd(OOtBu)+, LPdCH2CH(OOtBu)Ph+ и LPdCH2COPh+. Предложены схемы механизмов окисления кислородом и Н2О2. В случае О2 предлагается традиционный механизм, состоящий в α → β-переносе гидрида или в более вероятном элиминировании Pd–D, объясняющем образование “енолятного” интермедиата (схема 21 ). Именно депротонирование α-фенилвинилового спирта приводит к енолятному фрагменту.
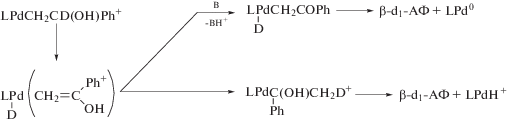
Схема 21 .
При окислении пероксидом водорода установлен двухмаршрутный механизм (схема 22 ), в рамках которого происходит традиционный перенос гидрида (дейтерида) в продукте присоединения к олефину палладия и нуклеофила ООН и возможный перенос Н(D) на ОН-группу, образующуюся при разрыве связи О–ОН. Результаты изотопных исследований, выполненных в этой работе [343], прекрасно согласуются с данными многочисленных экспериментов, представленных в разд. 3.
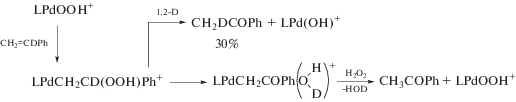
Схема 22 .
6.3. Кинетика и механизм реакций окисления этилена и циклогексена п-бензохиноном в растворах комплексов ${\text{Pd(AN}}{{)}_{x}}({{{\text{H}}}_{{\text{2}}}}{\text{O}})_{{4 - x}}^{{2 + }}$ в системе AN–Н2О
Кинетические закономерности и механизмы реакций окисления олефинов исследовались в водно-ацетонитрильных растворах сотрудниками МИТХТ им. М.В. Ломоносова [147, 344–350]. Кинетика реакций окисления циклогексена (ЦГ) [344] и этилена [345, 346] п-бензохиноном и КИЭ в реакциях окисления [347, 348] были изучены в бинарном растворителе AN–Н2О с различным содержанием ацетонитрила. Изучали и состояние палладия в системах Pd(OAc)2–AN и Pd(OAc)2–HClO4–AN–H2O [349], а также некоторые закономерности окисления циклогексена (ЦГ) и этилена [147, 350], включая фазовые равновесия жидкость–жидкость в системах AN–Н2О–ЦГ и AN–Н2О–ЦГ–циклогексанон [351].
В результате изучения механизмов формирования катионных комплексов палладия(II) ${\text{Pd(AN}}{{)}_{x}}({{{\text{H}}}_{{\text{2}}}}{\text{O}})_{{4 - x}}^{{2 + }}$ методами электронной и 1Н- и 13С-ЯМР-спектроскопии в системе Pd3(OAc)6–HClO4–AN–H2O были сделаны следующие выводы [349].
1). Процесс разрушения тримера ацетата палладия ацетонитрилом без добавления воды и хлорной кислоты происходит чрезвычайно медленно и требует 7 сут при комнатной температуре для достижения равновесия и образования мономерных и олигомерных нитрилацетатных комплексов [Pd(AN)2(OAc)2]n.
2). Состав комплексов ${\text{Pd(AN}}{{)}_{x}}({{{\text{H}}}_{{\text{2}}}}{\text{O}})_{{4 - x}}^{{2 + }}$ после добавления воды и кислоты к раствору тримерного ацетата палладия в AN зависит от времени выдерживания этого раствора.
3). После 7-сут выдерживания системы Pd3(OAc)6–AN добавление кислоты и воды с последующим 20-мин перемешиванием смеси приводит к образованию преимущественно комплекса ${\text{Pd(AN}})({{{\text{H}}}_{{\text{2}}}}{\text{O}})_{3}^{{2 + }},$ поглощающего при λmax = = 360–365 нм (система AN-IV).
4). Без предварительного выдерживания раствора Pd3(OAc)6 в AN и после 20-мин перемешивания смеси с водой и кислотой в растворе формируется комплекс ${\text{Pd(AN}}{{)}_{2}}({{{\text{H}}}_{{\text{2}}}}{\text{O}})_{2}^{{2 + }},$ поглощающий при λmax = 335–340 нм (система AN-III).
5). После 7-сут выдерживания сиcтемы AN-III и AN-IV переходят в систему AN-V (при содержании воды 0.74 м. д.) с образованием комплекса Pd(AN)3(H2O)2+, поглощающего при λmax = 305–310 нм.
6). В системе AN-III при содержании воды в растворителе AN–H2O в интервале от 0.5 до 0.95 м. д. преобладает комплекс ${\text{Pd(AN}}{{)}_{2}}({{{\text{H}}}_{{\text{2}}}}{\text{O}})_{2}^{{2 + }}.$
7). Катионные комплексы палладия, полученные описанным выше способом, идентичны комплексам, полученным растворением ацетонитрила в растворе перхлората палладия в хлорной кислоте при тех же составах растворов.
В интервале концентраций палладия 2.5–10 мМ при содержании воды 0.74 м. д., [HClO4] = 0.2 M и [LiClO4] = 0.3 M выполняется закон Бугера–Ламберта–Бера, что свидетельствует об отсутствии смесей мономерных и димерных (олигомерных) комплексов палладия в кислой водно-ацетонитрильной среде. При использовании рассчитанных по спектральным данным констант равновесия реакций последовательного замещения молекулы AN молекулами воды в комплексе ${\text{Pd(AN}})_{4}^{{2 + }}$ с образованием “метастабильных” комплексов (lg K1 = 3.30 ± 0.14, lg K2 = 0.03 ± 0.001, lg K3 = 0.91 ± ± 0.04, lg K4 = 2.60 ± 0.11) и значений коэффициентов экстинции всех комплексов при разных длинах волн было показано хорошее соответствие экспериментальных и теоретических зависимостей оптического поглощения растворов от длины волны со среднеквадратичной ошибкой 4.3%. (Неопубликованные результаты, полученные И.В. Мартыновым и Е.А. Кацманом, описаны в магистерской диссертации И.В. Мартынова. М.: МИТХТ, 2017).
Каталитическая активность систем AN–III и AN–IV в реакции окисления этилена оказалась весьма близкой, а активность системы AN–V (равновесная система), которая получается через 7 сут после добавления кислоты и воды из систем AN–III и AN–IV, ниже на 38–50% активности этих систем.
Описанные наблюдения указывают на заметную кинетическую инертность катионных комплексов палладия в отношении реакций обмена лигандами и на вероятное влияние самого каталитического процесса с участием в качестве лигандов олефина и п-бензохинона на формирование близких по составу комплексов независимо от состава исходных систем на старте процесса. Важным фактором, влияющим на состав комплексов, является содержание воды. Кинетические закономерности и основные черты механизмов в системах AN–III и AN–IV оказались также близкими.
1). В обоих процессах окисления олефинов п-бензохиноном на кинетических кривых убыли ЦГ и на кривых поглощения этилена по времени видны заметно различающиеся по скоростям реакций участки (А и Б).
2). Начальные скорости (участок А) обоих процессов имеют экстремальные зависимости от содержания воды – бимодальную зависимость в случае ЦГ [344, 350] и один максимум в интервале 0.2–0.3 м. д. воды в случае этилена [147].
3). Кинетику окисления ЦГ изучали при содержании ~0.3 м. д. воды в системе AN–III, а кинетику окисления этилена – при 0.67 м. д. воды в системе AN–IV.
4). Начальные скорости реакций растут с увеличением [Н+]: в случае этилена – это кривая насыщения, а в случае ЦГ кривая имеет слабый максимум. Экстремальные зависимости начальных скоростей от [Q] наблюдаются в обоих процессах.
5). Кинетические изотопные эффекты в системе H2O/D2O в обоих процессах [347] и в системе C2H4/C2D4 в реакции окисления этилена [348] близки к 1.
Адекватность кинетической модели при описании двух участков кинетических кривых была достигнута только при использовании предположения о переходе в ходе процесса каталитически активных комплексов Pd(II) в менее активные комплексы Pd(I) [344, 346]. Эта гипотеза позволила объяснить независимость скорости реакции окисления ЦГ на втором участке (Б) от времени реакции, исходной концентрации палладия и концентрации бензохинона [Q]t [344]. Аналогичное наблюдение о появлении двух участков на кинетических кривых с подтверждением образования комплекса Pd(I)–Pd2L2(OAc)2 (L = = 4,5-диазафлуорен-9-он) – было сделано при исследовании окислительного ацетоксилирования аллилбензола в работе Ш. Сталла и сотр. [352].
Рассмотрим подробнее результаты изучения кинетики окисления этилена и предложенные механизмы [345, 346]. Исследование зависимостей начальных скоростей (R0) от начальных концентраций реагентов и компонентов каталитической системы (участок А) в условиях существования комплекса ${\text{Pd(AN}})({{{\text{H}}}_{{\text{2}}}}{\text{O}})_{3}^{{2 + }}$ при постоянной ионной силе (I) раствора ([HClO4] + [LiClO4] = = 0.5 M) и 30°С позволило описать зависимости R0 от [Pd]∑, [Q]∑, [HClO4] и ${{P}_{{{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}}}}.$
Предложенный 10-стадийный механизм [345] включает 6 кинетически необратимых стадий (2)–(4), (7), (8) и (10), влияние которых на процесс и на кинетику реакции определяется их вкладом в материальный баланс по катализатору. Стадии (3), (4) и (8) при описании начальной скорости процесса рассматривались как “быстрые необратимые”.
Увеличение R0 при повышении [HClO4] и равенство КИЭ единице при замене Н2О тяжелой водой [347] является указанием на отсутствие квазиравновесной стадии диссоциации координированной палладием молекулы воды до первой медленной стадии включительно. Рост R0 при увеличении [Q]∑ свидетельствует о существовании, по крайней мере, еще одной медленной стадии, связанной с участием хиноновых комплексов Pd(0) в механизме процесса окисления. Поэтому, вероятно, скорость стадии депротонирования (3) не отражается на величине начальной скорости.
Анализ различных кинетических моделей показал, что без учета стадий образования хиноновых комплексов Pd(0) и вклада в материальный баланс по [Pd]∑ комплексов Pd(0) и гидрохинолятных комплексов описать экстремальную зависимость скорости реакции от концентрации п-бензохинона невозможно. Поэтому в квазистационарных условиях при допущениях о квазиравновесности стадий (1), (5), (6) и (9), равенстве скорости стадии (2) сумме скоростей стадий (7) и (10) и о структуре материального баланса по катализатору (17)
(17)
$\begin{gathered} {{\left[ {{\text{Pd}}} \right]}_{\sum }} = {\text{\;}}\left[ {{\text{P}}{{{\text{d}}}^{{2 + }}}} \right] + \left[ {{{\pi }_{1}}} \right] + \left[ {{\text{P}}{{{\text{d}}}^{0}}} \right] + \left[ {{\text{Pd}}{{{\text{Q}}}^{0}}} \right] + \\ + \,\,\left[ {{\text{PdQ}}_{2}^{0}} \right] + \left[ {{\text{Pd}}\left( {\text{Q}} \right){{{\left( {{\text{QH}}} \right)}}^{ + }}} \right], \\ \end{gathered} $(18)
${{R}_{0}} = \frac{{{{{{\text{[Pd]}}}}_{\Sigma }}{{P}_{{{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}}}}{{{[{\text{Q]}}}}_{{\text{0}}}}[{{{\text{H}}}^{ + }}][{{{\text{H}}}_{{\text{2}}}}{\text{O][AN](}}{{A}_{1}} + {{A}_{2}}{{{[{\text{Q]}}}}_{{\text{0}}}}[{{{\text{H}}}^{ + }}][{{{\text{H}}}_{{\text{2}}}}{\text{O])}}}}{D},$(19)
$\begin{gathered} D = {{A}_{3}}{{[{\text{Q]}}}_{{\text{0}}}}[{{{\text{H}}}^{ + }}][{{{\text{H}}}_{{\text{2}}}}{\text{O][AN]}} + \\ + \,\,{{A}_{4}}[{\text{Q]}}_{{\text{0}}}^{{\text{2}}}{{[{{{\text{H}}}^{ + }}]}^{2}}{{[{{{\text{H}}}_{{\text{2}}}}{\text{O]}}}^{{\text{2}}}}[{\text{AN]}} + \\ + \,\,{{A}_{5}}{{P}_{{{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}}}}{{[{\text{Q]}}}_{{\text{0}}}}[{{{\text{H}}}^{ + }}][{\text{AN]}} + \\ + \,\,{{A}_{6}}{{P}_{{{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}}}}[{\text{Q]}}_{{\text{0}}}^{{\text{2}}}{{[{{{\text{H}}}^{ + }}]}^{2}}[{{{\text{H}}}_{2}}{\text{O][AN]}} + \\ + \,\,{{A}_{7}}{{P}_{{{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}}}}{{[{{{\text{H}}}_{{\text{2}}}}{\text{O]}}}^{{\text{3}}}}[{\text{AN]}} + \\ + \,\,{{A}_{8}}{\text{[}}{{P}_{{{{{\text{C}}}_{{\text{2}}}}{\text{H}}{}_{{\text{4}}}}}}{\text{][Q}}{{{\text{]}}}_{{\text{0}}}}{\text{[}}{{{\text{H}}}_{{\text{2}}}}{\text{O][AN]}} + \\ + \,\,{{A}_{9}}{{P}_{{{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}}}}{\text{[Q]}}_{{\text{0}}}^{{\text{2}}}{\text{[}}{{{\text{H}}}_{{\text{2}}}}{\text{O] + }}{{A}_{{10}}}{{P}_{{{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}}}}[{{{\text{H}}}^{ + }}{\text{][}}{{{\text{H}}}_{{\text{2}}}}{\text{O}}{{{\text{]}}}^{{\text{2}}}}{\text{,}} \\ \end{gathered} $Из полученных значений концентраций интермедиатов следует, что основными состояниями катализатора являются исходный комплекс Pd2+ (36%) и гидрохинолятный комплекс [Pd(Q)(QH)+] (54%), а концентрации хиноновых комплексов Pd(0) вследствие их быстрого окисления близки к нулю. В кинетическое уравнение входят константы скорости стадии (2) (присоединение молекулы воды к катионному π-комплексу), стадий окисления хинонового комплекса Pd0 (7) и протолиза гидрохинолятного комплекса Pd(II) с хиноном (10). Заметим также, что в ацетонитрильной системе частота оборотов катализатора в реакции окисления этилена в присутствии катионных комплексов палладия (${{Р}_{{{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{4}}}}}}}$ = 0.87 атм, [HClO4] = 0.2 М, [Pd]∑ = 0.0005 М, [Q]0 = 0.2 М) примерно в 130 раз выше, чем в растворах хлоридных комплексов (${{Р}_{{{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{4}}}}}}}$ = 0.87 атм, [HCl] = 0.05 М, [Pd]∑ = 0.02 М, [Q]0 = 0.1 М).
Образование комплексов PdQ0 на участке А может привести к появлению в результате взаимодействия PdQ0 с Pd2+ комплекса Pd2Q2+ и, соответственно, маршрута с каталитическим циклом, который реализуется на комплексах Pd2Q2+ типа Pd2SO4(Q) [327]. Такой путь синтеза комплексов палладия(I) является достаточно распространенным [353, 354]. Например, в работе [354] описан комплекс Pd2(AN)6(BF4)2, полученный из ${\text{Pd(AN)}}_{{\text{4}}}^{{{\text{2}} + }}$ и комплекса ${\text{P}}{{{\text{d}}}_{{\text{2}}}}({\text{dba)}}_{{\text{3}}}^{{\text{0}}}$ в системе AN−СН2Сl2. Нельзя исключить и появления на участке Б маршрута, в котором с участием комплекса Pd(Q)(С2Н4)0 образуется ацетальдегид [323, 325, 326]. Таким образом, по аналогии с механизмом окисления ЦГ, для описания кинетики окисления этилена на участках А и Б следует учитывать возможность протекания реакции по третьему маршруту, который начинается на участке А и развивается на участке Б. Полный анализ кинетической модели рассматриваемой реакции в нестационарном режиме представлен в работе [346]. Механизм со стадиями (1)–(10) был дополнен стадиями (11)–(17) третьего маршрута:
Граф механизма с тремя маршрутами представлен на рис. 2. При записи систем дифференциальных уравнений для 17 стадий предполагаемого механизма [346] учитывали, что концентрации этилена, кислоты и ацетонитрила (11.2 М) не меняются во времени. Концентрация воды (22.3 М) в ходе процесса остается практически постоянной. Уравнения закона действия масс для стадий с участием молекул воды или ацетонитрила записывали через суммарные концентрации этих компонентов растворителя.
Рис. 2.
Кинетический граф трехмаршрутного механизма окисления этилена п-бензохиноном с обратимой нелинейной стадией (11) (квадратная вершина двудольного графа 11 [7]).
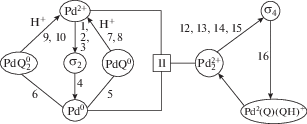
Средняя квадратичная ошибка описания 14 кривых изменения концентрации поглощенного этилена во времени при различных условиях составляет 10.36%. Проведенный анализ материального баланса по палладию ([Pd]Σ = 0.0005 М) в ходе процесса на участках А (1–15 мин) и В (13–45 мин) при учете интермедиатов, концентрация которых больше 5 мол. %, от [Pd]Σ показал следующее (мол. % палладия, связанного в σ4, указан с учетом присутствия двух атомов палладия в σ4).
1). Основные формы катализатора: через 1–3 мин – Pd2+ (26–34%), PdQ2 (13–26%), PdQ(QH)+ (13–26%) и σ4 (${{[{\text{P}}{{{\text{d}}}_{2}}{{\left( {{\text{AN}}} \right)}_{2}}{\text{Q}}({\text{C}}{{{\text{H}}}_{2}}{\text{C}}{{{\text{H}}}_{2}}{\text{OH}})\left( {{{{\text{H}}}_{2}}{\text{O}}} \right)]}^{ + }}$) (7–17%); в конце процесса через 40–45мин – Pd2+ (8–12%), PdQ2 (до 9%), PdQ(QH)+ (до 9%) и σ4 (63–84%).
2). Концентрации всех интермедиатов, кроме σ4, по ходу процесса понижаются, и палладий накапливается в форме σ4, что-наблюдается при завершении быстрого участка А кинетических кривых, т.е. через 13–15 мин.
3). На быстром участке А концентрация Pd2+ понижается на 70–80% и появляется третий маршрут реакции, где палладий накапливается в форме σ4.
Из анализа скоростей стадий образования ацетальдегида (4) и (16) следует, что скорости W4 и RΣ снижаются, начиная с первой минуты, по мере понижения [Pd2+] и перехода его в ${\text{Pd}}_{{\text{2}}}^{{{\text{2}} + }}.$ При максимальном значении W16 вклад третьего маршрута составляет 12–28% в зависимости от условий процесса ([Q], ${{P}_{{{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}}}}$).
Поскольку ранее для водных растворов катионных комплексов палладия предполагалась [325, 326] возможность катализа реакции окисления этилена комплексами [Pd(Q)]0 и [Pd(C2H4)(Q)0, был рассмотрен дополнительно и четвертый маршрут, включающий 6 стадий. Этот маршрут интересен тем, что окисление комплекса [Pd(C2H4)(Q)0 в реакции с Н3О+ сразу приводит к гидрохинолятному π-комплексу Pd(II), взаимодействующему далее с молекулой воды с образованием палладоэтанольного интермедиата. Анализ показал, однако, что качество описания с введением четвертого маршрута практически не улучшилось (ошибка 10.26%).
С целью уточнения состава координационной сферы палладия ключевых интермедиатов были проведены квантово-химические расчеты энергий Гиббса стадий (1), (4), (5), (11) и (12), а также моделирование стадий механизма (2) и (3) с расчетом соответствующих активационных барьеров.
В стадии присоединения этилена к комплексу [Pd(AN)(H2O)3]2+ возможно вытеснение молекулы воды (стадия (1)) или ацетонитрила (стадия (1.1). Моделирование этой стадии с учетом дикатионного комплекса палладия и двух перхлорат-ионов, сольватированных двумя молекулами воды и молекулой АN (разделенных ионных пар), показало, что, согласно значениям энергии Гиббса (ΔG298 = = –4.3 и –2.2 ккал/моль) для реакций (1) и (1.1) при координации этилена более вероятно вытеснение молекулы воды (1). Отсутствие противоионов при моделировании стадии (1) приводит к неверной энергетике реакции. Например, для реакции (1.1) значение ΔG298 становится равным 16.8 ккал/моль.
Для выявления наиболее вероятного состава комплексов Pd0, формирующихся в стадии (4), были рассчитаны комплексы различного состава – ${\text{Pd}}{{({\text{AN}})}_{x}}({{{\text{H}}}_{{\text{2}}}}{\text{O}})_{y}^{0}.$ Согласно расчетам, наибольшей энергетической устойчивостью обладает динитрильный комплекс ${\text{Pd}}({\text{AN}})_{{\text{2}}}^{0}.$ Тетраэдрический ${\text{Pd}}({\text{AN}})_{{\text{4}}}^{0}$- и плоский ${\text{Pd}}({\text{AN}})_{{\text{3}}}^{0}$-комплексы, имеющие минимумы на поверхности потенциальной энергии (ППЭ), термодинамически невыгодны (на 23.9 и 11.8 ккал/моль соответственно). Замена молекулы АN на H2O в ${\text{Pd}}({\text{AN}})_{{\text{2}}}^{0}$ с образованием двухлигандного комплекса Pd(AN)(H2O)0 приводит к повышению энергии Гиббса на 11.5 ккал/моль.
Присоединение молекулы п-бензохинона (Q) к ${\text{Pd}}({\text{AN}})_{{\text{2}}}^{0}$ по η2-типу (вариант стадии (5)) весьма выгодно (ΔG298 = –13.8 ккал/моль) с величиной ΔH298 = –25.0 ккал/моль. Присоединение второй молекулы Q, протекающее с вытеснением лиганда АN, также весьма выгодно (ΔG298 = –7.2 ккал/моль).
Взаимодействие комплексов Pd(II) и Pd(0) в стадии (11) термодинамически благоприятно и протекает безбарьерно:
На следующей стадии биядерный комплекс Pd(I) может достраивать координационную сферу молекулами AN или H2O, причем вариант с АN энергетически выгоднее на 10.5 ккал/моль.
Молекула Q может координироваться комплексами Pd(I) моно- или бидентатно (стадия (12)). Второй вариант энергетически предпочтительнее (на 3.0 ккал/моль). Можно предположить, что молекулы воды из первичного аддукта [Pd2(AN)4(H2O)2]2+ при взаимодействии с Q покинут координационную сферу биядерного комплекса как “слабые” лиганды и, в конечном счете, сформируется комплекс Pd(I) состава [Pd2Q(AN)4]2+. При этом молекула Q может быть терминальной с почти η2,η2-координацией или центральной с μ-η2,η2-координацией.
Для выяснения возможных гипотез о механизме стадий ГП (2) и (3) было проведено квантово-химическое моделирование ППЭ этих стадий, которое выявило важную роль ассоциатов из молекул воды и ионных пар, включающих катионный π-комплекс этилена и перхлорат-анионы, разделенные некоторым количеством молекул воды. Расчеты без учета сольватированных перхлорат-ионов показали, например, что присоединение молекулы воды к этилену в π-комплексе происходит безбарьерно (стадия (2)), но депротонирования полученного сольватированного МКИ не происходит – вторая молекула воды присоединяется к первой водородными связями. И только участие третьей молекулы воды с одновременным депротонированием приводит к образованию σ‑палладий органического интермедиата (σ2) и катиона ${{{\text{Н}}}_{{\text{5}}}}{\text{О}}_{{\text{2}}}^{{\text{ + }}}$ (стадия (3.1)).
Отметим, что участие трех молекул воды в стадии типа (2) образования σ-палладоэтанола ранее рассматривалось при моделировании механизма окисления олефинов в хлоридных системах [180, 185]. Известно (см. [7 (Ch. 8)]), что добавление уже 0.3 м. д. органических полярных растворителей в воду приводит к разрушению трехмерной сетки воды и образованию набора ассоциатов. Поэтому использование ассоциатов воды в элементарных стадиях механизмов следует считать вполне правдоподобным. К тому же кинетические эксперименты, описанные выше, проведены при содержании воды 0.67 м. д. В связи с этим были проведены дополнительные расчеты стадии (2) с использованием ассоциата из трех молекул воды (тример).
Расчеты показали, что присоединение тримера воды к этилену не требует энергии активации и может приводить к двум разным продуктам. В случае атаки этилена в π-комплексе атомом кислорода терминальной молекулы воды тримера, присоединение тримера сопровождается депротонированием и вытеснением ${{{\text{H}}}_{{\text{5}}}}{\text{O}}_{{\text{2}}}^{ + }$ из тримера без образования минимума на ППЭ. В этой элементарной стадии (3.2) получается продукт, идентичный продукту стадии (3.1).
Если “активная” молекула воды тримера является центральной, то образование продукта не сопровождается выбросом протона или молекул воды во внешнюю сферу (стадия (2.1)):
Депротонирование полученного интермедиата с фрагментом (XXVIII) становится возможным в результате его перестройки в продукт, содержащий палладоцикл (XXIX) и выбросом иона ${{{\text{Н}}}_{{\text{5}}}}{\text{О}}_{{\text{2}}}^{{\text{ + }}}$ в раствор.
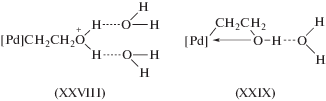
На следующем этапе было проведено моделирование стадий (2) и (3) с учетом перхлорат-анионов, сольватированных 6 молекулами воды. Добавление противоионов при рассмотрении стадии (2) с участием даже двух молекул воды приводит к присоединению атома кислорода димера воды к этилену с одновременным депротонированием образующегося интермедиата и появлением иона ${{{\text{Н}}}_{{\text{5}}}}{\text{О}}_{{\text{2}}}^{{\text{ + }}}{\text{,}}$ т.е. к объединению стадий (2) и (3). Такой процесс сопровождается двумя эффектами с появлением небольшого барьера в 3.6 ккал/моль и со структурными изменениями, обусловленными удалением протона от палладоэтанольного фрагмента на весьма большое расстояние 5.1 Å. (рис. 3). Присутствие гидратированных анионов приводит, по существу, к отрыву двух молекул воды из сольватационной сетки для присоединения к этилену с переносом протона по цепочке водородных связей и с образованием σ-палладоэтанольного комплекса.
Рис. 3.
Структура комплекса [Pd(AN)(H2O)2{σ-C2H4OH(H2O)}][ClO4{H2O}6]{H5O2}+[ClO4{H2O}4]–. Молекулы воды, составляющие сольватную оболочку перхлорат-ионов, окрашены серым цветом. Протон в форме {H5O2}+ указан стрелкой.
По результатам моделирования стадий (2) и (3) получены вероятные механизмы ключевых стадий, однако использование предложенных гипотез для построения и экспериментальной проверки кинетической модели процесса связано с решением важной проблемы, касающейся теории растворов. При анализе стадий типа (2) в реакциях, в которых молекулы ассоциированных растворителей (Н2О, ROH, RCOOH, PhOH и ROOH) являются реагентами, возникает вопрос, как выразить концентрацию свободной молекулы (например, Н2О или нужного ассоциата, например, (H2O)n) в кинетическом уравнении. Концентрацию молекулы ассоциата (Н2О)3 через [H2O]Σ можно выразить, если известен вид зависимости концентрации свободной молекулы воды от ее суммарной концентрации: [H2O] = f([H2O]Σ). Подходы к решению аналогичных проблем, связанных с образованием ассоциатов или многоядерных комплексов металлов, изложены в монографии [7 (Ch. 2)].
7. НЕКОТОРЫЕ ДОПОЛНИТЕЛЬНЫЕ АСПЕКТЫ ХИМИИ И ТЕХНОЛОГИИ ПРОЦЕССОВ ОКИСЛЕНИЯ ОЛЕФИНОВ
В данном обзоре не обсуждаются реакции винильного, аллильного и гомоаллильного окисления олефинов, детально рассмотренные в обзоре [10]. Обсудим кратко еще ряд новых направлений применения “вакеровских” механизмов нуклеопалладирования олефинов с последующей окислительной трансформацией палладийорганических интермедиатов, которые также не являются предметом этого обзора. Это синтез 1,2-продуктов окисления олефинов в системах, близких к вакеровским, использование гетерогенных катализаторов, двухфазных систем и других методов синтеза, облегчающих технологию выделения продуктов из реакционных систем, а также использование комплексов других переходных металлов в синтезе карбонильных соединений из олефинов.
7.1. Синтезы 1,2-продуктов окисления олефинов
В разд. 3 уже обсуждалась проблема синтеза этиленхлоргидрина и даже асимметрический синтез хиральных алкенхлоргидринов. При окислении олефинов катионными комплексами таллия(III), кроме карбонильных продуктов, образуются гликоли (разд. 3, [123]). В работе [79] обсуждался механизм превращения продукта ацетоксипалладирования алкена в 1,2-диацетаты и в моноацетаты алкенгликолей. В отличие от механизма образования 1,1-алкилидендиацетатов, предполагается, что вместо 1,2-сдвига (или β-[Pd]–H-элиминирования) в продукте ацетоксипалладирования олефина может происходить внутримолекулярное нуклеофильное замещение палладия с образованием Pd(0) и весьма стабильного иона карбения, например, (ХХХ).
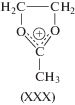
Реакции этого катиона с АсО– или с Н2О и приводят к 1,2-продуктам окисления [79]. При изучении синтезов 1,2-диацетатов и моногликольацетатов в системах PdCl2–CuCl2–AcOH, Pd(AN)2(NO3)mCln–AcOH, PdCl2–CuCl2–Fe(NO3)3–LiNO2 и др. [153] рассматривали также возможность гетеролиза связи Pd–С в рамках гетерометаллического комплекса [Cu](μ-OAc)[Pd]CH2CH(OAc)R. В работах Джианга (Jiang H.) [355, 356] предполагается возможность окисления Pd(II) в Pd(IV) в σ-Pd-β-ацетатном интермедиате с последующим восстановительнным элиминированием 1,2-продукта окисления. Показана высокая стереоспецифичность диацетоксилирования стиролов до син- и анти-1,2-диацетатов в системе Pd(OAc)2–KI–AcOH, достигающая 96–99% к син(цис)-диастереомеру при 100°С и 8 бар О2. При использовании добавок Na2CO3 вместо KI в тех же условиях [357] наблюдается стереоселективное образование цис-диолов. Синтез диолов происходит и в системе Pd(OAc)2–p-TSA–H2O при 100°С и 8 бар О2, но в этой системе гликоли расщепляются до альдегидов или кетонов с выходами 23–89%. Предполагается образование диолятного комплекса палладия и его расщепление с участием О2 (другие окислители – CuCl2, Cu(OAc)2 и BQ – не приводят к расщеплению С–С-связи и образованию карбонильных продуктов [356]).
В разд. 3 обсуждался механизм образования ацеталей и кеталей – продуктов 1,1-окисления олефинов в растворах спиртов. В работе [358] было установлено, что в условиях образования катионных комплексов палладия в системе Pd(OAc)2–MeSO3H–QH2–HPA в этаноле происходит эффективная ацетализация винильных соединений CH2=CHX (X = COOEt, CHO, C(OEt)2, CN), т.е. образование ацеталей в результате анти-М-окисления винильных производных. Неожиданно оказалось [16, 359], что в системе PdCl2(AN)2–CuCl2 в спиртах в присутствии молекулярных сит (3 Å) при высоком давлении кислорода происходит 1,2-алкоксилирование интернальных олефинов с образованием syn, anti-диастереомеров в соотношении (3–6 : 1). При этом в случае о-аллилфенолов в процессе реакции происходит изомеризация субстрата с образованием о-пропенилфенола и его 1,2-алкоксилированием до ArCH(OR)CH(OR)CH3. В присутствии хиральных лигандов [16, 360] алкоксилирование о-пропенилфенола происходит с образованием хирального син-диастереомера с величиной энантиомерного избытка ее ≈ 84%. Показано, что с ростом концентрации хлорида меди энантиоселективность понижается вследствие связывания хирального лиганда хлоридом меди. В работах [16, 359, 360] обсуждается специфическая роль о-ОН-группы в достижении высокой диастерио- и энантиоселективности в процессе образования 1,2-алкоксипроизводных.
7.2. Реакции Вакер-окисления в гетерофазных системах
С целью создания технологий, облегчающих отделение продуктов окисления олефинов от каталитических систем, внимание исследователей было обращено на разработку гетерогенных катализаторов, с которых, собственно, и начались работы по созданию Вакер-процесса (разд. 2).
Исследовали гетерогенные катализаторы, работающие в жидкой фазе и в газофазных условиях [13]. Палладийсодержащие системы (Na2PdCl4, PdCl2, Pd(OAc)2, PdSO4, PdCl2–CuCl2 и даже Pd0) наносят на γ-Al2O3, TiO2, SiO2, V2O5, силикалит, полимеры, окисленные полипиррол и полианалин и на активированный уголь [13]. В качестве окислителей используют О2 и Н2О2, а в качестве со-окислителей – CuCl2, VOSO4 и ГПК. Приведем только два примера использования гетерогенных катализаторов окисления олефинов. Так, катализатор Pd/C эффективно катализирует окисление различных стиролов пероксидом водорода в растворах AN–H2O–H2SO4 при 65°С с выходом кетонов 65–90% [361]. Катализатор легко отделялся от жидкой фазы и был использован 5 раз без значительной потери активности (см. также [226]). Использование ГПК на SiO2 вместе с PdSO4 или палладиевых солей ГПК (Pd1.5PMo12O40, Pd2.5PV2Mo10O40, Pd3PV3Mo9O40) с неполным замещением протонов ионом Pd2+, позволило получить гетерогенный катализатор для газофазного окисления бутена-1 кислородом воздуха (при отношении ${{{{Р}_{{{{{\text{Н}}}_{{\text{2}}}}{\text{О}}}}}} \mathord{\left/ {\vphantom {{{{Р}_{{{{{\text{Н}}}_{{\text{2}}}}{\text{О}}}}}} {{{Р}_{{{{{\text{С}}}_{{\text{4}}}}{{{\text{Н}}}_{{\text{8}}}}}}}}}} \right. \kern-0em} {{{Р}_{{{{{\text{С}}}_{{\text{4}}}}{{{\text{Н}}}_{{\text{8}}}}}}}}}$ = 7 и температурах 313–375 К) [362]). Селективность процесса 98%. Отмечается, что начальная активность катализатора понижается в ~10 раз при выходе процесса на стационарный режим вследствие недостаточно высокой скорости регенерации ГПК кислородом воздуха.
Достоинства гомогенных каталитических систем наталкиваются на проблему выделения продуктов из реакционной среды и рецикла катализатора. В реакциях окисления олефинов в бинарных водно-органических растворителях в случае высококипящих органических компонентов иногда возможно выделение продуктов простой дистилляцией с сохранением катализатора (или каталитической системы) в органическом растворителе (NMP, DMA, адиподинитрил (ADN), PhCH2CN и др.). Очевидно, что наиболее экономичным способом рецикла катализаторов и выделения продуктов является использование двухфазных систем с иммобилизацией катализатора в одной из двух фаз, т.е. в так называемом “жидком носителе” (liquid support). Различные подходы к организации таких реакционных систем давно рассмотрены в монографической литературе [363–368], в весьма полных обзорах [369, 370] и даже в учебниках [371]. В качестве фаз, содержащих каталитическую систему, используют Н2О, органический растворитель, ионные жидкости (ИЖ) и даже сверхкритический СО2 (sc-СО2). Используют также влияние температуры на взаимную растворимость фаз, т.е. на переход системы в одну фазу при повышении температуры и в две фазы при охлаждении системы. Рассмотрим здесь несколько примеров организации двухфазных систем в Вакер-окислении олефинов.
В работах Р. Шелдона и сотр. [221, 222] использовали водорастворимые диаминные лиганды для растворения Pd(OAc)2 (в присутствии NaOAc) в водной фазе при окислении кислородом гексена-1 и других олефинов, образующих органическую фазу. При 100°С и давлении 30 бар с лигандами фенантролином, дипиридилсульфонатом натрия и батофенантролиндисульфонатом натрия достигнута высокая селективность (97–99%) в реакции синтеза кетонов.
Второе хорошо разработанное направление в дизайне двухфазных систем – использование водорастворимых циклических углеводов циклодекстринов (ЦД) или циклических олигомерных продуктов поликонденсации фенолов и формальдегида (каликсаренов), содержащих заместители с комплексообразующими группами (–PPh2, –СN). Такие молекулы образуют водорастворимые комплексы с катализатором (например, с Pd(II)) и способны распознавать субстраты (олефины) и переносить их из органической фазы в водную [333, 369, 372, 373]. Эти системы обеспечивают высокую активность и селективность процессов окисления и позволяют сохранить катализатор в водной фазе.
Отметим еще и третье направление, связанное с использованием “альтернативных” растворителей [365, 366, 369, 370] – использование сверхкритического СО2 [374–376], ионных жидкостей [147, 370, 376, 377] и перфторуглеродных жидкостей [369, 370]. Ионные жидкости с органической фазой (субстрат или субстрат плюс органический растворитель) могли бы при удачном подборе ИЖ и каталитической системы служить идеальной двухфазной системой с иммобилизацией катализатора в ИЖ, удобной для разделения каталитической системы и продуктов реакции, а при необходимости и для дистилляции продуктов и реагентов. В качестве примера рассмотрим результаты работы [376], в которой исследовали двухфазную систему ИЖ–sc-CO2 для окисления гексена-1 кислородом. Окисление проводили в каталитической системе PdCl2–CuCl2–${\text{BMI}}{{{\text{M}}}^{ + }}{\text{PF}}_{{\text{6}}}^{ - }$–MeOH под давлением 125 бар при 25 бар О2 и температуре 333 К. При этом PdCl2 и CuCl2 не растворяются в sc-CO2 и существуют в ионной жидкости, которая также не растворяется в sc-CO2. После завершения процесса снижение давления приводит к полному извлечению (экстрагированию) всех веществ, растворимых в sc-CO2. Сравнение трех видов растворителей (sc-CO2, sc-CO2–${\text{BMI}}{{{\text{M}}}^{ + }}{\text{PF}}_{{\text{6}}}^{ - }$ и чистой ионной жидкости) показало, что наивысшая селективность при близкой к 100%-ной конверсии гексена-1 получается только для бинарного растворителя sc-CO2–${\text{BMI}}{{{\text{M}}}^{ + }}{\text{PF}}_{{\text{6}}}^{ - },$ как и лучшая рециклизуемость катализатора.
Опыт использования трех ИЖ (${\text{BMI}}{{{\text{M}}}^{ + }}{\text{PF}}_{{\text{4}}}^{ - },$ ${\text{BMI}}{{{\text{M}}}^{ + }}{\text{PF}}_{{\text{6}}}^{ - }$ и ТEA+Tf–) в реакции окисления циклогексена п-бензохиноном при 30°С показал, что в растворах катионных комплексов палладия в ИЖ {Pd(OAc)2–HClO4–Н2О–ИЖ} селективность реакции образования циклогексанона невысока с заметной олигомеризацией циклогексена, вероятно, вследствие суперкислотных свойств системы в использованных ИЖ [147].
7.3. Другие переходные металлы в катализе окисления олефинов до карбонильных соединений
Ограничив этот обзор развитием каталитической химии процессов окисления олефинов, катализируемых комплексами палладия, нельзя не отметить и успехи в создании каталитических систем для окисления олефинов на основе комплексов других переходных металлов. Некоторые комплексы металлов платиновой группы были протестированы в реакции окисления этилена еще в первой статье Смидта и сотр. [31]. Начиная с 1972 г., когда была установлена каталитическая активность катализатора Уилкинсона RhCl(PPh3)3 в реакции окисления олефинов кислородом до кетонов (одновременно с окислением PPh3 до PPh3=O) [378], комплексы других переходных металлов стали использовать для катализа окисления любых олефинов до кетонов (см. обзоры [13, 244, 245]). Весьма детально изучены каталитические системы на основе комплексов Rh(I) и Rh(III) [379–387], RuCl2 и RuCl3 [381, 384], Ir(I) и Ir(III) [388–390], Co(II) [391] и [Pt(III)]2 [392]. Недавно обнаружили, что даже комплексы Fe(II) в системе FeCl2–R3SiH–EtOH катализируют окисление различных олефинов воздухом при 80°С с высокими выходами [393]. Для большинства исследованных металлов обоснованы монооксигеназные или пероксидазные механизмы переноса атома кислорода на олефин, причем вторыми субстратами в монооксигеназных механизмах служат спирты [383] и Н2 [388].
8. ЗАКЛЮЧЕНИЕ
Одной из парадигм современной теории механизмов каталитических реакций является представление о множественности механизмов практически любого процесса. Разные механизмы реализуются и в рамках многомаршрутных каталитических реакций для одного и того же итогового уравнения маршрута, и в случае одномаршрутных механизмов одной и той же реакции при использовании различных каталитических систем. Так, например, анализ механизмов “простой” реакции димеризации алкинов [394] продемонстрировал разнообразие механизмов, зависящих от природы металла-катализатора, состава координационной сферы, степеней окисления одного и того же металла и от природы заместителей в алкине. Новый механизм димеризации олефинов (в добавление к трем известным) появился, как мы видели выше (разд. 6.1), при переходе к катионным комплексам палладия(II) и платины(II).
Аналогичная ситуация сложилась и в теории механизмов Вакер-окисления. Даже в классических условиях реакции окисления в хлоридных системах в зависимости от условий реакции обнаружено несколько механизмов (разд. 3). Механизмы реакций окисления олефинов зависят от степеней окисления палладия (II, I, 0), природы окислителя и со-окислителя, заряда комплекса, природы лигандов, растворителя, играющего роль лиганда, иногда катализатора процесса, и нередко от условий процесса. Как было показано в разд. 6.3, кинетические закономерности и результаты ряда изотопных исследований (разд. 6.2 и 6.3), принципиально различаются в случае классических хлоридных систем Вакер-окисления и в водных и водно-органических растворах катионных комплексов Pd(II) и Pd(I).
За последние 60 лет заметно продвинулось понимание деталей механизмов образования связи С–О в продуктах окисления олефинов, механизмов превращения σ-палладийорганических интермедиатов. Интересно, что редко обсуждаемый тип интермедиата (“енолятный”) был обнаружен даже в 2017 г. [343]. Прояснился вопрос об источниках атома кислорода в карбонильных продуктах. Кроме молекулы воды в классичесом Вакер-окислении, атом кислорода может поступать в молекулу кетона или альдегида из O2 ([Pd]O2, μ2-O гетерометаллических комплексов и через [Pd]OOH, разд. 4) и из Н2О2, ROOH и ${\text{NO}}_{{\text{2}}}^{ - }.$
Здесь уместно отметить, что научная школа И.И. Моисеева внесла существенный вклад и в понимание механизмов эпоксидирования олефинов гидропероксидами ROOH в растворах комплексов Mo(VI) [7 (Ch. 2, example 2.20), 395, 396] и окисления перфторолефинов, олефинов, антрацена и алканов [397–399] пероксидом водорода с участием промежуточных пероксокомплексов ванадия(V), включая ${\text{V}}({{{\text{O}}}_{{\text{2}}}})_{{\text{3}}}^{ - }$ [400].
Существенный прогресс достигнут и в понимании механизмов окисления комплексов Pd(0) п-хинонами и кислородом, окисления гидридных комплексов [Pd]–H кислородом [14, 175, 176, 395, 396, 401, 402]. Появилась новая информация о важной роли гетерометаллических комплексов PdCl2–CuCl2, PdCl2(CuCl2)n, Pd(OAc)2–Cu(OAc), двухядерных комплексов Pd(I) в механизмах процессов окисления олефинов (разд. 3, 4 и 6). В результате успешного развития химии кластеров палладия в исследованиях Моисеева и сотр., включая пионерские работы по синтезу гигантских кластеров, было установлено, что кластеры палладия катализируют также процессы окисления олефинов (и другие реакции) [7 (Ch. 5), 10, 403, 404].
Проведенный анализ квантово-химических исследований механизмов реакций окисления олефинов выявил ряд методологических проблем, затрудняющих использование результатов этих теоретических работ для дискриминации гипотез о возможных элементарных стадиях процесса окисления (разд. 3.6). Результаты этого анализа и собственный опыт (разд. 6.3, [346]) дают все основания согласиться с мнением известного специалиста и участника многих теоретических работ по механизму Вакер-окисления Дж. Кейта [49]: “Nucleophilic attacкs such as this (cis- or trans-hydroxypalladation) certainly must be reinvestigated when better methodology has been established to treat deprotonations in aqueous solution”.
Управление региоселективностью реакций Вакер-окисления и окислительной этерификации олефинов [10] (разд. 5) является существенным достижением последних 20 лет. Показано, что контролирующими региоселективность факторами являются:
1) “катализатор” – природа со-окислителя (или окислителя), состав координационной сферы активного комплекса и растворитель,
2) “субстрат” – природа олефина,
3) “механизм реакции” – в первую очередь, источник атома кислорода и природа интермедиатов.
Важную роль в дизайне каталитических систем для анти-М-окисления α-олефинов до альдегидов сыграло использование полифункциональных каталитических систем (ПФКС), в которых в качестве нуклеофилов вместо Н2О выступают мягкие нуклеофилы, например tBuOH и iPrOH, являющиеся так же, как и молекула воды, катализаторами процесса окисления. Образующиеся при этом в качестве интермедиатов алкениловые эфиры RCH=CHOtBu легко гидролизуются in situ с образованием tBuOH и альдегидов. Аналогичный подход был использован нами ранее [7, 405, 406] при создании ПФКС для анти-М-гидратации алкинов до альдегидов. Тройной катализатор (CuCl–RSH–HCl) в водном растворе NH4Cl в результате замены жесткого нуклеофила (Н2О) мягким и стерически нагруженным меркаптаном RSH позволил получить пропионовый альдегид из метилацетилена с выходом 70%.
Использование хиральных функциональнозамещенных олефинов или хиральных лигандов привело к появлению методов синтеза хиральных кетонов и альдегидов. Примеры асимметрического катализа и синтеза хиральных продуктов приведены в обзорах [13–15, 17, 18] и статьях [216, 218, 275]. Многочисленные результаты получены при исследовании внутримолекулярной окислительной реакции циклизации замещенных фенолов в соответствии с механизмом Вакер-окисления [13, 15–17, 216]. Активное использование реакции окисления олефинов в тонком органическом синтезе успешно продолжается. Значение для промышленного органического синтеза процессов окисления высших олефинов и циклоолефинов до кетонов и процесса синтеза винилацетата (реакция Моисеева) по-прежнему важно.
Весьма поучительная история развития каталитической химии реакций окисления олефинов (и других молекул) красноречиво демонстрирует, насколько сложной является задача выяснения механизма только одной реакции. Казалось бы, такие “однозначные” тесты на механизм, как определение стереохимии продуктов 1,2-окисления или окислительного карбонилирования олефинов, сохранение или перенос хиральных центров в процессах окисления и изомеризации замещенных аллиловых спиртов, оказались совсем не однозначными. Анализ этих исследований свидетельствует о необходимости применения комплекса методов с обязательным корректным изучением кинетики и использованием изотопных методик. Принципиально важным является также применение процедур дискриминации гипотез, какими бы очевидными ни казались полученные отдельные результаты [7 (Ch. 7)]. Очень хорошим примером в связи с этим является – изменение механизма Вакер-окисления в хлоридных системах при варьировании всего лишь двух параметров – [Cl]– и [H+] (разд. 3).
Проведенный анализ достижений в области синтеза кетонов и альдегидов, 1,1- и 1,2-продуктов окисления олефинов, синтеза сложных виниловых эфиров, хиральных карбонильных соединений и других продуктов свидетельствует о неисчерпаемых возможностях современного металлокомплексного катализа, в создании основ которого важное место занимает открытие и изучение процессов окисления олефинов в воде, спиртах и карбоновых кислотах, катализируемых комплексами палладия.
Список литературы
Моисеев И.И. π-Комплексы олефинов в жидкофазном окислении. М.: Наука, 1970. 270 с.
Maitlis P.M. The Organic Chemistry of Palladium. V. I, II. N.Y.: Acad. Press, 1971.
Henry P.M. Palladium-Catalyzed Oxidation of Hydrocarbons. Dordrecht: D. Reidel, 1980.
Tsuji J. Organic Synthesis with Palladium Compounds. Berlin–Heidelberg: Springer–Verlag, 1980.
Parshall G.W., Ittel S.D. Homogeneous Catalysis. N.Y.: Wiley, 1992.
Tsuji J. Palladium Reagents and Catalysts. Innovations in Organic Synthesis. Chichester: Wiley, 1995. 549 p.
Temkin O.N. Homogeneous Catalysis with Metal Complexes. Kinetic Aspects and Mechanisms. Chichester: Wiley, 2012. 802 p.
Moiseev I.I. Fundamental Research in Homogeneous Catalysis / Ed. Shilov A.E. N.Y.: Gordon and Breach, 1986. P. 12.
Jira R. Applied Homogeneous Catalysis with Organometallic Compounds. V. 1 / Eds. Cornils B., Herrmann W.A. Weinheim: VCH, 2002. P. 386–405.
Kozitsyna Yu., Vargaftik M.N., Moiseev I.I. // J. Organomet. Chem. 2000. V. 593–594. P. 274–291.
Henry P.M. Handbook of Organopalladium Chemistry for Organic Synthesis / Ed. Negishi E. N.Y.: Wiley & Sons, 2002. P. 2119.
Hintermann L. Transition Metals for Organic Synthesis. V. 2 / Eds. Beller M., Bolm C., 2004. P. 379–388.
Takacs J.M., Jiang X.-tian. // Curr. Org. Chem. 2003. V. 7. № 4. P. 369–396.
Stahl S.S. // Angew. Chem. Int. Ed. 2004. V. 43. P. 3400–3420.
Muzart J. // Tetrahedron. 2007. V. 63. P. 7505–7521.
Gligorich K.M., Sigman M.S. // Chem. Commun. 2009. P. 3854–3867.
McDonald R.I., Liu G., Stahl S.S. // Chem. Rev. 2011. V. 111. P. 2981–3019.
Sigman M.S., Werner E.W. // Accounts of Chem. Res. 2012. V. 45. P. 874–884.
Dong J.J., Browne W.R., Feringa B.L. // Angew. Chem. Int. Ed. 2015. V. 54. № 3. P. 734–744.
Wright J.A., Gaunt M.J., Spenser J.B. // Chem. Eur. J. 2006. V. 12. P. 949.
Morandi B., Wickens Z.K., Grubbs R.H. // Angew. Chem. Int. Ed. 2013. V. 52 № 10. P. 2944.
Iwabuchi Y. Green Oxidation in Organic Synthesis / Eds. Jiao N., Stahl S.S. Chichester: Wiley, 2019. P. 35–79.
Jira R. Acetaldehyde. Ullmann’s Encyclopedia of Industrial Chemistry. 1985. V. A1. P. 31.
Темкин О.Н. Каталитические реакции. Современное естествознание. Энциклопедия. Т. 1. Физическая химия. М.: Наука-Флинта, 1999. С. 137–145.
Темкин О.Н. Гомогенный катализ. Большая российская энциклопедия. Т. 7. M.: БРЭ, 2007. С. 382.
Catalysis from A to Z. Concise Encyclopedia. / Eds. Cornils B., Herrmann W.A., Wong C.-H., Zanthoff H.-W. Weinheim: Wiley-VCH, 2013. P. 1499, 2388.
Jira R. // Angew. Chem. Int. Ed. 2009. V. 48. P. 9034.
Моисеев И.И. Из истории катализа: люди, события, школы / Под ред. Кальнера В.Д. M.: Калвис, 2005. С. 361.
Phillips F.C. // Am. Chem. J. 1894. V. 16. P. 255.
Anderson J.S. // J. Chem. Soc. 1934. P. 971.
Smidt J., Hafner W., Jira R., Sedlmeier J., Sieber R., Kojer H., Rüttinger R. // Angew. Chem. 1959. B. 71. S. 176.
Kharasch M.S., Seyler R.C., Mayo F.R. // J. Amer. Chem. Soc. 1938. V. 60. P. 882.
Темкин О.Н., Флид Р.М., Малахов А.И. // Кинетика и катализ. 1963. Т. 4. С. 270.
Варгафтик М.Н., Моисеев И.И., Сыркин Я.К. // Докл. АН СССР. 1960. Т. 130. С. 820.
Fischer E.O., Hafner W. // Zt. Naturforschungen. 1954. 9b. S. 503.
Fischer E.O., Jira R. // Zt. Naturforschungen. 1954. 9b. S. 618.
Fischer E.O., Jira R. // Zt. Naturforschungen. 1954. 10b. S. 355.
Smidt J., Hafner W. // Angew. Chem. 1959. B. 71. P. 284.
Моисеев И.И., Федоровская Э.А., Сыркин Я.К. // Журн. неорган. химии. 1959. Т. 4. С. 2641.
Моисеев И.И., Варгафтик М.Н., Сыркин Я.К. // Докл. АН СССР. 1960. Т. 133. С. 377.
Smidt J., Hafner W., Jira R., Sieber R., Sedlmeier J., Sabel A. // Angew. Chem. 1962. B. 74. S. 93.
Темкин О.Н., Флид Р.М. Каталитические превращения ацетиленовых соединений в растворах комплексов металлов. М.: Наука, 1968. 212 с.
Варгафтик М.Н., Моисеев И.И., Сыркин Я.К. // Докл. АН СССР. 1962. Т. 147. С. 399.
Моисеев И.И., Варгафтик М.Н., Сыркин Я.К. // Докл. АН СССР. 1963. Т. 153. С. 140.
Henry P.M. // J. Amer. Chem. Soc. 1964. V. 86. P. 3246.
Леванда О.Г., Моисеев И.И. // Кинетика и катализ. 1971. Т. 12. № 3. С. 567.
Moiseev I.I., Levanda O.G., Vargaftik M.N. // J. Amer. Chem. Soc. 1974. V. 96. P. 1003.
Henry P.M. // J. Org. Chem. 1973. V. 38. P. 2415.
Keith J.A., Henry P.M. // Angew. Chem. Int. Ed. 2009. V. 48. P. 9038.
Jira R., Sedlmeier J., Smidt J. // Ann. Chem. 1966. B. 693. S. 99.
Варгафтик М.Н., Моисеев И.И., Сыркин Я.К. // Докл. АН СССР. 1966. Т. 166. С. 370.
Варгафтик М.Н., Моисеев И.И., Сыркин Я.К. // Докл. АН СССР. 1961. Т. 139. С. 1396.
Hafner W., Jira R., Sedlmeier J., Smidt J. // Chem. Ber. 1962. B. 95. S. 1575.
Osada H., Hashimoto H. // J. Chem. Soc. Jpn. 1966. V. 69. P. 2137.
Katsuyama Y., Hashimoto H. // Bull. Chem. Soc. Jpn. 1968. V. 41. № 6. P. 1395.
Захарова Л.М., Варгафтик М.Н., Моисеев И.И., Кацман Е.А. // Кинетика и катализ. 1969. Т. 10. № 4. С. 901.
Захарова Л.М., Варгафтик М.Н., Моисеев И.И., Кацман Е.А. // Изв. АН СССР. Сер. хим. 1970. № 3. С. 700.
Мартынова Л.М., Кацман Е.А. // Кинетика и катализ. 1990. Т. 31. № 4. С. 844.
Koĉovsky P., Bäckvall J.-E. // Chem. Eur. J. 2015. V. 21. P. 36–56.
Henry P.M., Marks O.W. // Inorg. Chem. 1971. V. 10. P. 373.
Sabel A., Smidt J., Jira R., Prigge H. // Chem. Ber. 1969. B. 102. S. 2939.
Henry P.M. // J. Am. Chem. Soc. 1971. V. 93. P. 3853.
Henry P.M. // J. Am. Chem. Soc. 1972. V. 94. P. 1527.
Henry P.M. // J. Am. Chem. Soc. 1972. V. 94. P. 5200.
Henry P.M. // J. Org. Chem. 1972. V. 37. P. 2443.
Henry P.M. // Inorg. Chem. 1972. V. 11. P. 1876.
Henry P.M. // J. Am. Chem. Soc. 1972. V. 94. P. 7311.
Henry P.M. // Accounts Chem. Res. 1973. V. 6. P. 16.
Henry P.M., Ward G.A. // J. Am. Chem. Soc. 1971. V. 93. P. 1494.
Henry P.M. // J. Am. Chem. Soc. 1972. V. 94. P. 7305.
Winstein S., McCaskie J., Lee H.-B., Henry P.M. // J. Am. Chem. Soc. 1976. V. 98. P. 6913.
Lee H.-B., Henry P.M. // Can. J. Chem. 1976. V. 54. P. 1726.
Белов А.П., Сацко Н.Г., Моисеев И.И. // Кинетика и катализ. 1972. Т. 13. С. 892.
Белов А.П., Моисеев И.И., Уварова Н.Г. // Изв. АН СССР. Сер.хим. 1965. № 12. С. 2224.
Белов А.П., Моисеев И.И., Уварова Н.Г. // Изв. АН СССР. Сер. хим. 1966. № 9. С. 1642.
Моисеев И.И., Белов А.П., Игошин В.А., Сыркин Я.К. // Докл. АН СССР. 1967. Т. 173. С. 863.
Моисеев И.И. π-Комплексы олефинов в жидкофазном окислении. М.: Наука, 1970. С. 123–144.
Белов А.П., Моисеев И.И. // Изв. АН СССР. Сер. хим. 1966. № 1. С. 139.
Schultz R., Gross D. Homogeneous Catalysis. Advances in Chemistry. V. 70 / Ed. Luberoff B.J. Washington: Amer. Chem. Soc., 1974. P. 97–125.
Pandey R.N., Henry P.M. // Can. J. Chem. 1974. V. 52. P. 1241.
Pandey R.N., Henry P.M. // Can. J. Chem. 1975. V. 53. P. 2223.
Majima T., Kurosawa H. // J. C. S. Chem. Commun. 1977. P. 610.
Stille J.K., Morgan K.A. // J. Am. Chem. Soc. 1966. V. 88. P. 5135.
Stille J.K., James D.E., Hines L.F. // J. Am. Chem. Soc. 1973. V. 95. P. 5064.
Stille J.K., Wong P.K. // J. Org. Chem. 1975. V. 40. P. 335.
Stille J.K., James D.E., Hines L.F. // J. Am. Chem. Soc. 1975. V. 97. P. 674.
Stille J.K., Divakaruni R. // J. Organomet. Chem. 1979. V. 169. P. 239.
Akermark B., Backvall J.-E., Siirula-Hansen K., Sjoberg K., Zellenberg K. // Tetrahedron Lett. 1974. P. 1362.
Akermark B., Backvall J.-E., Siirula-Hansen K., Sjoberg K., Zellenberg K. // Tetrahedron Lett. 1975. P. 819.
Backvall J.-E., Akermark B., Ljunggren S.O. // J. Am. Chem. Soc. 1979. V. 101. P. 2411.
Backvall J.-E., Nordberg R.E. // J. Am. Chem. Soc. 1981. V. 103. P. 4959.
Andell O.S., Backvall J.-E. // J. Organomet. Chem. 1983. V. 244. P. 401.
Backvall J.-E. // Acc. Chem. Res. 1983. V. 16. P. 335.
Hall S.S., Akermark B. // Organometallics. 1984. P. 1745.
Akermark B., Soderberg B.C., Hall S.S. // Organometallics. 1987. P. 2608.
Stangl H., Jira R. // Tetrahedron Lett. 1970. P. 3589.
Akermark B., Almemark M., Almlof J., Backvall J.-E., Ross B., Stogard A. // J. Am. Chem. Soc. 1977. V. 99. P. 4617.
Eisenstein O., Hoffmann R. // J. Am. Chem. Soc. 1981. V. 103. P. 4308.
Backvall J.-E., Bjorkman E.E., Petersson L., Siegbahn P. // J. Am. Chem. Soc. 1984. V. 106. P. 4369.
Брайловский С.М., Темкин О.Н., Шестакова В.С., Куперман А.Ф. // Кинетика и катализ. 1981. Т. 23. № 6. С. 1446.
Алексеева Н.Ф., Темкин О.Н., Флид Р.М. // Кинетика и катализ. 1970. Т. 11. № 6. С. 1580.
Светлова А.Б., Брайловский С.М., Темкин О.Н. // Кинетика и катализ. 1978. Т. 19. № 6. С. 1588.
Пшеничникова А.Б., Брайловский С.М., Темкин О.Н. Химическая кинетика в катализе. Кинетические модели жидкофазных реакций. Сб. / Под ред. Кипермана С.Л. Черноголовка: Изд-во АН СССР, 1985. С. 67.
Пшеничникова А.Б., Брайловский С.М., Темкин О.Н. // Кинетика и катализ. 1989. Т. 30. № 1. С. 92.
Gregor N., Henry P.M. // J. Am. Chem. Soc. 1981. V. 103. P. 681.
Zao K., Lautens M., Henry P.M. // Organometallics.1983. V. 2. P. 197.
Zao K., Lautens M., Henry P.M. // Organometallics. 1985. V. 4. P. 1286.
Wan W.K., Zao K., Henry P.M. // Organometallics. 1988. V. 7. P. 1677.
Gregor N., Zao K., Henry P.M. // Organometallics. 1984. V. 3. P. 1251.
Whitesides G.M., Goasch J.F., Stedrousky E.R. // J. Am. Chem. Soc. 1972. V. 94. P. 5258.
Hosokawa T., Sugafuji T., Yamanaka T., Murahashi S.-I. // J. Organomet. Chem. 1994. V. 470. P. 253.
Zhang Z., Lu X., Xu Z., Zhang Q., Han X. // Organometallics. 2001. V. 20. P. 3724.
Francis J.W., Henry P.M. // Organometallics. 1991. V. 10. P. 3498.
Francis J.W., Henry P.M. // Organometallics. 1992. V. 11. P. 2832.
Francis J.W., Henry P.M. // J. Mol. Catal. 1995. V. 99. P. 77.
Francis J.W., Henry P.M. // J. Mol. Catal. 1996. V. 112. P. 317.
Hamed O., Thompson C., Henry P.M. // J. Org. Chem. 1997. V. 62. P. 7082.
Hamed O., Henry P.M. // Organometallics. 1997. V. 16. P. 4903.
Hamed O., Henry P.M., Thompson C. // J. Org. Chem. 1999. V. 64. P. 7745.
Hamed O., Henry P.M. // Organometallics. 1998. V. 17. P. 5184.
El-Qisairi A., Henry P.M. // J. Organomet. Chem. 2000. V. 603. P. 50.
El-Qisairi A., Qaseer H.A., Henry P.M. // J. Organomet. Chem. 2002. V. 656. P. 168.
Henry P.M. Homogeneous Catalysis. Advances in Chemistry. V. 70 / Ed. Luberoff B.J. Washington: Amer. Chem. Soc., 1974. P. 126–154.
Моисеев И.И., Варгафтик М.Н. // Изв. АН СССР. Сер. хим. 1965. № 4. С. 759.
Stern E.W. // Proc. Chem. Soc. 1963. P. 111.
Brookhart M., Green M.L.H. // J. Organomet. Chem. 1983. V. 250. P. 395.
Митченко С.А., Шубин А.А. Металлоорганические производные ацидокомплексов платины: синтез, реакционная способность и дизайн каталитических реакций. Донецк: ДонГУЭТ, 2004. 217 с.
Матвеев К.И., Бухтияров И.Ф., Шульц Н.Н., Емельянова О.А. // Кинетика и катализ. 1964. Т. 5. С. 572.
Francois P., Trambouaz Y. // Bull. Soc. Chim. Fr. 1969. P. 31.
Francois P. // Ann. Chim. (Paris). 1969. V. 14. № 4. P. 370.
Моисеев И.И., Пестриков С.В. // Докл. АН СССР. 1966. Т. 171. С. 151.
Моисеев И.И., Пестриков С.В., Сверж С.В. // Кинетика и катализ. 1969. Т. 10. № 1. С. 74.
Bruk L.G., Temkin O.N., Goncharova Z.V., Flid V.R. // React. Kinet. Catal. Lett. 1973. V. 9. P. 303.
Rappoport Z., Sleezer P.D., Winstein S., Young W.G. // Tetrahedron Lett. 1965. P. 3719.
Шлапак М.С., Брайловский С.М., Темкин О.Н. // Кинетика и катализ. 1983. Т. 24. № 6. С. 1380.
Скумов М.Я., Брайловский С.М., Темкин О.Н. // Изв. АН СССР. Сер. хим. 2002. № 4. С. 572.
Скумов М.Я., Брайловский С.М., Темкин О.Н. // Журн. орган. химии. 2003. Т. 39. № 2. С. 199.
Брайловский С.М., Элефтериу Л., Чернышова О.Н., Белов А.П., Темкин О.Н. // Кинетика икатализ. 1982. Т. 23. № 1. С. 54.
Backvall J.-E., Hopkins R.B., Grennberg H., Mader M.M., Awasthi A.K. // J. Am. Chem. Soc. 1990. V. 112. P. 5160.
Backvall J.-E., Bystrom S.E., Nordberg P.E. // J. Org. Chem. 1984. V. 49. P. 4619.
Backvall J.-E., Awasthi A.K., Renko Z.D. // J. Am. Chem. Soc. 1987. V. 109. P. 4752.
Grennberg H., Faizo S., Backvall J.-E. // Angew. Chem. Int. Ed. 1993. V. 32. № 2. P. 263.
Grennberg H., Backvall J.-E. // J.C.S. Chem. Commun. 1993. P. 1331.
Grennberg H., Gogoll A., Backvall J.-E. // Organometallics. 1993. V. 12. P. 1790.
Milani B., Anzilietti A., Vicentini L. // Organometallics. 1997. V. 16. P. 5064.
Калабин С.М., Белов А.П. // Кинетика и катализ. 1984. Т. 25. № 2. С. 287.
Ефремов Г.Е., Бовырина Е.А., Подтягина А.В., Ошанина И.В., Темкин О.Н. // Кинетика и катализ. 2019. Т. 60. № 1. С. 54.
Жир-Лебедь Л.Н., Темкин О.Н. // Кинетика и катализ. 1984. Т. 25. С. 316.
Жир-Лебедь Л.Н., Темкин О.Н. // Кинетика и катализ. 1984. Т. 25. С. 325.
Stern E.W., Spector M.L. // Proc. Chem. Soc. 1961. P. 370.
Tsutsui M., Ori M., Fransis J. // J. Am. Chem. Soc. 1972. V. 94. P. 1414.
Beck I.E., Gusevskaya E.V., Stepanov A.G., Likholobov V.A. // J. Mol. Catal. A. Chem. 1992. V. 73. P. 115.
Gusevskaya E.V. // Quim. Nova. 2003. V. 26. № 2. P. 242.
Heck R. // J. Am. Chem. Soc. 1968. V. 90. P. 5518.
Heck R. // J. Am. Chem. Soc. 1968. V. 90. P. 5526.
Heck R. // J. Am. Chem. Soc. 1968. V. 90. P. 5531.
Темкин О.Н., Калия О.Л., Шестаков Г.К., Флид Р.М. // Докл. АН СССР. 1970. Т. 190. С. 398.
Rodenheaver G.T., Hunt D.F. // Chem. Commun. 1971. P. 818.
Backvall J.-E., Akermark B. // J. C. S. Chem. Commun. 1975. P. 82.
Heumann A., Backvall J.-E. // Angew. Chem. 1985. B. 97. № 3. S. 228.
Мартынова Л.М., Кацман Е.А. // Кинетика и катализ. 1989. Т. 30. № 6. С. 1489.
Uemura S., Zush K., Okano M. // J. C. S. Chem. Commun. 1972. P. 234.
Kabbe J. // Lieb. Annalen. 1962. B. 656. S. 204.
Брайловский С.М., Брук Л.Г., Костюшин А.С., Темкин О.Н., Флид Р.М. // Кинетика и катализ. 1973. Т. 14. № 5. С. 1222.
Брайловский С.М., Темкин О.Н., Костюшин А.С., Одинцов К.Ю. // Кинетика и катализ. 1990. Т. 31. № 6. С. 1371.
Брайловский С.М., Темкин О.Н., Ходакова С.И., Флид Р.М. // Журн. орган. химии. 1975. Т. 11. С. 672.
Navon N., Golub G., Cohen H., Meyerstein D. // Organometallics. 1995. V. 14. P. 5670.
Naumann D., Roy T., Tebbe K.-F., Crump W. // Angew. Chem. Int. Ed. 1993. V. 32. № 10. P. 1482.
Furuta H., Ishizuka T., Osuka A., Uwatoko Y., Ishikawa Y. // Angew. Chem. Int. Ed. 2001. V. 40. № 12. P. 2323.
Bartholomew E.R., Bertz S.H., Cope S., Murphy M., Ogle C. // J. Am. Chem. Soc. 2008. V. 130. P. 11 244.
King A., Huffman L.M., Casitas A., Costas M., Rifus X., Stahl S.S. // J. Am. Chem. Soc. 2010. V. 132. P. 12 068.
Giannoccaro P., Ravasio N., Aresta M. // J. Organomet. Chem. 1993. V. 451. P. 243.
Larock R. // J. Org. Chem. 1976. V. 41. P. 2241.
Шестакова В.С., Омельченко Т.Н., Брайловский С.М., Темкин О.Н. // Журн. орган. химии. 1978. Т. 14. С. 2346.
Konnick M.M., Stahl S.S. // J. Am. Chem. Soc. 2008. V. 130. P. 5753.
Konnick M.M., Popp B.V., Stahl S.S. // J. Am. Chem. Soc. 2011. V. 133. P. 13 268.
Backvall J.-E., Bjorkman E.E., Pettersson L., Siegbahn P. // J. Am. Chem. Soc. 1985. V. 107. P. 7265.
Siegbahn P. // J. Am. Chem. Soc. 1995. V. 117. P. 5409.
Kragten D.D., Van Santen O., Lerou J.J. // J. Phys. Chem. 1999. V. 103. P. 80.
Siegbahn P.E.M. // J. Phys. Chem. 1996. V. 100. P. 14 672.
Beyramabadi S.A., Eshtiagh-Hosseini H., Housaindokht M.R., Morsali A. // Organometallics. 2008. V. 27. P. 72.
Keith J.A., Nielsen R.J., Oxgaard J., Goddard III W.A., Henry P.M. // Organometallics. 2009. V. 28. P. 1618.
Keith J.A., Oxgaard J., Goddard W.A. // J. Am. Chem. Soc. 2006. V. 128. P. 3132.
Keith J.A., Nielsen R.J., Oxgaard J., Goddard W.A. // J. Am. Chem. Soc. 2007. V. 129. P. 12 342.
Anderson B.J., Keith J. A., Sigman M.S. // J. Am. Chem. Soc. 2010. V. 132. P. 11 872.
Comas-Vives A., Stirling A., Liedos A., Ujaque G. // Chem. Eur. J. 2010. V. 16. P. 8738.
Kovacs G., Stirling A., Liedos A., Ujaque G. // Chem. Eur. J. 2012. V. 18. P. 5612.
Imandi V., Kunnikurun S., Nair N.N. // Chem. Eur. J. 2013. V. 19. P. 4724.
Темкин О. О различных взаимосвязях кинетики и термодинамики. Saarbrücken: Lambert Acad. Publ., 2016. 119 с.
Clement W.H., Selwitz C.M. // J. Org. Chem. 1964. V. 29. P. 241.
Fahey D.R., Zuech E.A. // J. Org. Chem. 1974. V. 39. № 22. P. 3276.
McQuillin F.J., Parker D.G. // J. C. S. Perkin I. 1974. P. 809.
Coe J.S., Unsworth J.B.J. // J. C. S. Dalton Trans. 1975. P. 645.
Tsuji J., Shimizu I., Yamamoto K. // Tetrahedron Lett. 1976. P. 2975.
Tsuji J. // Synthesis. 1984. P. 369.
Hosokawa T., Nomura T., Murahashi S.-I. // J. Organomet. Chem. 1998. V. 551. P. 387.
Mitsudome T.L., Umetani T., Nosaka N., Mori K., Mizugaki T., Ebitani K., Kaneda K. // Angew. Chem. Int. Ed. 2006. V. 45. № 3. P. 481.
Fernandes R.A., Chaudhari D.A. // J. Org. Chem. 2014. V. 79. № 12. P. 5787.
Wang J.-L., He L.-N., Miao C.-X., Li Y.-N. // Green Chem. 2009. V. 11. P. 1317.
Lloyd W.G., Luberoff B.I. // J. Org. Chem. 1969. V. 34. P. 3949.
Wang Y.-F., Gao Y.-R., Mao S., Zhang Y.-L., Guo D.-D., Yan Z.-L., Guo S.-H., Wang Y.-Q. // Org. Lett. 2014. V. 16. P. 1610.
Michel B.W., Camelio A.M., Cornell C.N., Sigman M.S. // J. Am. Chem. Soc. 2009. V. 131. P. 6076.
Григорьев А.А., Моисеев И.И., Клименко М.Я., Липина Ю.Н. // Хим. пром-сть. 1972. Т. 48. С. 14.
Nishimura T., Kakiuchi N., Onoue T., Ohe K., Uemura S. // J. C. S. Perkin Trans. 1. 2000. P. 1915.
Ogura T., Kamimura R., Shiga A., Hosokawa T. // Bull. Chem. Soc. Jpn. 2005. V. 78. P. 1555.
Conti F., Donati M., Pregaglia G.F.J. // Organomet. Chem. 1971. V. 30. P. 421.
Кислина И., Сысоева С., Темкин О.Н. // Изв. АН СССР. Сер. хим. 1994. № 6. С. 1025.
Кислина И.C., Сысоева С., Либрович Н.Б., Темкин О.Н., Еременко И.Л., Нефедов С.Е. // Докл. АН. 1998. Т. 360. № 5. С. 649.
Hosokawa T., Takano M., Murahashi S.-I. Ozaki H., Kitagawa Y., Sakagichi K.-I., Katsube Y. // J. C. S. Chem. Commun. 1994. P. 1433.
Hosokawa T., Takano M., Murahashi S.-I. // J. Am. Chem. Soc. 1996. V. 118. P. 3990.
Holland P.L., Tolman W.B. // Coordin. Chem. Rev. 1999. V. 190–192. P. 855–869.
Straub B.F., Rominger F., Hofmann P. // Chem. Commun. 2000. P. 1611.
Wikens Z.K., Skakuj K., Morandi B., Grubbs R.H. // J. Am. Chem. Soc. 2014. V. 136. № 3. P. 890.
Ning Y.-S., Wang M.-M., Yao C.-Z., Chen X.-M., Kang Y.-B. // Org. Lett. 2016. V. 18. № 11. P. 2700.
Roussel M., Mimoun H. // J. Org. Chem. 1980. V. 45. P. 5387.
Hosokawa T., Uno T., Inui S., Murahashi S.-I. // J. Am. Chem. Soc.1981. V. 103. P. 2318.
Semmelhack M.F., Kim C.R., Dobler W., Meier M. // Tetrahedron Lett. 1989. V. 30. № 37. P. 4925.
Van Benthem R.A.T.M., Hiemstra H., Michels J.J., Speckamp W.N. // J. C. S. Chem. Commun. 1994. P. 357.
Van Benthem R.A.T.M., Hiemstra H., Van Leeuwen P.W.N.M., Geus J.W., Speckamp W.N. // Angew. Chem. Int. Ed. 1995. V. 14. № 4. P. 457.
Yokota T., Fujibayashi S., Nishiyama Y., Sakaguchi S., IshiiY. // J. Mol. Catal. A. Chem. 1996. V. 114. P. 113.
Ten Brink G.-J., Arends I.W.C.E., Papadogianakis S.G., Sheldon R.A. // Chem. Commun. 1998. P. 2359.
Ten Brink G.-J., Arends I.W.C.E., Papadogianakis S.G., Sheldon R.A. // Appl. Catal. A. 2000. V. 194–195. P. 435.
Chaudhari D.A., Fernandes R.A. // J. Org. Chem. 2016. V. 81. № 5. P. 2113.
Smith A.B.III., Cho Y.S., Friestad G.K. // Tetrahedron Lett. 1998. V. 39. P. 8765.
Fernandes R.A., Bethi V. // Tetrahedron. 2014. V. 70. P. 4760.
Kulkarni M.G., Shaikh Y.B., Borhade A.S., Chavhan S.W., Dhondge A.P., Gaikwad D.D., Desai M.P., Birhade D.R., Dhatrak N.R. // Tetrahedron Lett. 2013. V. 54. № 19. P. 2293.
Smidt J., Jira R. // Chem. Ber. 1960. B. 93. S. 162.
Andrews M.A., Kelly K.P. // J. Am. Chem. Soc. 1981. V. 103. P. 2894.
Heumann A., Reglier M., Waegell B. // Angew. Chem. Int. Ed. 1979. V. 18. P. 866.
Heumann A., Chauvet F., Waegell B. // Tetrahedron Lett. 1982. V. 23. № 27. P. 2767.
Andrews M.A., Cheng C.-W. // J. Am. Chem. Soc. 1982. V. 104. P. 4268.
Diamond S.E., Mares F., Szalkiewicz A., Muccigrosso D.A., Solar J.P. // J. Am. Chem. Soc. 1982. V. 104. P. 4266.
Andrews M. A., Chang T. C.-T., Cheng C.-W.F., Emge T.F., Kelly K.P., Koetzle T.F. // J. Am. Chem. Soc. 1984. V. 106. P. 5913.
Andrews M. A., Chang T. C.-T., Cheng C.-W.F., Kelly K.P. // Organometallics. 1984. V. 3. № 12. P. 1777.
Andrews M.A., Chang T.C.-T., Cheng C.-W.F. // Organometallics. 1985. V. 4. P. 268.
Kozhevnikov I.V. Catalysis by Polyoxometalates. Chichester: Wiley, 2002.
Матвеев К.И., Жижина Е.Г., Шитова Н.Б., Кузнецова Л.И. // Кинетика и катализ. 1977. Т. 18. С. 380.
Zhizhina E.G., Simonova M.V., Odyakov V.F., Matveev K.I. // Appl. Catal. A. Gen. 2007. V. 319. P. 91.
Grate J.H., Hamm D.R., Mahajan S. // Molecular Engineering. 1993. V. 3. P.205.
Lambert A., Derouane E.G., Kozhevnikov I.V. // J. Catal. 2002. V. 211. P. 445.
Kuznetsova N.I., Fedotov M.A., Likholobov V.A., Yermakov Y.I. // J. Mol. Catal. 1986. V. 38. P. 263.
Tsuji J., Nagashima H., Hori K. // Chem. Lett. 1980. P. 251.
Mimoun H., Charpentier R., Mitschler A., Fisher J., Weiss R. // J. Am. Chem. Soc. 1980. V. 102. P. 1047.
Mimoun H. // Pure Appl. Chem. 1981. V. 53. P. 2389.
Mimoun H. // Angew. Chem. Int. Ed. 1982. V. 21. P. 734.
Cornell C.N., Sigman M.S. // Org. Lett. 2006. V. 8. № 18. P. 4117.
Hosokawa T., Ohkata H., Moritani I. // Bull. Chem. Soc. Jpn. 1975.V. 48. P. 1533.
Hosokawa T., Yamashita S., Murahashi S.-I., Sonoda A. // Bull. Chem. Soc. Jpn. 1976. V. 49. P. 3663.
Hosokawa T., Hirata M., Murahashi S.-I., Sonoda A. // Tetrahedron Lett. 1976. P. 1821.
Hosokawa T., Shimo N., Maeda K., Sonoda A., Murahashi S.-I. // Tetrahedron Lett. 1976. P. 383.
Hosokawa T., Miyagi S., Murahashi S.-I., Sonoda A. // J. Chem. Soc. Chem. Commun. 1978. P. 687.
Hosokawa T., Miyagi S., Murahashi S.-I., Sonoda A. // J. Org. Chem. 1978. V. 43. P. 2752.
Hosokawa T., Uno T., Murahashi S.-I. // J. C. S. Chem. Commun. 1979. P. 475.
Hegedus L S., Allen G.F., Bozell J.J., Waterman E.L. // J. Am. Chem. Soc. 1978. V. 100. P. 5800.
Larock R.C., Hightower T.R. // J. Org. Chem. 1993. V. 58. P. 5298.
Larock R.C., Wei L., Hightower T.R. // Synlett. 1998. P. 522.
Roshchin A.I., Kel’chevski S.V., Bumagin N.A. // J. Organomet. Chem. 1996. V. 560. P. 163.
Trend R.M., Ramtohul Y.R., Ferreira E.M., Stoltz B.M. // Angew. Chem. Int. Ed. 2003. V. 42. P. 2892.
Muzart J. // Eur. J. Org. Chem. 2010. P. 3779.
Gao W., He Z., Qian Y., Zhao J., Huang Y. // Chem. Sci. 2012. V. 3. P. 883.
Лихолобов В.А., Ермаков Ю.И. // Кинетика и катализ. 1980. Т. 21. № 4. С. 904.
Зудин В.Н., Лихолобов В.А., Ермаков Ю.И. // Кинетика и катализ. 1977. Т. 18. № 4. С. 921.
Брук Л.Г., Ошанина И.В., Городский С.Н., Темкин О.Н. // Рос. хим. журн. (Ж. Рос. xим. об-ва им. Д.И. Менделеева). 2006. Т. 50. № 4. С. 103.
Feringa B.L. // J. Chem. Soc. Chem. Commun. 1986. P. 909.
Kiers N.H., Feringa B.L., Kooijman H., Spek A.L., van Leeuwen P.W.N.M. // J. C. S. Chem. Commun. 1992. P. 1169.
Kiers N.H., Feringa B.L., van Leeuwen P.W.N.M. // Tetrahedron Lett. 1992. V. 33. P. 2403.
Meulemans T.M., Kiers N.H., Feringa B.L., van Leeuwen P.W.N.M. // Tetrahedron Lett. 1994. V. 35. P. 455.
Wenzel T.T. // J. C. S. Chem. Commun. 1993. P. 862.
Keinan E., Seth K.K., Lamed R. // J. Am. Chem. Soc. 1986. V. 108. P. 3474.
Gaunt M.J., Yu J., Spenser J.B. // Chem. Commun. 2001. P. 1844.
Wright J.A., Gaunt M.J., Spenser J.B. // Chem. Eur. J. 2006. V. 12. P. 949.
Темкин О.Н. // Кинетика и катализ. 2014. Т. 55. № 2. С. 181.
Ho T.L., Chang M.Y., Chen C. // Tetrahedron Lett. 2003. V. 44. P. 6955.
Cornell C.N., Sigman M.S. // Inorg. Chem. 2007. V. 46. P. 1903.
Weiner B., Baeza A., Jerphagnon T., Feringa B.L. // J. Am. Chem. Soc. 2009. V. 131. P. 9473.
Teo P., Wickens Z.K., Dong G., Grubbs R.H. // Org. Lett. 2012. V. 14. № 13. P. 3237.
Wickens Z.K., Morandi B., Grubbs R.H. // Angew. Chem. Int. Ed. 2013. V. 52. P. 11 257.
Kim K.E., Li J., Grubbs R.H., Stoltz B.M. // J. Am. Chem. Soc. 2016. V. 138. P. 13 179.
Dong G., Teo P., Wickens Z.K., Grubbs R.H. // Science. 2011. V. 333. P. 1609.
Dong J.-J., Browne W.R., Feringa B.L. // Angew. Chem. Int. Ed. 2014. V. 53. № 3. P. 734.
Jiang Y.-Y., Zhang O., Yu H., Fu Y. // ACS Catalysis. 2015. V. 5. № 3. P. 1414.
Crabtree R. // Acc. Chem. Res. 1979. V. 12. P. 331.
Landis C.K., Halpern J. // J. Organomet. Chem. 1983. V. 250. P. 485.
Шилов А.Е., Зефирова А.К., Тихомирова Н.М. // Журн. физ. химии. 1959. Т. 33. С. 2113.
Дьячковский Ф.С., Шилов А.Е. // Успехи химии. 1966. Т. 35. № 4. С. 699.
Ittel S.D., Johnson L.K., Brookhart M. // Chem. Rev. 2000. V. 100. P. 1169.
Taube R., Silvester G. Applied Homogeneous Catalysis with Organometallic Compounds. / Eds. Cornils B., Hermann W.A. Weinheim: Wiley–VCH, 2000. P. 298.
Nifant’ev I.E., Ustynyuk L.Yu., Besedin D.V., Pasabu A. // Organometallics. 2013. V. 12. P. 173.
Устынюк Л.Ю., Фушман Э.А., Разави А. // Кинетика и катализ. 2006. Т. 47. № 2. С. 215.
Ustynyuk L.Yu., Fushman E.A. // Mendeleev Commun. 2009. V. 19. P. 182.
Ustynyuk L.Yu., Fushman E.A., Lalayan S.S., Nifantev I.E. // J. Organomet. Chem. 2012. V. 700. P. 166.
Hansen S.M., Volland M.A.O., Rominger F., Eisentrager F., Hofman P. // Angew. Chem. Int. Ed. 1999. V. 38. P. 1273.
Chow D., Robson J.H., Wright G.F. // Can. J. Chem. 1965. V. 43. P. 312.
Соколов В.И., Устынюк Ю.А., Реутов О.А. // Докл. АН СССР. 1967. Т. 173. № 5. С. 1103.
Соколов В.И., Башилов В.В., Реутов О.А. // Докл. АН СССР. 1969. Т. 188. № 1. С. 127.
Brown H.C., Kurek J.T. // J. Am. Chem. Soc. 1969. V. 91. P. 5647.
Kabbe H.J. // Lieb. Ann. 1962. B. 656. S. 204.
Beller M., Seayad J., Tillack A., Jiao H. // Angew. Chem. Int. Ed. 2004. V. 43. P. 3368.
Olah G.A., Clifford P.K. // J. Am. Chem. Soc. 1971. V. 93. P. 1261.
Соколов В.И., Башилов В.В., Реутов О.А. // Докл. АН СССР. 1973. Т. 213. № 5. С. 1103.
Темкин О.Н., Есикова И.А., Могилянский А.И., Флид Р.М. // Кинетика и катализ. 1971. Т. 12. С. 915.
Kawai I., Nakajima R., Hara T. // Bull. Chem. Soc. Jpn. 1970. V. 43. P. 749.
Charavel B., Metzger J. // Bull. Soc. Chim. Fr. 1968. P. 4102.
Есикова И.А. Диc. … канд. хим. наук. М.: МИТХТ, 1970.
Харченко А.А., Идлис Г.С., Гурфейн Н.С. // Журн. орг. химии. 1970. Т. 6. № 7. С. 137. (и ссылки в статье).
Ichikawa K., Fijita K., Ouchi H. // J. Amer. Chem. Soc. 1959. V. 81. P. 5316.
Henry P.M. // J. Am. Chem. Soc. 1966. V. 88. № 8. P. 1597.
Henry P.M. // J. Am. Chem. Soc. 1965. V. 87. № 5. P. 990.
Henry P.M. // J. Org. Chem. 1973. V. 38. № 13. P. 2415.
Sen A., Lai T.-W. // J. Am. Chem. Soc. 1981. V. 103. P. 4627.
Sen A., Lai T.-W. // Organometallics. 1982. V. 1. P. 415.
Lai T.-W., Sen A. // Organometallics. 1984. V. 3. P. 866.
Hegedus L.S., Mulhern T.A., Asada H. // J. Am. Chem. Soc. 1986. V. 108. P. 6224.
Hahn C., Vitagliano A., Giordao F., Taube R. // Organometallics. 1998. V. 17. P. 2060.
Hahn C., Morvillo P., Vitagliano A. // Eur. J. Inorg. Chem. 2001. P. 419.
Hahn C., Morvillo P., Herdtweck E., Vitagliano A. // Organometallics. 2002. V. 21. P. 1807.
Cucciolito M., D’Amora A., Vitagliano A. // Organometallics. 2005. V. 24. P. 3359.
Hahn C., Cucciolito M., Vitagliano A. // J. Am. Chem. Soc. 2002. V. 124. P. 9038.
Hahn C. // Chem. Eur. J. 2004. V. 10. P. 5888.
Cochran B.M., Michael F.E. // J. Am. Chem. Soc. 2008. V. 130. P. 2786.
Матвеев К.И., Шитова Н.Б. // Кинетика и катализ. 1969. Т. 10. № 4. С. 717.
Шитова Н.Б., Матвеев К.И., Елизарова Г.Л. // Кинетика и катализ. 1970. Т. 11. № 5. С. 1152.
Шитова Н.Б., Матвеев К.И., Обыночный А.А. // Кинетика и катализ. 1971. Т. 12. № 6. С. 1417.
Шитова Н.Б., Матвеев К.И., Кузнецова Л.И. // Изв. СО АН СССР. Сер. хим. 1973. Вып. 1. № 2. С. 25.
Матвеев К.И., Шитова Н.Б., Жижина Е.Г. // Кинетика и катализ. 1976. Т. 17. № 4. С. 893.
Жижина Е.Г., Шитова Н.Б., Матвеев К.И. // Кинетика и катализ. 1981. Т. 22. № 6. С. 1451.
Шитова Н.Б., Кузнецова Л.И., Юрченко Э.Н., Овсянникова И.А., Матвеев К.И. // Изв. АН СССР. Сер. хим. 1973. № 7. С. 1453.
Tsuji J., Minato M. // Tetrahedron Lett. 1987. V. 28. № 32. P. 3683.
Miller D.G., Wayner D.D.M. // J. Org. Chem. 1990. V. 55. P. 2924.
Backvall J.-E., Hopkins R.B. // Tetrahedron Lett. 1988. V. 29. № 23. P. 2885.
Skumov M., Balbolov E. // Catal. Lett. 2000. V. 69. P. 103.
Melgo M.S., Lindner A., Schuchardt U. // Appl. Catal. A. Gen. 2004. V. 273. P. 217.
Monflier E., Blouet E., Barbaux Y., Mortreux A. // Angew. Chem. Int. Ed. 1994. B. 33. № 20. S. 2100.
Kolb M., Bratz E., Dialer K. // J. Mol. Catal. 1977. V. 2. P. 399.
Davidson J.M., Triggs C. // J. Chem. Soc. A. 1968. № 6. P. 1324.
Clark F.R.S., Norman R.O.C., Tomas C.B., Willson J.S. // J. C. S. Perkin Trans 1. 1974. № 11. P. 1289.
Старчевский М.К., Варгафтик М.Н., Моисеев И.И. // Изв. АН СССР. Сер. хим. 1979. № 1. С. 242.
Wenzel T.T. // J. C. S. Chem. Commun. 1989. P. 932.
Hayashi T., Yamasaki K., Mimura H., Uozumi Y. // J. Am. Chem. Soc. 2004. V. 126. P. 3036.
Michel B.W., Camelio A.M., Cornell C.N., Sigman M.S. // J. Am. Chem. Soc. 2009. V. 131. P. 6076.
DeLuca R.J., Edwards J.L., Steffens L.D., Michel B.W., Oiao X., Zhu C., Coek S.P., Sigman M.S. // J. Org. Chem. 2013. V. 78. P. 1682.
Bigi M.A., White M.C. // J. Am. Chem. Soc. 2013. V. 135. P. 7831.
Walker K.L., Dornan L.M., Zare R.N., Waymouth R.M., Muldoon M.J. // J. Am. Chem. Soc. 2017. V. 139. P. 12 495.
Темкин О.Н., Брук Л.Г., Захарова Д.С., Одинцов К.Ю., Кацман Е.А., Петров О.В., Истомина О.Ю. // Кинетика и катализ. 2010. Т. 51. № 5. С. 715.
Мартынов И.В., Ефремов Г.Е., Бовырина Е.А., Кацман Е.А., Темкин О.Н. // Кинетика и катализ. 2018. Т. 59. № 4. С. 427.
Ефремов Г.Е., Бовырина Е.А., Кацман Е.А., Шамсиев Р.С., Темкин О.Н. // Изв. АН. Сер. хим. 2019. № 6. С. 1366.
Захарова Д.С., Черткова О.А., Чёлкин А.А., Брук Л.Г., Темкин О.Н. // Изв. АН. Сер. хим. 2013. № 3. С. 843.
Мартынов И.В., Ефремов Г.Е., Темкин О.Н. // Изв. АН. Сер. хим. 2017. № 5. С. 922.
Захарова Д.С., Мартынов И.В., Носова В.М., Темкин О.Н. // Тонкие хим. технологии. 2016. Т. 11. № 2. С. 57.
Захарова Д.С., Семеняка А.Н., Черткова О.А., Фролкова А.В., Кацман Е.А., Брук Л.Г., Темкин О.Н. // Тонкие хим. технологии. 2015. Т. 10. № 3. С. 77.
Frolkova A., Zakharova D.C., Frolkova A., Balbenov S. // Fluid Phase Equilibria. 2016. V. 408. P. 11.
Jawarski J.N., Mc Cann S.D., Guzei I.A., Stahl S.S. // Angew. Chem. Int. Ed. 2017. V. 56. P. 3605.
Темкин О.Н., Брук Л.Г. // Успехи химии. 1983. Т. 52. С. 206.
Murahashi T., Nagai T., Okano T., Matsutani T., Kurosawa H. // J. C. S. Chem. Commun. 2000. P. 1689.
Wang A.-Z., Jiang H.-F. // J. Am. Chem. Soc. 2009. V. 131. P. 3846.
Wu W., Jiang H.-F. // Acc. Chem. Res. A. 2012. V. 45. № 10. S. 1736.
Wang A.-Z., Jiang H.-F. // J. Org. Chem. 2010. V. 75. P. 2321.
Yokota T., Sakaguchi S., Ishii Y.J. // Jpn. Petroleum Inst. 2003. V. 46. № 1. P. 15.
Schultz M.J., Sigman M.S. // J. Am. Chem. Soc. 2006. V. 128. P. 1460.
Zhang Y., Sigman M.S. // J. Am. Chem. Soc. 2007. V. 129. P. 3076.
Xia X., Gao X., Xu J., Hu C., Peng X. // Synlett. 2017. V. 28. P. 607.
Stobbe-Kreemers A.W., Van der Lans G., Makkee M., Scholten J.J.F. // J. Catal. 1995. V. 154. P. 187.
Joo F. Aqueous Organometallic Catalysis. Dord: Kluwer Academic Publishers, 2002.
Aqueous Phase Organometallic Catalysis – Concepts and Applications / Eds. Cornils B., Herrmann W.A. Weinheim: Wiley–VCH, 2004.
Adams D.J., Dyson P.J., Tave S.J. Chemistry in Alternative Reaction Media. Weinheim: Wiley–VCH, 2004.
Multiphase Homogeneous Catalysis / Ed. Herrmann W.A. Weinheim: Wiley-VCH, 2004.
Catalyst Separation, Recovery and Recycling – Chemistry and Process Design / Eds. Cole-Hamilton D., Tooze R. Berlin–Heidelberg: Springer–Verlag, 2006.
Recoverable and Recyclable Catalysts / Ed. Benaglia M. Weinheim: Wiley–VCH–Verlag GmbH, 2009.
Караханов Э.А., Максимов А.Л., Рунова Е.А. // Успехи химии. 2005. Т. 74. № 1. С. 104–119.
Караханов Э.А., Максимов А.Л. // Рос. хим. журн. (Ж. Рос. хим. об-ва им. Д.И. Менделеева). 2008. Т. 52. № 4. С. 125–135.
Hagen J. Industrial Catalysis. A Practical Approach. Weinheim: Wiley–VCH, 2015.
Караханов Э.А., Кардашева Ю.С., Максимов А.Л., Рунова Е.А. // Рос. хим. журн. (Ж. Рос. хим. об-ва им. Д.И. Менделеева). 2002. Т. 46. № 2. С. 53.
Karakhanov E., Buchneva T., Maximov A., Zavertyeva M. // J. Mol. Catal. A. 2002. V. 184. P. 11.
Jiang H., Jia L., Li J. // Green Chem. 2000. V. 2. P. 161.
Maksimov A.L., Sakharov D.A., Filippova T.Yu., Zhuchkova A.Ya., Karakhanov E.A. // Ind. Eng. Chem. Res. 2005. V. 44. № 23. P. 8644.
Hou Z., Han B., Gao L., Jiang T., Liu Z., Chang Y., Zhang X., He J. // New J. Chem. 2002. V. 26. P. 1246.
Namboodiri V.V., Varma R.S., Sahle-Demessie E., Pillai U.R. // Green Chemistry. 2002. V. 4. P. 170.
Dudley C., Read G. // Tetrahedron Lett. 1972. V. 52. P. 5273.
Mimoun H., Machirant M.M.P., Roch I.S. // J. Am. Chem. Soc. 1978. V. 100. P. 5437.
Nyberg E.D., Pribitch D.C., Drago R.S. // J. Am. Chem. Soc. 1983. V. 105. P. 3538.
Januszkiewicz K., Alper H. // Tetrahedron Lett. 1983. V. 24. P. 5163.
Faraj M., Martin J.C.M., Bregeault J.-M. // J. Mol. Catal. 1985. V. 31. P. 57.
Bressan M., Morandini F., Morvillo A. // J. Organomet. Chem. 1985. V. 280. P. 139.
Taqui Khan M.M., Rao A.P. // J. Mol. Catal. 1988. V. 44. P. 95.
Read G. // J. Mol. Catal. 1988. V. 44. P. 15.
Krom M., Coumans R.G.E., Smits J.M.M., Gal A.W. // Angew. Chem. Int. Ed. Engl. 2001. V. 40. P. 2106.
Bruin B., Verhagen J.A.W., Schouten C.H.J., Gal A.W., Feichtinger D., Plattner D.A. // Chem. Eur. J. 2001. V. 7. P. 416.
Atlay M.T., Preece M., Strukul G., James B.R. // J. Chem. Soc. Chem. Commun. 1982. P. 406.
Woerpel K.A., Bergman R.G. // J. Am. Chem. Soc. 1993. V. 115. P. 7888.
Ritter J.C.M., Bergman R.G. // J. Am. Chem. Soc. 1997. V. 119. P. 2580.
Hamilton D.E., Drago R.S., Zombeck A. // J. Am. Chem. Soc. 1987. V. 109. P. 374.
Matsumoto K., Nagai Y., Matsunami J., Mizuno K., Abe T., Somazawa R., Kinoshita J., Shimura H. // J. Am. Chem. Soc. 1998. V. 120. P. 2900.
Liu B., Jin F., Wang T., Han W. // Angew. Chem. Int. Ed. 2017. V. 56. P. 12712.
Тeмкин О.Н. // Кинетика и катализ. 2019. Т. 60. № 6. С. 683.
Гавриленко В.А., Евзирихин Е.И., Колосов В.А., Моисеев И.И. // Изв. АН СССР. Сер. хим. 1977. № 1. С. 29.
Гавриленко В.А., Евзирихин Е.И., Моисеев И.И., Фиш И.Ш. // Изв. АН СССР. Сер. хим. 1977. № 8. С. 1746.
Моисеева Н.И., Гехман А.Е., Сахаров С.Г., Скибида И.П., Моисеев И.И. // Изв. АН СССР. Сер. хим. 1986. № 10. С. 2396.
Гехман А.Е., Шишкин Д.И., Моисеев И.И. // Изв. АН СССР. Сер. хим. 1987. № 6. С. 1436.
Гехман А.Е., Амеличкина Г.Е., Моисеева Н.И., Варгафтик М.Н., Моисеев И.И. // Кинетика и катализ. 2001. Т. 42. № 4. С. 549.
Марков А.А., Долин С.П., Моисеева Н.И., Гехман А.Е., Моисеев И.И. // Журн. неорган. химии. 2011. Т. 56. № 5. С. 778.
Konnick M.M., Guzei. I.A., Stahl S.S. // J. Am. Chem. Soc. 2004. V. 126. P. 10 212.
Landis C.R., Morales C.M., Stahl S.S. // J. Am. Chem. Soc. 2004. V. 126. P. 16 302.
Vargaftik M.N., Zagorodnikov V.P., Stolyarov I.P., Moiseev I.I., Likholobov V.A., Kochubey D.I., Chuvilin A.L., Zamaraev K.I. // J. Mol. Catal. 1989. V. 53. № 3. P. 315–348.
Moiseev I.I., Vargaftik M.N. Catalysis by Di- and Polynuclear Meal Complexes / Eds. Cotton F.A., Adams R. N.Y.: Wiley–VCH, 1998. P. 395.
Шестаков Г.К., Темкин О.Н., Всесвятская Н.Ю., Степанов А.М. // Журн. орган. химии. 1979. Т. 15. № 2. С. 245.
Всесвятская Н.Ю., Шестаков Г.К., Темкин О.Н. // Кинетика и катализ. 1986. Т. 27. С. 1330.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ