

Кинетика и катализ, 2021, T. 62, № 2, стр. 198-207
Взаимодействие глутатиона с ресвератролом в присутствии пероксида водорода. Кинетическая модель
К. М. Зинатуллина a, *, О. Т. Касаикина a, Н. П. Храмеева b, М. И. Индейкина b, А. С. Кононихин b
a ФГБУН Федеральный исследовательский центр химической физики им. Н.Н. Семенова РАН
119991 Москва, ул. Косыгина, 4, корп. 1, Россия
b ФГБУН Институт биохимической физики им. Н.М. Эмануэля РАН
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: karinazinat11@gmail.com
Поступила в редакцию 13.10.2020
После доработки 01.12.2020
Принята к публикации 03.12.2020
Аннотация
Исследованы кинетические закономерности взаимодействия глутатиона (GSH) c ненасыщенным фенолом ресвератролом (RVT) в деионизированной воде в присутствии пероксида водорода (H2O2). GSH, содержащий две карбоксильных группы, при физиологической концентрации (0.1–10 мМ) образует кислые растворы (рН 3–4); молекулы GSH ассоциируются в димеры. В этих условиях GSH относительно медленно окисляется кислородом воздуха, а реакция GSH с H2O2 сопровождается выходом радикалов. Скорость генерирования тиильных радикалов (Wi) составляет доли процента от скорости расходования GSH, но ее достаточно для инициирования цепной тиол-ен реакции GSH с RVT. На основании полученных экспериментальных данных по кинетике процесса и составу продуктов, а также литературных сведений о реакциях GSH с H2O2 и тиильных радикалов, предложена кинетическая модель сложного процесса взаимодействия GSH с RVT в присутствии H2O2 в водной среде при 37°С. Модель включает 19 квазиэлементарных реакций с соответствующими константами скорости, в том числе, формирование промежуточных комплексов GSH–H2O2 и GSH–GSH, образование радикалов и их последующие превращения в реакциях с RVT и GSH в конечные продукты. Компьютерное моделирование на основе разработанной модели удовлетворительно описывает особенности кинетики процесса в широком диапазоне концентраций реагентов.
ВВЕДЕНИЕ
Глутатион (GSH) – природный эндогенный тиол, который содержится в биологических тканях и жидкостях в высоких концентрациях (0.1–10 мМ), на порядки превосходящих концентрации других компонентов антиоксидантной системы. Считается, что GSH регулирует функции белков и экспрессию генов, реагирует с гидроксильными и пероксильными радикалами, восстанавливает гидропероксиды, дисульфидные связи, предотвращает окисление протеинов [1–3]. В литературе отмечают существенные изменения в содержании глутатиона при развитии многих патологий, в том числе при болезнях Альцгеймера, Паркинсона, сердечно-сосудистых и онкологических заболеваниях [4–9].
Ранее мы детально исследовали механизм реакции GSH с H2O2 в среде бидистиллированной деионизированной воды [10–14]. Установлено, что взаимодействие GSH и H2O2 сопровождается образованием радикалов [10, 11]. Методом ингибиторов с использованием оригинального акцептора радикалов [10, 15] было показано, что в деионизированной воде выход радикалов в реакции с Н2О2 наблюдается и в случае других тиолов: цистеина, гомоцистеина, ацетилцистеина. В [10] на основе своих и литературных данных была построена кинетическая модель взаимодействия GSH с H2O2, включающая 13 квазиэлементарных реакций с соответствующими константами скорости, которая удовлетворительно описывала кинетические кривые расходования GSH и инициирования радикалов (расходования акцептора радикалов). В [16] методом спиновых ловушек с использованием 5,5-диметил-1-пирролин-N-оксида (DMPO) было показано, что при взаимодействии GSH с H2O2 действительно образуются тиильные радикалы. Выход радикалов небольшой, но даже в таком количестве они могут инициировать цепные процессы. В [13, 14] установлено, что в присутствии Н2О2 в водных растворах инициируются цепные тиол-ен реакции GSH с ненасыщенными фенолами ресвератролом и кофейной кислотой. Реакции тиолов с олефинами (тиол-ен реакции, гидротиолирование алкенов) с образованием тиоэфиров известны давно с 1905 г. [17]. Но в последние десятилетия им уделяется большое внимание в связи с возможностью селективного и стереоселективного синтеза разнообразных соединений в полимерной и медицинской химии [18–20]. Ресвератрол и кофейная кислота – растительные полифенолы, метаболиты биосинтеза лигнина, содержат ненасыщенную связь в боковых заместителях ароматического каркаса. В последнее время эти фенолы, в особенности ресвератрол (RVT, 3,5,4'-тригидроксистильбен), привлекают внимание медиков и биохимиков в связи с так называемым “французским парадоксом” – необычно низким уровнем сердечно-сосудистых и онкологических заболеваний при высококалорийном питании с обилием жиров, наблюдаемом в некоторых регионах Франции на фоне регулярного потребления красного вина [21, 22]. Благодаря наличию ненасыщенной связи, сопряженной с двумя фенольными фрагментами (схема 1 ), RVT может существовать в транс- и цис-форме, и активно реагировать с тиильными радикалами, которые с высокими константами скорости (∼105 М–1 с–1) обратимо присоединяются к двойным связям и катализируют цис–транс-изомеризацию олефинов [23, 24].
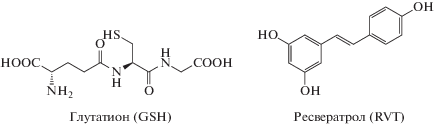
Схема 1 . Структурные формулы глутатиона и ресвератрола.
В настоящей работе экспериментально исследованы кинетические закономерности взаимодействия GSH с ресвератролом в присутствии Н2О2, методом масс-спектрометрии (MS-электроспрей положительных ионов) изучен состав продуктов, образующихся в реакциях GSH с Н2О2 и с RVT. C учетом полученных данных о тиол-ен реакции GSH с RVT, а также литературных сведений о реакциях GSH, H2O2 и тиильных радикалов предложена уточненная кинетическая модель сложного процесса взаимодействия GSH и H2O2 и тиол-ен реакции с RVT (в водной среде при 37°С), которая хорошо описывает особенности кинетики процесса в широком диапазоне концентраций реагентов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Глутатион (GSH), реактив Эллмана (DTNB, 5,5'-дитиобис-(2-нитробензойная кислота))-“Sigma-Aldrich”, пероксид водорода, Н2О2, “PanReac AppliChem”, транс-ресвератрол (RVT), “abcrGmbH”, использовали без очистки.
В качестве реакционной среды использовали деионизированную воду.
Базовый раствор RVT (13.3 мМ) готовили в этаноле (“Медхимпром”), который добавляли к реакционной смеси. Концентрацию H2O2 (в отсутствие GSH) контролировали методом йодометрии. Концентрацию GSH определяли с применением реактива Эллмана спектрофотометрически при λmax = 412 нм и ε = 0.14 × 105 М–1 см–1 [25, 26].
Тиол-ен реакцию GSH с RVT проводили при температуре 37°С непосредственно в термостатируемой кювете спектрофотометра СФ-2000 (ООО “ОКБ Спектр”, Россия), в которой регистрировали расходование RVT ε = 0.3 × 105 М–1 см–1 при λmax = 304–308 нм, а также в стеклянной термостатируемой ячейке, снабженной устройствами для отбора проб и барботажа воздухом. По ходу реакции из реакционного сосуда отбирали аликвоты по 90 мкл для анализа RVT и GSH. Аликвоты добавляли соответственно к 3 мл деионизированной воды и натрий-фосфатного буферного раствора (PBS, рН 7.4), содержащего 0.3 мМ DTNB, и записывали УФ-спектры.
GSH, содержащий две карбоксильных группы, при физиологической концентрации (0.1–10 мМ) образует кислые растворы (рН 3–4). В работе [27] нами были выявлены существенные различия в кинетике и механизме реакции GSH с Н2О2 в деионизированной воде и в фосфатных буферных системах с рН ≥ 7, часто применяемых в биохимических исследованиях. Поэтому в каждом опыте измеряли pH растворов рН-метром-милливольтметром рН-410 (“Аквилон”, Россия). Ошибка в измерении рН составляла ±0.02. Ошибка в измерении скоростей расходования GSH и RVT не превышала 15%.
Исследования молекулярных продуктов реакции проводили методом масс-спектрометрии в Центре коллективного пользования “Новые материалы и технологии” ИБХФ РАН на тандемном масс-спектрометре LTQ FT Ultra (“Thermo Finnigan”, Германия) методом электроспрейной ионизации в режиме измерения положительных ионов. Непосредственно перед вводом в масс-спектрометр образец разбавляли в 20 раз 50%-ным раствором ацетонитрила с добавлением 0.1% муравьиной кислоты.
Компьютерное моделирование кинетических кривых расходования реагентов в реакции GSH с RVT в присутствии H2O2 и оптимизацию констант скоростей 19-ти квази-элементарных реакций, составляющих кинетическую модель процесса осуществляли с использованием программы [28].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Кинетические особенности тиол-ен реакции глутатиона с ресвератролом в присутствии пероксида водорода в деионизированной воде
Выше отмечалось, что концентрация GSH в биологических тканях и жидкостях на порядки выше микромольной концентрации других компонентов антиоксидантной системы. Поэтому в экспериментах, как правило, [GSH] варьировали в диапазоне 0.1–10 мМ, а [RVT] были порядка 1–100 мкМ. На рис. 1 представлены кинетические кривые расходования RVT в отсутствие (кривая 1) и в присутствии Н2О2 при разных концентрациях GSH (кривые 2–5). Видно, что расходование RVT, который является эффективным акцептором радикалов, наблюдается только при совместном присутствии GSH и Н2О2 (кривые 3–5). Необходимо отметить, что введение GSH в реакционную среду приводит к снижению рН (табл. 1).
Рис. 1.
Кинетические кривые расходования 0.03 мМ RVT в реакции с GSH в отсутствие (1) и в присутствии 4.55 мМ Н2О2 (2–5); концентрация GSH (мМ): 1 – 25 (концентрация RVT – 0.033 мМ); 2 – 0; 3 –2.5; 4 – 5; 5 – 10. Точки – экспериментальные данные, сплошные линии – расчетные данные по кинетической модели (табл. 2).
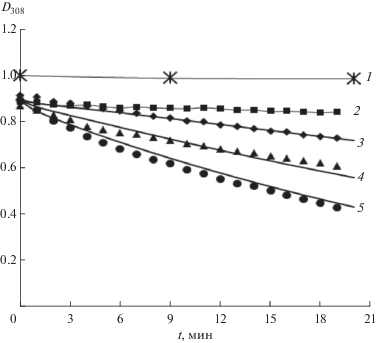
Таблица 1.
Кинетические характеристики расходования 0.03 мМ RVT при разных концентрациях GSH в присутствии 4.55 мМ Н2О2 в деионизированной воде при 37°С
| [GSH], мM | рН | WGSH × 107, М/с | WRVT × 109, М/с | *Wi × 109, М/с |
|---|---|---|---|---|
| 0 | 6.70 | 0 | 0 | 0 |
| 2.0 | 3.28 | 1.62 | 4.3 | 2.1 |
| 2.5 | 3.23 | 1.72 | 5.5 | 2.5 |
| 5.0 | 3.10 | 2.14 | 8.2 | 4.2 |
| 7.0 | 3.03 | 2.37 | 11.6 | 5.4 |
| 10.0 | 3.00 | 2.63 | 17.5 | 7.1 |
Из рис. 2 следует, что начальная скорость расходования RVT (WRVT) линейно возрастает с ростом его начальной концентрации. Ранее в [10] нами были получены эмпирические зависимости для скорости расходования глутатиона (WGSH) и скорости инициирования радикалов (Wi) при взаимодействии GSH и Н2О2, измеренной методом ингибиторов:
(1)
$\begin{gathered} {{W}_{{{\text{GSH}}}}} \cong {\text{const}}\left[ {{\text{GSH}}} \right]_{0}^{{0.3}}\left[ {{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}}} \right]_{0}^{{1.2}}, \\ {\text{где}}\,\,{\text{сonst}} = (1.7 \pm 0.2) \times {{10}^{{ - 3}}}\,\,{{{\text{М}}}^{{ - 0.5}}}\,\,{{{\text{с}}}^{{ - 1}}}. \\ \end{gathered} $(2)
$\begin{gathered} {{W}_{{\text{i}}}} \cong {\text{const[GSH}}{{{\text{]}}}^{{0.75}}}{{[{{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}}]}^{{0.75}}}, \\ {\text{где}}\,\,{\text{const}} = (1.3 \pm 0.2) \times {{10}^{{ - 5}}}\,\,{{{\text{М}}}^{{ - 0.5}}}\,\,{{{\text{с}}}^{{ - 1}}}. \\ \end{gathered} $Рис. 2.
Зависимости скоростей расходования RVT (WRVT) от концентрации RVT в реакционной смеси 4.55 мМ Н2О2 с разными концентрациями GSH (мМ): 1 – 10; 2 – 5; 3 – 2.5.
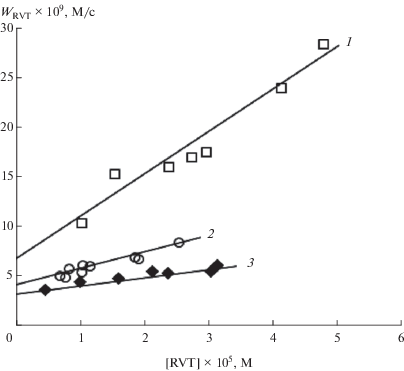
Скорость расходования RVT (WRVT), как и WGSH [10], нелинейно зависит от концентраций GSH и Н2О2. В табл. 1 представлены значения WRVT, экспериментально измеренные при разных концентрациях GSH в присутствии 4.55 мМ Н2О2, и скорости инициирования радикалов (*Wi), рассчитанные по уравнению (2). Примечательно, что значения скоростей, отсекаемые линейными зависимостями WRVT – [RVT] (рис. 2) на оси ординат, практически (в пределах ошибки) совпадают с расчетными значениями *Wi для соответствующих концентраций GSH. Длина цепи в расходовании ресвератрола (WRVT/Wi), как можно видеть из табл. 1, невелика, порядка 2-х звеньев, а выход радикалов меньше 1% (Wi/WGSH < 0.01).
Скорость расходования RVT удовлетворительно описывается уравнением (3) для цепных реакций окисления и полимеризации с квадратичным обрывом цепей на ведущих цепи радикалах [29]. В [13, 14] мы предположили, что в цепной реакции ненасыщенных фенолов с GSH в присутствии Н2О2 обрыв происходит на тиильных радикалах GS•, а лимитирующей стадией является реакция радикала с RVT:
(3)
${{W}_{{{\text{RVT}}}}} = {{W}_{{\text{i}}}} + а\left[ {{\text{RVT}}} \right]W_{{\text{i}}}^{{0.5}}.$Здесь параметр а ≅ 3.5 М–0.5 с–0.5. аналогичен отношению констант скорости реакций продолжения (kp) и обрыва цепей (kt)
Анализ продуктов
Исследования молекулярных продуктов реакции проводили методом масс-спектрометрии с применением электроспрейной ионизации в режиме измерения положительных ионов. На рис. 3а приведен масс-спектр исходного образца GSH, в котором наряду с молекулярным ионом МН+ 308.09 присутствуют ионы М'Н+ 615.17, свидетельствующие о наличии в образце достаточно устойчивых димеров GSH–GSH. В работе [30] было отмечено, что при исследовании масс-спектров GSH методом электроспрея отрицательных ионов в водном растворе наряду с ионами GSH обнаруживаются ионы димера, тогда как в фосфатном буферном растворе (0.1 М, рН ∼ 7) димер не регистрируется. Очевидно, одноименно отрицательно заряженные вследствие диссоциации карбоксильных групп ионы глутатиона не образуют димеров при рН ≥ 7.
Рис. 3.
Масс-спектры исходного глутатиона (a), продуктов реакции 10 мМ GSH с 2 мМ Н2О2 (б) и продуктов, образующихся в смеси 2.3 мМ GSH, 1.3 мМ RVT и 3.2 мМ Н2О2 (в) в деионизированной воде.
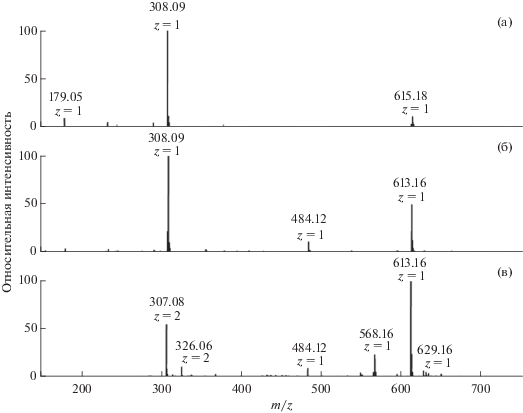
Основным продуктом окисления GSH в реакции с Н2О2 является соответствующий дисульфид GSSG (M''Н+ 613.16) (рис. 3б). Реакция происходит в соответствии с известным и многократно подтвержденным [30–34] стехиометрическим уравнением:
На рис. 3в в масс-спектре продуктов, полученных в реакционной смеси 2.3 мМ GSH, 1.3 мМ RVT и 3.2 мМ Н2О2 в исходно деионизированной воде видно, что основной продукт – дисульфид GSSG (MН+ 613.16). Наряду с GSSG образуется продукт МН+ 568.16 с массой, соответствующей гидропероксиду (PO2H), который может получиться в результате последовательного присоединения тиильного радикала GS• и кислорода к RVT:
Кинетическая модель взаимодействия глутатиона c ресвератролом
Для анализа кинетики взаимодействия GSH с RVT в присутствии Н2О2 использовано компьютерное моделирование с использованием программы [28]. Ранее в [10] мы представили кинетическую модель взаимодействия GSH с Н2О2, которая включала 13 квазиэлементарных реакций. С учетом дополнительных экспериментальных и уточненных в литературе данных для описания взаимодействия GSH с Н2О2 оставлено 10 реакций (табл. 2, реакции (I)–(X)).
Таблица 2.
Кинетическая модель взаимодействия GSH с RVT в присутствии Н2О2 в водной среде при 37°С
| №№ | Реакции | Константы скорости | Значение ki, М–1 с–1 |
|---|---|---|---|
| I | GSH + H2O2 → K | k1 | 5 |
| II | K → GSH + H2O2 | k2 | *4 × 10–3 |
| III | K + GSH → GSSG + 2H2O | k3 | 6 × 10–2 |
| IV | GSH + GSH → C | k4 | 1.3 |
| V | C → GSH + GSH | k5 | *9 × 10–4 |
| VI | C + H2O2 → GSSG + 2H2O | k6 | 1.5 × 10–3 |
| VII | C + H2O2 → 2GS• + 2H2O | k7 | 2 × 10–5 |
| VIII | GSH + H2O2 → •OH + GS• + H2O | k8 | 1 × 10–3 |
| IX | •OH + GSH → H2O + GS• | k9 | 1 × 106 |
| X | GS• + GS• → GSSG | k10 | 1 × 109 |
| XI | GS• + RVT → P• | k11 | 2.5 × 105 |
| XII | P•→ GS• + RVT | k12 | *1 × 104 |
| XIII | P• + GSH → GS• + PH | k13 | 5 × 104 |
| XIV | P• + (O2) → ${\text{PO}}_{{\text{2}}}^{\centerdot }$ | k14 | **1 × 106 |
| XV | ${\text{PO}}_{{\text{2}}}^{\centerdot }$ + GSH → POOH + GS• | k15 | 5 × 103 |
| XVI | GS• + ${\text{PO}}_{{\text{2}}}^{\centerdot }$ → GSH + O2 | k16 | 1 × 109 |
| XVII | GSH + RVT → Y | k17 | 5 |
| XVIII | Y → GSH + RVT | k18 | 2 × 10–4 |
| XIX | Y + GS• → P• + GSH | k19 | 1 × 106 |
Примечание. К – комплекс GSH–H2О2; С – комплекс GSH–GSH; Y – комплекс GSH с RVT; P• – алкильный радикал, образующийся в результате присоединения тиильного радикала GS• к двойной связи RVT; ${\text{PO}}_{2}^{ \bullet }$ и POOH – соответствующие пероксильный радикал и гидропероксид. * Константа скорости имеет размерность с–1. ** k14 = k [O2] имеет размерность с–1; [O2] = 1 × 10–4 M.
Поскольку в серии работ [35–38] приводятся достаточно убедительные результаты спектроскопических (УФ и ИК) исследований и теоретического анализа [36] появлению комплексов GSH–Н2О2 не только в буферных растворах с физиологическим рН, но и в чистой воде [37], которая при добавлении GSH имеет рН 2, мы сохранили в кинетической модели реакции (I)–(III) образования комплекса К (GSH–Н2О2) и окисления его в дисульфид GSSG, хотя в масс-спектрах продуктов реакции (рис. 3б) не обнаруживается соответствующий комплексу ион МН+ 342.
Примечательно, что измеренная в [37] методом время-разрешенной рамановской спектроскопии скорость расходования GSH при концентрациях реагентов 1 М, равная 2 × 10–3 М/с, практически совпадает с величиной скорости WGSH = = 1.8 × 10–3 M/c, рассчитанной по уравнению (1).
В [27] мы показали, что в фосфатных буферных системах при рН ≥ 7 усиливается реакция окисления GSH кислородом воздуха и резко снижается скорость инициирования радикалов в реакции GSH с Н2О2 по сравнению с Wi в деионизованной воде. При рН ≥ 7 не образуются димеры глутатиона. Поэтому при конструировании модели было принято, что выход радикалов происходит, в основном, в реакции (VII) при взаимодействии димера GSH–GSH (С, табл. 2) с Н2О2. Реакции (VII) и (VIII), которые поставляют радикалы, практически не влияют на скорость расходования GSH (WGSH). Тиоловая группа –SH в комплексах К [35] и С определяется реактивом Эллмана так же, как в свободном GSH. Величина k10 = 109 М–1 с–1 известна для быстрой рекомбинации тиильных радикалов [34].
Реакции XI–XVI имеют место при добавках RVT и вместе с остальными реакциями описывают кинетические кривые расходования RVT. Известно, что тиильные радикалы с высокими константами скорости обратимо (∼105 М–1 с–1) присоединяются к двойным связям –С=С– [23, 40], поэтому в модель введены реакции (XI) и (XII). Образующийся в результате присоединения GS• к RVT алкильный радикал P• может прореагировать с GSH (k13 ≈ 105–106 М–1 с–1 [23, 40]) или с кислородом, поскольку опыты проводили в аэробных условиях (k14 ≈ 109–1010 М–1 с–1 [41]). В табл. 3 приведены рассчитанные по модели значения квазистационарных концентраций радикалов, из которых видно, что в присутствии О2 доминируют пероксильные радикалы ${\text{PO}}_{2}^{\centerdot }$ и увеличивается содержание молекулярных продуктов присоединения радикалов GS• к RVT. Определяющая роль О2 в кинетике присоединения тиильных радикалов к олефинам при изучении ее методом флеш-фотолиза отмечена и проанализирована в работах [41, 42].
Таблица 3.
Влияние кислорода на тиол-ен реакцию GSH с RVT в присутствии Н2О2*
| Условия | [GS●], М | [P●], М | [${\text{PO}}_{2}^{ \bullet }$], М | [PH], М | [POOH], М | ΔGSH, М |
|---|---|---|---|---|---|---|
| Без RVT, Реакции (I)–(X) |
7.3 × 10–10 | – | – | – | – | 3.37 × 10–4 |
| C RVT без O2 Реакции (I)–(XIII) |
7.3 × 10–10 | 8.6 × 10–12 | – | 1.9 × 10–6 | – | 3.39 × 10–4 |
| С RVT и О2 Реакции (I)–(XVI) |
9.96 × 10–11 | 1.9 × 10–14 | 5.06 × 10–9 | 4.0 × 10–9 | 9.2 × 10–5 | 4.2 × 10–4 |
| C Y (GSH–RVT) Реакции (I)–(XIX) |
4.76 × 10–11 | 3.2 × 10–14 | 9.6 × 10–9 | 5.3 × 10–9 | 1.54 × 10–4 | 2.2 × 10–4 |
* Рассчитанные по модели для рис. 4 значения концентраций компонентов при t = 150 мин. Прочерки означают, что в расчете (см. Условия) не учитывались реакции с участием этих частиц.
На рис. 4 представлены экспериментально многократно повторенные и воспроизводимые кинетические кривые расходования глутатиона и ресвератрола, взятых при сопоставимых по масштабу концентрациях. Вопреки нашим ожиданиям, в присутствии RVT скорость расходования GSH не увеличивается за счет дополнительного расходования в цепной реакции с RVT, а уменьшается. Чтобы получить такой эффект, модель дополнили обратимым связыванием RVT с GSH в комплекс Y (реакции (XVII)–(XIX)).
Рис. 4.
Кинетические кривые расходования 1.9 мМ GSH (1, 2, 3) и 0.53 мМ RVT (3) в присутствии 2.1 мМ Н2О2: 1 – GSH (◆) с RVT; 2 – GSH (△) без RVT; водная среда, 37°С. Точки – экспериментальные данные, сплошные линии – расчетные данные по кинетической модели (табл. 2).
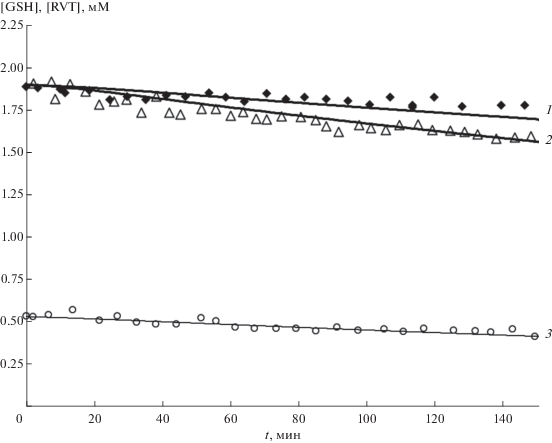
Представленная кинетическая модель с оптимизированными константами скоростей вполне удовлетворительно описывает экспериментальные концентрационные зависимости для WRVT и WGSH (рис. 2 и табл. 1), а также экспериментальные кинетические кривые расходования RVT и GSH в реакции GSH с RVT в присутствии Н2О2 (рис. 1 и 4).
ЗАКЛЮЧЕНИЕ
Опираясь на экспериментально полученные кинетические кривые и концентрационные зависимости скорости расходования глутатиона и ресвератрола в присутствии H2O2 от концентраций реагентов (в водной среде при 37°С) и данные по составу продуктов, разработана кинетическая модель взаимодействия GSH с RVT, инициированного тиильными радикалами, образующимися в реакции GSH с H2O2. Скелетная модель включает 19 реакций с соответствующими оптимизированными для условий эксперимента значениями констант скоростей. Реакции образования комплексов GSH–H2O2 и GSH–GSH позволили описать нетривиальные концентрационные зависимости скорости расходования глутатиона и инициирования радикалов при взаимодействии GSH с H2O2, реакции (XIV) и (XV) отражают важную роль кислорода воздуха в радикально-цепном расходовании RVT. Дополнение модели обратимыми реакциями (XVII)–(XIX) образования комплексов RVT с компонентами процесса позволило описать нетривиальный эффект заметного уменьшения скорости расходования GSH при повышенных концентрациях RVT. Подавляющее большинство исследований по биохимии GSH проводят в условиях, близких к физиологическим в животных организмах, т.е. в буферных растворах, обеспечивающих рН 7.2–7.4. В таких условиях радикалы в реакции GSH с H2O2 не образуются [27], и, следовательно, нет расходования RVT. Возможно, обнаруженное нами образование радикалов при взаимодействии GSH с H2O2 и другими пероксидами, а также реакции GSH с ненасыщенными фенолами имеют место и играют роль в физиологии растений, в которых внутри- и межклеточные жидкости характеризуются более низкими по сравнению с фауной значениями рН, при использовании тиолов в косметике, фармацевтике, приготовлении БАДов и в виноделии.
Список литературы
Mironczuk-Chodakowska I., Witkowska A.M. // Adv. Med. Sci. 2018. V. 63. № 3. P. 68. https://doi.org/10.1016/j.advms.2017.05.005
Sporer A.J., Kahl L.J., Price-Whelan A., Dietrich L.E.P. // Annu. Rev. Biochem. 2017. V. 86. P. 777. https://doi.org/10.1146/annurev-biochem-061516-044453
Hartl J., Kiefer P., Kaczmarczyk A., Mittelviefhaus M., Meyer F., Vonderach T., Hattendorf B., Jenal U., Vorholt J.A. // Nat. Metab. 2020. V. 2. P. 153. https://doi.org/10.1038/s42255-019-0166-0
Elgawish M.S., Kishikawab N., Kurodab N. // Analyst. 2015. V. 140. P. 8148. https://doi.org/10.1039/c5an01604e
Chen Y., Han M., Matsumoto A., Wang Y., Thompson D.C., Vasiliou V. // Adv. Exp. Med. Biol. 2018. V. 1032. P. 37. https://doi.org/10.1007/978-3-319-98788-0_3
Burtis A.C., Ashwood E.R., Saunders W.B., Tietz N.W. Fundamentals of Clinical Chemistry, Philadelphia, 4th edn, 1996.
Estrela J.M., Ortega A., Obrador E. // Crit. Rev. Clin. Lab Sci. 2006. V. 43. № 2. P. 143. https://doi.org/10.1080/10408360500523878
Toyokuni S. // Front. Pharmacol. 2014. V. 5. № 200. P. 1. https://doi.org/10.3389/fphar.2014.00200
Guo R., Yang G., Feng Z., Zhu Y., Yang P., Song H., Wang W., Huang P., Zhang J. // Biomater. Sci. 2018. V. 6. № 5. P. 1238. https://doi.org/10.1039/c8bm00094h
Зинатуллина К.М., Касаикина О.Т., Кузьмин В.А., Храмеева Н.П. // Кинетика и катализ. 2019.Т. 60. № 3. С. 281. [Zinatullina, K.M., Kasaikina, O.T., Kuzmin, V.A., Khrameeva, N.P. // Kinet. Catal. 2019. V. 60. № 3. Р. 266.] https://doi.org/10.1134/S0023158419030169
Зинатуллина К.М., Касаикина О.Т., Кузьмин В.А., Храмеева Н.П., Шапиро Б.И. // Изв. АН. Сер. хим. 2017. № 7. С. 1300. [Zinatullina K.M., Kasaikina O.T. Kuzmin V.A., Khrameeva N.P. Shapiro B.I. // Russ. Chem. Bull. 2017. V. 66. № 7. P. 1300.] https://doi.org/10.1134/S0023158417050093
Зинатуллина К.М., Храмеева Н.П., Касаикина О.Т., Кузьмин В.А. // Изв. АН. Сер. хим. 2018. № 4. С. 726. [Zinatullina K.M., Khrameeva N.P., Kasaikina O.T., Kuzmin V.A. // Russ. Chem. Bull. 2018. V. 67. № 4. P. 726.] https://doi.org/10.1007/s11172-018-2129-0
Зинатуллина К.М., Храмеева Н.П., Касаикина О.Т., Кузьмин В.А., Шапиро Б.И. // Изв. АН. Сер. хим. 2017. № 11. С. 2145. [Zinatullina K.M., Khrameeva N.P., Kasaikina O.T., Kuzmin V.A., Shapiro B.I. // Russ.Chem. Bull. 2017. V. 66. № 11. P. 2145.] https://doi.org/10.1007/s11172-017-1995-1
Zinatullina K.M., Khrameeva N.P., Kasaikina O.T. // Bulg. Chem. Comm. 2018. V. 50. Special Issue C. P. 25.
Зинатуллина К.М., Касаикина О.Т., Кузьмин В.А., Храмеева Н.П., Шапиро Б.И. // Изв. АН. Сер. хим. 2016. № 12. С. 2825. [Zinatullina K.M., Kasaikina O.T., Kuzmin V.A., Khrameeva N.P., Shapiro B.I. // Russ.Chem. Bull. 2016. V. 65. № 12. P. 2825.] https://doi.org/10.1007/s11172-016-1663-x
Зинатуллина К.М., Касаикина О.Т., Мотякин М.В., Ионова И.С., Дегтярев Е.Н., Храмеева Н.П. // Изв. АН. Сер. хим. 2020. № 10. С. 1865. [Zinatullina K.M., Kasaikina O.T., Motyakin M.V., Ionova I.S., Degtyarev E.N., Khrameeva N.P. Russ. Chem. Bull. 2020. V. 69. № 10. P. 1865.] https://doi.org/10.1007/s11172-020-2971-8
Posner T. // Ber. Dtsch. Chem. Ges. 1905. V. 38. № 1. P. 646. https://doi.org/10.1002/cber.190503801106
Nilsson C., Simpson N., Malkoch M., Johansson M., Malmström E. // J. Polym. Sci. A: Polym. Chem. 2008. V. 46. № 4. P. 1339. https://doi.org/10.1002/pola.22474
Liu Y., Hou W., Sun H., Cui C., Zhang L., Jiang Y., Wu Y., Wang Y., Li J., Sumerlin B.S., Liu Q., Tan W. // Chem. Sci. 2017. V. 8. P. 6182. https://doi.org/10.1039/c7sc01447c
Biermann U., Metzger J.O. // Eur. J. Org. Chem. 2018. № 6. P. 730. https://doi.org/10.1002/ejoc.201701692
Salehi B, Mishra A.P., Nigam M., Sener B., Kilic M., Sharifi-Rad M., Fokou P.V.T, Martins N., Sharifi-Rad J. // Biomedicines. 2018. V. 6. № 3. P. 91. https://doi.org/10.3390/biomedicines6030091
Yu W., Fu Y.C., Wang W. // J. Cell. Biochem. 2012. V. 113. № 3. P. 752. https://doi.org/10.1002/jcb.23431
Chatgilialoglu C., Ferreri C. // Acc. Chem. Res. 2005. V. 38. № 6. P. 441. https://doi.org/10.1021/ar0400847
Samadi A., Andreu I., Ferreri C., Dellonte S., Chatgilialoglu C. // J. Am. Oil Chem. Soc. 2004. V.81. № 8. P. 753. https://doi.org/10.1007/s11746-004-0974-8
Ellman G.L. // Arch. Biochem. Biophys. 1959. V. 82. P. 70.
Pereira C.D., Minamino N., Takao T. // Anal. Chem. 2015. V. 87. P. 10 785. https://doi.org/10.1021/acs.analchem.5b03431
Зинатуллина К.М., Касаикина О.Т., Кузьмин В.А., Храмеева Н.П., Писаренко Л.М. // Изв. АН. Сер. хим. 2019. № 7. С. 1441. https://doi.org/10.1007/s11172-019-2574-4
Sirick A.V., Pliss R.E., Rusakov A.I., Pliss E.M. // Oxid. Commun. 2014. V. 37. № 1. P. 37.
Denisov E.T., Denisova T.G. Handbook of antioxidants: bond dissociation energies, rate constants, activation energies and enthalpies of reactions. Boca Raton: CRC press. 2000. 289 p.
Deutsch J.C., Santhosh-Kumar C.R., Kolhouse J.F. // J. Chromatogr. A. 1999. № 862. P. 161. https://doi.org/10.1016/S0021-9673(99)00932-2
Winterbourn C.C., Metodiewa D. // Free RadicBiol Med. 1999. V. 27. P. 322. https://doi.org/10.1016/S0891-5849(99)00051-9
Petzolda H., Sadler P.J. // Chem. Commun. 2008. P. 4413. https://doi.org/10.1039/b805358h
Singh B., Das R.S., Banerjee R., Mukhopadhyay S. // Inorg. Chim. Acta. 2014. № 418. P. 51. https://doi.org/10.1016/j.ica.2014.03.003
Chatgilialoglu C., Bowry V.W. // J. Org. Chem. 2018. V. 83. № 16. P. 9178. https://doi.org/10.1021/acs.joc.8b01216
Abedinzadeh Z., Gardes-Albert M., Ferradini C. // Can. J. Chem. 1989. V. 67. P. 1247. https://doi.org/10.1139/v89-190
Berges J., Caillet J., Langlet J., Abedinzadeh Z. // Theor. Claim. Acta. 1993. V. 85. P. 87 99. https://doi.org/10.1007/BF01374579
Picquart M., Grajcar L., Baron M.H., Abedinzadeh Z. // Biospectroscopy. 1999. V. 5. P. https://doi.org/10.1002/(SICI)1520-6343(1999)5:6<328: :AID-BSPY2>3.0.CO;2-J
Abedinzadeh Z. // Can. J. Physiol. Pharmacol. 2001. V. 79. P. 166. https://doi.org/10.1139/cjpp-79-1-166
Hellwege K.-H., Madelung O., Martienssen W. Landolt-Bornstein: Numerical Data and Functional Relationships in Science and Technology. Springer-Verlag. 1983. V. 13. 308 p.
Chatgilialoglu C., Studer A. Encyclopedia of Radicals in Chemistry, Biology and Materials. John Wiley & Sons, 2012. https://doi.org/10.1002/9781119953678
Ito O., Matsudo M. // J. Amer. Chem. Soc. 1979. V. 101. № 7. P. 1815. https://doi.org/10.1021/ja00501a031
Ito O., Matsudo M. // J. Amer. Chem. Soc. 1979. V. 101. № 19. P. 5732. https://doi.org/10.1021/ja00513a045
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ