

Кинетика и катализ, 2020, T. 61, № 6, стр. 848-854
Каталитическое гидрометоксилирование ацетилена на поверхности механоактивированной соли K2PdCl4
Т. В. Краснякова a, Д. В. Никитенко a, О. В. Хазипов b, С. А. Митченко a, b, *
a Институт физико-органической химии и углехимии им. Л.М. Литвиненко
83114 Донецк, ул. Р. Люксембург, 70, Украина
b ФГБОУ ВО Южно-Российский государственный политехнический университет (НПИ) им. М.И. Платова
346428 Новочеркасск, ул. Просвещения, 132, Россия
* E-mail: samit_RPt@mail.ru
Поступила в редакцию 09.02.2020
После доработки 23.04.2020
Принята к публикации 30.04.2020
Аннотация
На поверхности предварительно механоактивированной соли K2PdCl4 протекает каталитическая реакция присоединения метилового спирта к тройной связи ацетилена с образованием диметилацеталя и побочного продукта винилхлорида. Присоединение спирта происходит региоселективно в соответствии с правилом Марковникова. Стереоселективность гидрохлорирования ацетилена отвечает транс-присоединению Н и Cl к тройной С≡С-связи ацетилена. Определены эффективные энергии активации маршрутов реакции. Предложен возможный механизм образования H3C–CH(ОСH3)2 и H2C=CHCl.
ВВЕДЕНИЕ
Каталитическое присоединение спиртов к алкинам представляет собой один из путей функционализации тройной связи [1]. Такая реакция является начальным этапом многих каскадных процессов с участием ненасыщенных соединений [2], приводящих к созданию сложных молекул [3–5]. Получаемые в результате присоединения двух молекул спирта к тройной С≡С-связи алкинов ацетали широко используются в качестве синтетических промежуточных соединений в медицинской химии и материаловедении, исходных реагентов в органическом синтезе [6], а также рекомендованы в качестве присадок к дизельному топливу для повышения цетанового числа [7].
Реакции присоединения RХ (Х = O, S, Se) к алкинам с образованием виниловых эфиров и ацеталей являются 100% атом-экономными и протекают в присутствии металлокомплексных катализаторов, как правило, в гомогенных условиях [1, 2, 8–18]. Гетерогенные каталитические системы гидроалкоксилирования тройной С≡С-связи алкинов представлены слабо [7, 19].
Ранее нами было обнаружено, что поверхность предварительно механоактивированных в атмосфере ацетилена или пропилена хлоридных солей платины K2PtCl4 [20, 21] и палладия K2PdCl4 [22, 23] проявляет каталитическую активность в реакции гидрохлорирования ацетилена газообразным хлороводородом. При обработке в атмосфере непредельных соединений на поверхности соли образуются активные центры катализатора – комплексы с дефицитом хлорид-лигандов в координационной сфере [${\text{MCl}}_{3}^{*}$]– (M = Pt, Pd) [21, 23, 24]. Механизм реакции (схема 1 ) включает π-координацию ацетилена к координационно ненасыщенным комплексам, последующее скоростьопределяющее хлорметаллирование под действием молекулы НCl при участии соседнего комплекса металла с генерированием σ-винильного производного соответствующего металла и регенерацией комплекса с координационной вакансией. Завершает цикл протодеметаллирование указанного интермедиата с образованием конечного продукта.
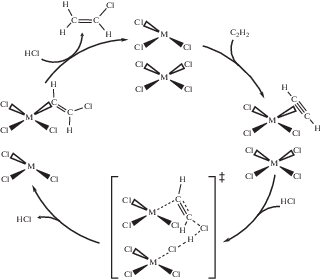
Схема 1 . Стадийный механизм каталитического гидрогалогенирования ацетилена на механоактивированных солях K2МCl4 (М = Pt, Pd) [20–23].
Если галоген в молекуле НCl заменить на метоксигруппу, то можно полагать, что в подобных условиях аналогичным образом будет протекать реакция гидрометоксилирования ацетилена – присоединение к тройной связи ацетилена молекулы метилового спирта. Целью настоящей работы являлась проверка этой гипотезы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Ацетилен получали методом [25], соль K2PdCl4 синтезировали по стандартной методике [26] и высушивали в сушильном шкафу при температуре 120–140°С в течение 3 сут. Навески соли массой 0.25 г подвергали предварительной механической обработке в атмосфере ацетилена в течение 1 ч. Механообработку образцов осуществляли в стеклянном виброреакторе объемом 13.7 мл, содержащем стеклянные мелющие тела, при помощи микровибромельницы ММVE-0.005 (“Гефест”, Россия) с удельной энергонапряженностью ~15 Вт/кг.
После предварительной механообработки соли реактор продували аргоном, герметизировали и через резиновую уплотняющую прокладку вводили микрошприцем 10 мкл122 СН3ОН (х. ч.), 1 мл ацетилена и метан (внутренний стандарт). Реактор, содержащий катализатор и газовую смесь, помещали в термостат. Пробы газовой фазы замкнутого реактора отбирали через определенные промежутки времени шприцем-дозатором фиксированного объема 0.121 мл, не нарушая герметичности реактора, и анализировали. Расходование ацетилена и накопление винилхлорида контролировали методом ГЖХ с применением хроматографа ЛХМ-8-МД (“Хроматограф”, Россия) с пламенно-ионизационным детектором и набивной колонкой, заполненной Силахромом С 120. Сбор и обработку данных осуществляли с помощью системы МультиХром фирмы “Амперсенд”.
Относительную концентрацию ацетилена φАс и винилхлорида определяли из отношения площадей соответствующих хроматографических пиков и метана. Количество вещества рассчитывали с использованием калибровочных зависимостей для ацетилена. Калибровку хроматографа проводили путем введения в пустой реактор известного количества С2Н2, после чего состав газовой фазы реактора подвергали хроматографическому анализу. Количество выделившегося винилхлорида оценивали с учетом различия в 1.26 раза чувствительности детектора ионизации пламени хроматографа к С2Н2 и С2Н3Сl [27].
Независимо выход продуктов реакции определяли ЯМР-спектрометрически. Для этого после проведения реакции ацетилена в реактор вводили 1 мл дейтерированного хлороформа, в полученную взвесь добавляли 1 мкл гексаметилдисилана в качестве внутреннего стандарта. Содержимое реактора помещали в пробирку и центрифугировали, надосадочную жидкость переносили в ЯМР-ампулу. ЯМР-спектры продуктов реакции регистрировали на приборе AVANCE-II-400 (“Bruker BioSpin GmbH”, Германия) с рабочей частотой 400 МГц.
Для оценки TON (количества каталитических циклов в расчете на один активный центр катализатора) после полного расходования ацетилена реактор продували сухим аргоном, заменяли герметизирующую прокладку и вновь вводили метанол, ацетилен и внутренний стандарт метан. Процесс осуществляли многократно.
Значения констант скорости накопления винилхлорида и диметилацеталя находили как произведение константы скорости расходования ацетилена на выход соответствующего продукта при данной температуре. Для определения кинетического изотопного эффекта реакцию проводили в присутствии CD3OD (“Merck”).
Оценку количества углеродистых отложений после проведения реакции осуществляли по потере массы путем сравнения отожженных на воздухе образцов механоактивированной соли K2PdCl4 до и после реакции. Предварительно в сушильном шкафу при температуре 100°С с поверхности образцов 4 ч десорбировали низкокипящие соединения. Затем навеску соли выдерживали в муфельной печи при температуре 400°С в течение 3 ч – времени, необходимого для прекращения потери массы.
ИК-спектры образцов соли получали с использованием ИК-Фурье-спектрофотометра FT/IR-4200 (“JASCO”, Япония). В агатовой ступке перетирали 10 мг катализатора с 200 мг KBr и прессовали таблетку.
Рентгеновские исследования катализаторов проводили при помощи дифрактометра ДРОН-3 (“Буревестник”, Россия), оснащенного автоматизированной системой сбора и обработки данных, в CuKα-излучении с использованием никелевого фильтра в диапазоне углов 2θ от 10° до 70° (геометрия съемки по Брэггу–Брентано).
ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ
Предварительно механоактивированная в атмосфере ацетилена соль K2PdCl4 проявляет активность в реакции присоединения метилового спирта к ацетилену:
(I)
${{{\text{C}}}_{2}}{{{\text{H}}}_{2}} + 2{\text{C}}{{{\text{H}}}_{3}}{\text{OH}} \to {\text{C}}{{{\text{H}}}_{3}}{\text{CH}}{{({\text{OC}}{{{\text{H}}}_{3}})}_{2}}.$На поверхности неактивированной соли реакция с заметной скоростью не протекает.
Кроме сигналов, принадлежащих 1,1-диметилацеталю, в 1Н-ЯМР спектре обнаружены сигналы, отнесенные к винилхлориду (рис. 1).
Рис. 1.
Фрагмент ЯМР-спектра продуктов H3C–CH(ОСH3)2 и H2C=CHCl реакции гидрометоксилирования ацетилена на поверхности механоактивированной соли K2PdCl4.
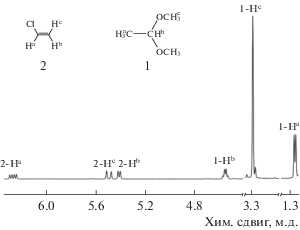
1H-ЯМР-спектр H3C–CH(ОСH3)2, δ, м. д.: 4.55 (q, JH–H = 5.3 Гц, Hb), 3.29 (s, Hc), 1.27 (d, JH–H = 5.3 Гц, Ha).
1H-ЯМР-спектр H2C=CHCl, δ, м. д.: 6.27 (dd, Jтранс(H–H) = 14.7 Гц, Jцис(H–H) = 7.1 Гц, Ha), 5.49 (d, Jтранс(H–H) = 14.7 Гц, Hc), 5.41 (d, Jцис(H–H) = 7.1 Гц, Hb).
Региоселективность образования диметилацеталя формально отвечает правилу Марковникова.
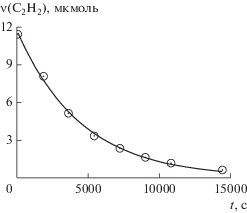
Каталитическая реакция протекает в отсутствие непрерывной механической обработки. При избытке метилового спирта расходование ацетилена из газовой фазы замкнутого реактора отвечает кинетическому уравнению реакции псевдопервого порядка (рис. 2):
(1)
${\nu }({{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{2}}}}) = {{{\nu }}_{0}}({{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{2}}}}) \times {{e}^{{ - {{k}_{{{\text{ef}}}}}t}}},$Рис. 2.
Типичная кинетика расходования ацетилена в системе механоактивированная соль K2PdCl4–СH3ОН–С2Н2 при 50°С. Точки отвечают экспериментальным значениям, огибающая кривая – расчет по уравнению (1).
Значения наблюдаемых констант скорости расходования ацетилена и выходов продуктов при различных температурах приведены в табл. 1. При многократном повторении реакции на одной загрузке катализатора существенного падения каталитической активности не наблюдалось.
Таблица 1.
Значения наблюдаемой константы скорости расходования ацетилена kef и выходов продуктов реакции – винилхлорида (YВХ) и диметилацеталя (YДМА) – в расчете на прореагировавший ацетилен
| T, °C | kef, 10–4 c–1 | YВХ, % | YДМА, % |
|---|---|---|---|
| 30 | 1.5 ± 0.1 | 5 ± 1 | 15 ± 2 |
| 50 | 2.1 ± 0.1 | 11 ± 1 | 27 ± 3 |
| 80 | 3.7 ± 0.1 | 20 ± 1 | 28 ± 3 |
| 103 | 7.9 ± 0.2 | 30 ± 2 | 27 ± 2 |
Количество загруженной соли палладия массой m = 0.25 г отвечает 767 мкмоль. Зная параметры решетки кристаллической структуры K2PdCl4 (a = b = 7.05 Å и с = 4.1 Å [28]) и удельную поверхность соли (Sуд ≈ 6.7 м2/г [23]), можно оценить верхний предел количества комплексных анионов палладия, находящихся на поверхности:
что составляет ~6 × 1018 анионов и соответствует ~10 мкмоль Pd на поверхности. В наших экспериментах максимальное количество ацетилена, переработанного на одной загрузке катализатора, было не менее 1.7 ммоль. В таком случае минимальное число каталитических циклов в расчете на комплексы палладия, находящиеся на поверхности соли, TON, равно ~170, что отвечает ~2.2 циклам в расчете на массивный палладий. При этом существенного падения каталитической активности не наблюдалось.Проведение гидрометоксилирования ацетилена при 50°C в присутствии дейтерированного метилового спирта CD3OD показало, что наблюдаемая константа скорости расходования ацетилена (2.0 ± 0.1) × 10–4 с–1 совпадает в пределах экспериментальной погрешности с константой, полученной в аналогичных условиях в реакции c СН3ОН (табл. 1). В 1Н-ЯМР-спектре продуктов обнаружена смесь недейтерированного и монодейтерированного (следы) изотопомеров винилхлорида. Стереохимия монодейтерированного винилхлорида отвечает транс-присоединению атомов дейтерия и хлора к тройной связи ацетилена.
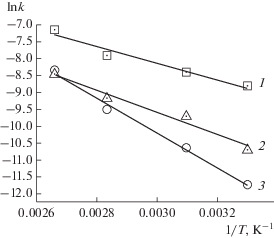
Температурная зависимость наблюдаемой константы скорости каталитической реакции (рис. 3) позволила определить значения эффективной энергии активации для протекающих в конкурентных условиях гидрометоксилирования (ГМ) и гидрохлорирования (ГХ) ацетилена, которые составляют ЕГМ = 27 ± 5 кДж/моль и ЕГХ = 43 ± 6 кДж/моль соответственно.
Рис. 3.
Температурные зависимости наблюдаемых констант скорости расходования ацетилена (1), накопления 1,1-диметилацеталя (2) и винилхлорида (3) в реакции ацетилена с метанолом в системе с механоактивированной солью K2PdCl4.
Протекание реакции сопровождается потемнением катализатора, а отжиг на воздухе возвращает ему первоначальный цвет. После отжига потеря массы в пересчете на загрузку катализатора 0.25 г для свежеобработанного образца (до проведения реакции при 50°C, образец 1) составляет ~4.5 мг, а для катализатора после проведения реакции (переработано около 1.7 ммоль ацетилена, образец 2) – 28.0 мг. Эти данные позволяют оценить количество углерода, отложившегося на поверхности соли палладия в ходе реакции – ~2.0 ммоль. Полученная величина практически совпадает с количеством углерода (~2.1 ммоль), содержавшемся в прореагировавшем ацетилене, но не давшим наблюдаемые продукты реакции (диметилацеталь и винилхлорид). В ИК-спектре образца 2 наблюдается полоса 1708 см–1, которую можно отнести к валентным колебаниям С=С-связи [29]. В спектре образца 1 эта полоса отсутствует, а в спектре образца 2 после отжига исчезает.
Исследования кристаллической структуры образцов загружаемой в реакцию механоактивированной соли палладия показали присутствие всех рентгеновских максимумов, отвечающих единственной фазе K2PdCl4 [28]. Проведение реакции не вносит изменения в структуру соли: дифрактограммы свежеобработанной соли и катализатора после осуществления реакции идентичны.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
По аналогии с реакцией каталитического гидрохлорирования ацетилена газообразным HCl [23] (схема 1 ), возможный стадийный механизм гидрометоксилирования ацетилена на поверхности механообработанной соли K2PdCl4 можно представить схемой 2 . В реакции принимают участие два соседних поверхностных комплекса палладия(II), расположенных на кристаллографической плоскости (100) [27], один из которых – комплекс с вакансией [${\text{PdCl}}_{3}^{*}$]– [24], способный координировать метанол. Реакция начинается с замещения по диссоциативному механизму молекулы спирта из координационной сферы металла ацетиленом с образованием π-ацетиленового комплекса. Подобное вытеснение наблюдалось для комплексов Pt(II) [30]. Последующее метоксипалладирование π-координированного ацетилена молекулой спирта при участии соседнего комплекса палладия приводит к σ-винильному производному Pd(II), образованию молекулы НCl и регенерирует комплекс с координационной вакансией. Протодеметаллирование σ-винильного производного палладия под действием молекулы хлороводорода (схема 2 , А) или метанола (схема 2 , В) дает метилвиниловый эфир и, соответственно, комплекс [PdCl4]2– или [PdCl3(ОСН3)]2–. Однако реакция не останавливается на стадии образования СН3ОСН=СН2 – последний быстро реагирует еще с одной молекулой спирта, давая диметилацеталь. Присоединение двух молекул спирта к алкинам с образованием ацеталей в присутствии металлокомплексных катализаторов на основе Au(I), Au(III), Pt(II), Pd(II), Zn(II) ранее отмечалось в гомогенных [8–12] и гетерогенных [7, 19] системах.
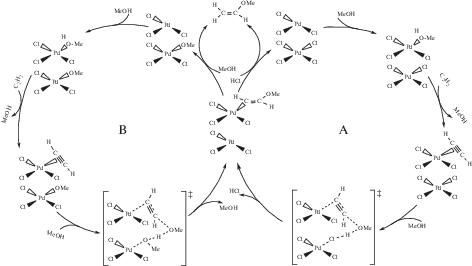
Схема 2 . Возможный механизм каталитической реакции присоединения метанола к ацетилену на поверхности механоактивированной соли K2PdCl4.
Доводами в пользу предложенного механизма являются следующие соображения. Во-первых, в реакции присоединения спиртов к алкинам зафиксированы промежуточные σ-винилэфирные производные металла для комплексов Pd(II) [11] и Pt(II) [30–32]. Во-вторых, промежуточные продукты реакции – виниловые эфиры – обнаружены в гомогенных условиях при катализе комплексами платины и палладия [9], а также в гетерогенной системе ZnO/SiO2 [7, 19]. В последнем случае диметилацеталь получается [7] при температурах 200–270°С в результате быстрой равновесной реакции между метанолом и метилвиниловым эфиром, причем с повышением температуры выход H3C–CH(ОСH3)2 уменьшается. Аналогично, при алкоксилировании этилвиниловых эфиров в интервале температур 15–120°С равновесие сильно сдвинуто в сторону образования ацеталей [33]. Возможно, по этой причине нам не удалось зафиксировать промежуточный продукт реакции – метилвиниловый эфир.
Считается, что присоединение второй молекулы спирта осуществляется быстро и катализируется кислотой [2, 7]. Действительно, в предложенном механизме на стадии гидрометоксилирования ацетилена выделяется молекула HCl, потенциально способная выступать катализатором. Однако в гетерогенной системе при отсутствии сольватации нет возможности образования свободных Н+-ионов. Гидрометоксилирование промежуточного продукта – метилвинилового эфира – вероятно осуществляется через π-координацию его двойной С=С-связи к комплексам палладия с последующим ее метоксипалладированием подобно стадийному механизму, приведенному на схеме 2 . Отметим, что способность активировать двойную связь олефинов комплексами [${\text{PdCl}}_{3}^{*}$]– была продемонстрирована в [23] при гидрохлорировании пропилена с получением изопропилхлорида.
Предложенный механизм предполагает участие молекулы СH3ОН на стадии метоксипалладирования π-координированного ацетилена с разрывом связи О–Н. В эксперименте с изотопномеченым спиртом кинетический изотопный эффект не был обнаружен, следовательно, эта стадия не является скоростьопределяющей. Поскольку протолиз обычно протекает быстро [34–37], более вероятно лимитирование реакции стадией замещения спирта в координационной сфере металла ацетиленом за счет его π-координации. Реакция гидрометоксилирования ацетилена включает такое лимитирующее замещение независимо от возможного маршрута А или В (схема 2 ): предложенные механизмы отличаются только стадией протодеметаллирования σ-винильного производного палладия и оба предполагают вытеснение спирта из координационной сферы металла ацетиленом с промежуточным образованием π-ацетиленового комплекса. Отметим, что в случае газофазного гидрохлорирования связывание ацетилена в π-комплекс осуществлялось быстро [37], и очевидной причиной замедления этой стадии в присутствии спирта является блокирование последним вакансии в координационной сфере металла. В реакции гидрохлорирования ацетилена молекулы субстратов адсорбируются на одних и тех же участках поверхности, не конкурируя между собой из-за разных механизмов взаимодействия с поверхностью [23]: ацетилен связывается в π-комплекс, оккупируя вакантное место в кооординационной сфере металла, а полярная молекула хлористого водорода удерживается вблизи этого активного центра электростатическими силами.
Расходование ацетилена осуществляется в параллельных процессах – с образованием диметилацеталя и винилхлорида. Соотношение выходов продуктов коррелирует с различием в значениях наблюдаемой энергии активации маршрутов реакции. Относительно небольшой суммарный выход ацеталя и винилхлорида может быть следствием конкурирующей олигомеризации ацетилена и продуктов его превращений, наблюдавшейся как в гомогенных [34], так и в гетерогенных [7, 38–40] условиях. Подтверждением такого предположения является близость количества углерода, потерянного образцом 2 при отжиге, и содержащегося в части прореагировавшего ацетилена, который не превратился в наблюдаемые продукты реакции диметилацеталь и винилхлорид. С этим также согласуется наличие в ИК-спектре образца 2 ожидаемой для продуктов олигомеризации полосы валентных колебаний С=С-связи, которая исчезает после отжига и отсутствует у свежеобработанной соли.
Источником атомов Cl в образующемся винилхлориде может быть только хлор из матрицы катализатора233. По данным ЯМР-спектроскопии стереоселективность продукта отвечает транс-присоединению атомов Н и Cl к ацетилену. Такая стереоселективность позволяет исключить внутрисферную атаку хлорид-лиганда на π-ацетиленовый комплекс металла [41]. Минимальное расстояние между соседними комплексами металла в кристалле K2PdCl4 составляет порядка 4.1 Ǻ [23], что при заторможенной подвижности в твердых телах делает невозможным присоединение галогена от соседнего металлокомплекса. Поэтому можно полагать, что в образовании винилхлорида принимает участие молекула хлороводорода, формирующаяся в процессе присоединения молекулы спирта к тройной связи алкина на стадии метоксипалладирования π-координированного ацетилена: HCl может приводить к протолизу метоксивинильного производного палладия (схема 2 , В) или хлорпалладированию π-координированного ацетилена (схема 1 ). Малое количество монодейтерированного винилхлорида, образующегося в присутствии дейтерированного спирта, может быть обусловлено высоким значением (4.2) изотопного эффекта стадии протодепалладирования промежуточного хлорвинильного производного Pd(II) при температуре 50°С.
ЗАКЛЮЧЕНИЕ
Предварительно механоактивированная соль K2PdCl4 проявляет каталитическую активность в реакции гидрометоксилирования ацетилена с получением диметилацеталя. Параллельно выделяется винилхлорид. Региоселективность образования диметилацеталя отвечает правилу Марковникова, а стереохимия винилхлорида – транс-присоединению атомов водорода и хлора к тройной С≡С-связи ацетилена. Возможный механизм присоединения молекулы метилового спирта к ацетилену предполагает: 1) вытеснение ацетиленом метанола из координационной сферы Pd(II) (лимитирующая стадия); 2) метоксипалладирование π-координированного ацетилена под действием молекулы СH3ОН при участии соседнего комплекса палладия с образованием σ-винильного производного Pd(II), молекулы хлороводорода и комплекса с координационной вакансией; 3) протодеметаллирование σ-винильного производного палладия с получением промежуточного метилвинилового эфира. Быстрое присоединение к последнему второй молекулы спирта дает конечный продукт СН3СН(ОСН3)2. Образование винилхлорида происходит при участии молекулы хлороводорода, генерируемой на стадии метоксипалладирования ацетилена.
Список литературы
Konkol M., Schmidt H., Steinborn D. // J. Mol. Catal. A Chem. 2007. V. 269. № 1–2. P. 119.
Roithová J., Janková Š., Jašíková L., Váňa J., Hybelbauerová S. // Angew. Chem. Int. Ed. 2012. V. 51. № 33. P. 8378.
Mizushima E., Sato K., Hayashi T., Tanaka M. // Angew. Chem. 2002. V. 114. № 23. P. 4745.
Lein M., Rudolph M., Hashmi S.K., Schwerdtfeger P. // Organometallics. 2010. V. 29. № 10. P. 2206.
Nolan S.P. // Acc. Chem. Res. 2011. V. 44. № 2. P. 91.
Ushimaru R., Nishimura T., Iwatsuki T., Naka H. // Chem. Pharm. Bull. 2017. V. 65. № 11. P. 1000.
Trimm D., Cant N., Lei Y. // Catal. Today 2009. V. 145. № 1–2. P. 163.
Fukuda Y., Utimoto K. // J. Org. Chem. 1991. V. 56. № 11. P. 3729.
Steinborn D., Nünthel R., Krause K. // J. Organomet. Chem. 1991. V. 414. № 2. P. 54.
Teles J.H., Brode S., Chabanas M. // Angew. Chem. Int. Ed. 1998. V. 37. № 10. P. 1415.
Kataoka Y., Matsumoto O., Ohashi M., Yamagata T., Tani K. // Chem. Lett. 1994. V. 23. № 7. P. 1283.
Hartman J.W., Sperry L. // Tetrahedron Lett. 2004. V. 45. № 19. P. 3787.
Corma A., Ruiz V.R., Leyva-Pérez A., Sabater M.J. // Adv. Synth. Catal. 2010. V. 352. № 10. P. 1701.
Ananikov V.P., Orlov N.V., Zalesskiy S.S., Beletskaya I.P., Khrustalev V.N., Morokuma K., Musaev D.G. // J. Am. Chem. Soc. 2012. V. 134. № 15. P. 6637.
Malyshev D.A., Scott N.M., Marion N., Stevens E.D., Ananikov V.P., Beletskaya I.P., Nolan S.P. // Organometallics. 2006. V. 25. № 19. P. 4462.
Chinchilla R., Nájera C. // Chem. Rev. 2014. V. 114. № 3. P. 1783.
Lam R.H., Walker D.B., Tucker M.H., Gatus M.R.D., Bhadbhade M., Messerle B.A. // Organometallics. 2015. V. 34. № 17. P. 4312.
Ketcham J.M., Biannic B., Aponick A. // Chem. Commun. 2013. V. 49. № 39. P. 4157.
Breuer K., Teles J.H., Demuth D., Hibst H., Schäfer A., Brode S., Domgörgen H. // Angew. Chem. Int. Ed. 1999. V. 38. № 10. P. 1401.
Митченко Р.С., Шубин А.А., Краснякова Т.В. // Теорет. и эксперим. химия. 2006. Т. 42. № 5. С. 306.
Mitchenko S.A., Krasnyakova T.V., Mitchenko R.S., Korduban A.N. // J. Mol. Catal. A: Chem. 2007. V. 275. № 1–2. P. 101.
Митченко С.А., Краснякова Т.В., Жихарев И.В. // Теорет. и эксперим. химия. 2008. Т. 44. № 5. С. 306.
Krasnyakova T.V., Zhikharev I.V., Mitchenko R.S., Burkhovetski V.I., Korduban A.M., Kryshchuk T.V., Mitchenko S.A. // J. Catal. 2012. V. 288. P. 33.
Митченко С.А., Краснякова Т.В., Жихарев И.В. // Теорет. и эксперим. химия. 2010. Т. 46. № 1. С. 32.
Рапопорт Ф.М., Ильинская А.А. Лабораторные методы получения чистых газов. Mосква: Госхимиздат, 1963. 362 с.
Черняева И.Н. Синтез комплексних соединений металлов платиновой группы. Справочник Mосква: Наука, 1964. 239 с.
Замащиков В.В., Литвиненко С.М., Шологон В.И. // Кинетика и катализ. 1987. Т. 28. № 5. С. 1059.
JCPDS: International Centre for Diffraction Data, № 9-367.
Беллами Л.Д. Инфракрасные спектры сложных молекул. Пер. с англ. Под ред. Пентина Ю.А. Москва: Изд-во Иностранной литературы, 1963. 592 с.
Chisholm M.H., Clark H.C. // Inorg. Chem. 1971. V. 10. № 11. P. 2557.
Avshu A., O’Sullivan R.D., Parkins A.W., Alcock N.W., Countryman R.M. // J. Chem. Soc. Dalton Trans. 1983. № 8. P. 1619.
Kataoka Y., Matsumoto O., Tani K. // Organometallics. 1996. V. 15. № 24. P. 5246.
Kankaanpera A., Salomaa P., Juhala P., Aaltonen R., Mattsen M. // J. Am. Chem. Soc. 1973. V. 95. № 11. P. 3618.
Брайловский С.М., Темкин О.Н., Климова Е.С. // Кинетика и катализ. 1983. Т. 24. С. 1091.
Митченко С.А., Краснякова Т.В., Жихарев И.В. // Кинетика и катализ. 2009. Т. 50. № 5. С. 764.
Zhdanko A., Maier M.E. // Chem. Eur. J. 2014. V. 20. № 7. P. 1918.
Митченко С.А., Краснякова Т.В. // Кинетика и катализ. 2014. Т. 55. № 6. С. 741.
Nkosi B., Adams M.D., Coville N.J., Hutchings G.J. // J. Catal. 1991. V. 128. P. 378.
Conte M., Davies C.J., Morgan D.J., Davies T.E., Carley A.F., Johnstonb P., Hutchings G.J. // Catal. Sci. Technol. 2013. V. 3. P. 128.
Malta G., Kondrat S.A., Freakley S.J., Davies C.J., Dawson S., Liu X., Lu L., Dymkowski K., Fernandez-Alonso F., Mukhopadhyay S., Gibson E.K., Wells P.P., Parker S.F., Kiely C.J., Hutchings G.J. // ACS Catal. 2018. V. 8. № 9. P. 8493.
Темкин О.Н., Шестаков Т.К., Трегер Ю.А. Ацетилен: химия, механизмы реакций и технология. Москва: Химия, 1991. 429 с.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ