

Кинетика и катализ, 2021, T. 62, № 6, стр. 718-732
Исследование никелевых катализаторов, нанесенных на смешанные оксиды MnOx–СеO2 в процессе углекислотной конверсии метана
М. В. Грабченко a, *, Н. В. Дорофеева a, И. Н. Лапин a, V. La Parola b, L. F. Liotta b, О. В. Водянкина a
a Томский государственный университет,
63405 Томск, просп. Ленина, 36, Россия
b Istituto per lo Studio dei Materiali Nanostrutturati (ISMN)-CNR
90146 Palermo, via Ugo La Malfa, 153, Italy
* E-mail: marygra@mail.ru
Поступила в редакцию 18.01.2021
После доработки 21.06.2021
Принята к публикации 18.07.2021
Аннотация
Ni-содержащие (10 мас. % Ni) нанесенные катализаторы на основе оксидных носителей
CeО2–MnOх с разным соотношением Ce/Mn (0.25, 1 и 9) были синтезированы и исследованы в процессе
углекислотной конверсии метана (УКМ). Методами низкотемпературной сорбции N2, РФА и ТПВ-H2 изучено влияние соотношения Ce/Mn на фазовый состав катализатора, размер и распределение
Ni-содержащего оксидного предшественника для определения роли фаз MnOx и твердого раствора Ce1– xMnxO2 – δ в формировании активной поверхности катализатора. Добавление оксида марганца способствует
внедрению катионов Nin+ в структуру поверхности с образованием твердых растворов, что приводит к снижению
размеров кристаллитов NiO на поверхности носителей. Приготовленные катализаторы были
исследованы в процессе УКМ в режиме длительных температурных испытаний в течение 24
ч. Катализатор, полученный на основе носителя с массовым соотношением Ce/Mn = 0.25,
продемонстрировал максимальную активность и стабильность в УКМ (X(CH4)/X(CO2) = 47/70), что обусловлено наличием развитой границы раздела MnOx и присутствием на поверхности высокодисперсных частиц Ni и MnNiOx.
Графический абстракт
ВВЕДЕНИЕ
Энергетическая проблема, возникшая на фоне интенсивного потребления минерального сырья, определила мировые тенденции снижения потребления природных ископаемых и необходимость поиска новых высокоэффективных процессов с использованием альтернативных источников энергии, в том числе синтез-газа. В настоящее время активно изучаются процессы получения синтез-газа с различным соотношением H2/CO из возобновляемого сырья – СО2 и СН4. Применение углекислого газа в качестве сырья способствует также решению экологических задач сокращения выбросов CO2 в рамках технологии “улавливания и использования углерода (CUU)” [1]. Основным источником метана являются запасы природного и попутного газа, в качестве перспективных ресурсов CO2 и CH4 выступают биогаз и свалочный газ. В связи с этим интерес к исследованию процесса углекислотной конверсии метана (УКМ), который не только позволяет уменьшить выбросы парниковых газов, но и получить промышленно важный синтез-газ – ценный ресурс для производства продуктов с высокой добавленной стоимостью [2] – постоянно растет. Несмотря на очевидные экономические и экологические преимущества, процесс углекислотной конверсии метана имеет большие трудности в практической реализации. Во-первых, углекислотная конверсия метана является высоко эндотермическим процессом (ΔH25°C = 247 кДж/моль), поэтому для достижения существенной конверсии целесообразно использование температуры >700°C [3–5]. Во-вторых, образование углерода и спекание частиц активного компонента приводит к быстрой дезактивации катализаторов на основе более экономически выгодных неблагородных переходных металлов (Co и/или Ni) [4].
Никелевые катализаторы – наиболее известные материалы в процессе УКМ, что связано с их высокой каталитической активностью и доступностью по сравнению с катализаторами на основе благородных металлов Rh, Ru, Pt и Pd [6, 7]. В литературе для решения проблемы снижения активности никельсодержащих катализаторов из-за зауглероживания поверхности и спекания Ni-частиц при высоких температурах предлагают различные подходы: использование носителей оснóвной природы [4, 8–10] и мезопористых носителей с развитой поверхностью [11], промотирование благородными металлами [12–15], создание биметаллических активных частиц [15–17].
На процесс дезактивации никелевых катализаторов вследствие зауглероживания оказывает влияние размер частиц активного компонента. Увеличение размера частиц Ni0 способствует формированию разных типов углерода и кокса, которые различаются по морфологии, структуре и реакционной способности [18]. Насыщение углеродом малых частиц никеля с образованием небольших углеродных кластеров происходит быстрее, однако данные кластеры менее стабильны, чем углеродные кластеры на больших никелевых частицах [19], и легче взаимодействуют с СО2. Таким образом, большинство современных исследований направлено на получение катализаторов с высокодисперсными частицами никеля. Однако количество и природа углеродных отложений зависят не только от активного металла и температуры реакции, но во многих случаях и от используемого носителя [4, 6, 20].
Среди перспективных носителей можно выделить CeO2, MnO2 и ZrO2 [21, 22]. CeO2 широко применяется в качестве промотора, а также индивидуального носителя для никелевых катализаторов в УКМ по причине его высокой кислородной емкости, окислительно-восстановительных свойств, а также оптимального значения удельной поверхности, что способствует стабилизации металла в высокодисперсном состоянии [23–26]. Помимо этого CeO2 обладает собственной активностью в отношении CH4 [27] и CO2 [28]. Таким образом, СеО2 в качестве промотора и/или носителя для никелевых катализаторов может обеспечивать их стабильность в процессе УКМ за счет диспергирования Ni-частиц, специфичного взаимодействия металла с носителем, собственных окислительно-восстановительных свойств.
Использование в качестве носителей двойных церийсодержащих оксидов CeO2–MnOx, CeO2–ZrO2, CeO2–La2O3 и т.д. позволяет решить проблему стабильности и устойчивости катализаторов к процессам углеотложения в УКМ благодаря комбинированию свойств оксидов (снижение кислотности, увеличение реакционной способности поверхностного/объемного кислорода, повышение термической стабильности и т.д.).
Среди оксидов переходных металлов в качестве модифицирующей добавки особый интерес представляют оксиды марганца, которые, будучи относительно дешевыми, весьма термостабильны и устойчивы. Так, внедрение MnOx в структуру CeO2 приводит к образованию твердого раствора CexMn1– xO2 с большим количеством поверхностных дефектов [22]. По сравнению с образцами на основе индивидуального CeO2 введение Mnn+ в состав катализатора усиливает взаимодействие Ni-наночастиц с носителем и, следовательно, уменьшает их спекание, в то время как взаимодействие между марганцем и оксидом церия увеличивает концентрацию кислородных вакансий и поверхностно-активных форм кислорода, что ведет к повышению скорости окисления осажденного углерода [29]. Более того, MnOx обладает оснóвными свойствами, которые способствуют росту количества адсорбированного CO2, что также позволяет исключить образование углеродсодержащих отложений на поверхности катализатора [30]. Очевидно, что изменение структуры и свойств поверхности смешанных оксидов СеO2–MnOx благодаря синергетическому эффекту между MnOx и CeO2 будет иметь решающее значение для углекислотной конверсии метана. В связи вышесказанным целью настоящей работы является изучение влияния массового соотношения Mn/Ce в составе катализаторов Ni/СеO2–MnOx на их фазовый состав, распределение Ni-содержащего активного компонента, а также на каталитическую активность и стабильность в реакции углекислотной конверсии метана.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез носителей и катализаторов
Носители СеO2–MnOx c различным массовым соотношением Сe/Mn (0.25, 1 и 9) были синтезированы золь–гель-методом с применением лимонной кислоты (C6H8O7 ⋅ H2O) как хелатирующего агента. В качестве прекурсоров Ce и Mn использовали Ce(NO3)3 ⋅ 6H2O и Mn(NO3)2 ⋅ 6H2O (х. ч.). Соли растворяли в минимальном объеме воды при интенсивном перемешивании. В круглодонной колбе отдельно в воде растворяли лимонную кислоту. Раствор нитратов Ce и Mn быстро добавляли к раствору лимонной кислоты в мольном соотношении C6H8O7 ⋅ H2O/(Ce + Mn) = = 1.2. Вышеуказанную смесь выдерживали на масляной бане при температуре 90°С при постоянном перемешивании в течение 3 ч, затем сушили в термошкафу при 120°С в течение 14 ч. Полученный порошок извлекали и прокаливали в статических условиях при 350°С 1 ч (5°С/мин) и затем при 700°С 4 ч (5°С/мин).
Ni наносили на поверхность приготовленных носителей методом пропитки по влагоемкости из водного раствора Ni(NO3)2 · 6H2O с номинальным содержанием 10 мас. %. Образцы были высушены при температуре 120°С и прокалены в изотермических условиях при 350°С в течение 1 ч (5°С/мин) и затем при 600°С (5°С/мин) в течение 4 ч. Синтезированные носители и катализаторы обозначены как CeMnOx (9 : 1), Ni/CeMnOx (9 : 1), CeMnOx (1 : 1), Ni/CeMnOx (1 : 1), CeMnOx (1 : 4), Ni/CeMnOx (1 : 4).
Исследование физико-химических свойств носителей и катализаторов
Элементный анализ катализаторов проводили методом рентгенофлюоресцентного анализа (РФлА) с помощью рентгеновского флуоресцентно-волнового дисперсионного спектрометра XRF-1800 (“Shimadzu”, Япония). Источник представлял собой рентгеновскую трубку с Rh-анодом, напряжением 40 кВ, током 95 мА и диафрагмой 10 мм.
Удельную поверхность и пористость образцов измеряли с помощью газо-адсорбционного анализатора Tristar 3020 (“Micromeritics”, США) с использованием азота в качестве газа-адсорбата. Навески образцов для анализа составляли ~0.1 г. Измерения выполняли после предварительной дегазации, которую осуществляли в условиях вакуума при температуре 200°С в течение 2 ч. Определение удельной поверхности (Sуд) производили многоточечным методом БЭТ. Распределение пор по размерам рассчитывали методом BJH по данным десорбционной ветви изотермы адсорбции–десорбции азота.
Исследование приготовленных образцов методом рентгенофазового анализа (РФА) проводили с помощью дифрактометра Shimadzu-6000 (“Shimadzu”, Япония) с применением CuKα-источника в диапазоне углов 2θ от 10° до 80°. Состав кристаллической фазы был установлен с использованием баз данных PCPDFWIN и ICSD и программного обеспечения POWDER CELL 2.4 для полного анализа профиля и расчета структурных параметров. Средние размеры кристаллитов обнаруживаемых фаз рассчитывали с помощью уравнения Шеррера. Для изучения устойчивости частиц Ni0 к спеканию были проведены исследования РФА in situ на дифрактометре Shimadzu-7000 (“Shimadzu”, Япония), оснащенном печью HTK-1200N (“Anton Paar”, Австрия). Перед экспериментом катализаторы восстанавливали в потоке 10% Н2/Ar при 750°С. Дифрактограммы регистрировали в вакууме (1.5 × 10–6 атм) в диапазоне от 42° до 59° (2θ) при 25 (перед нагреванием и после охлаждения камеры), 400, 650 и 800°С. Скорость нагрева печи от 25 до 650°С составляла 10°С/мин, с 650 до 800°С – 2°С/мин.
Спектры комбинационного рассеяния (КР) для уточнения состава образцов получали на конфокальном рамановском дисперсионном спектрометре InVia (“Renishaw”, Великобритания). Возбуждение осуществляли лазером при длине волны 785 нм. Спектры записывали в диапазоне 190–1000 см–1 со спектральным разрешением 2 см–1.
Особенности восстановления образцов изучали методом температурно-программированного восстановления (ТПВ-Н2). Эксперименты проводили на хемосорбционном анализаторе Chemisorb 2750 (“Micromeritics”, США) с детектором по теплопроводности, используя газовую смесь H2/Ar (10 об. % Н2). Для удаления воды из газовой смеси, образующейся в ходе реакции восстановления, применяли ловушку со смесью жидкого азота с изопропиловым спиртом (температура смеси –86°С). Температурный диапазон при проведении ТПВ-Н2 составлял 30–850°С, скорость потока газа – 20 мл/мин, скорость нагрева – 10°С/мин. Перед каждым экспериментом образцы подвергали окислительной предобработке в воздухе (20 мл/мин) в режиме температурно-программированного окисления (ТПО) до 400°С, выдержкой при данной температуре в течение 20 мин и последующим охлаждением.
Термогравиметрический анализ (ТГА) осуществляли с использованием системы TGA/DSC1 STAR (“Mettler Toledo”, Швейцария) в потоке воздуха при нагревании от 100 до 1000°C для оценки количества частиц углерода, осажденных на Ni-катализаторах после каталитических испытаний. Выделение CO2, происходящее во время экспериментов ТГА, контролировали с помощью анализатора QM (“Balzers, Quadstar”, Лихтенштейн).
Исследование каталитических свойств синтезированных Ni-содержащих катализаторов в реакции УКМ
Реакцию углекислотной конверсии метана (УКМ) проводили в проточном кварцевом реакторе с внутренним диаметром 12 мм. Перед реакцией катализаторы предварительно обрабатывали in situ в токе O2 (5 об. % в He, 50 мл/мин) при 350°C в течение 0.5 ч. После охлаждения до комнатной температуры образцы восстанавливали в потоке H2 (5 об. % в He, 30 мл/мин), повышая температуру до 750°C со скоростью линейного нагрева 10°C/мин и временем выдержки 1 ч. Поток реагентов состава 15 об. % CH4 + 15 об. % CO2 в He пропускали через катализатор (100 мг) со скоростью 100 мл/мин, что эквивалентно объемной скорости газа 60000 мл г–1 ч–1. Испытания на стабильность выполняли при 650°C в течение 24 ч. Состав газа на входе и выходе проанализировали с помощью газового хроматографа Agilent 7890B (“Agilent Technologies”, США), оснащенного капиллярной колонкой DB-1 и молекулярным ситом для анализа смеси реагентов/продуктов, включая CH4, CO, CO2, H2, O2, с использованием пламенно-ионизационного детектора и детектора по теплопроводности. На выходе из реактора конденсировали воду. Конверсии метана и CO2 и выход водорода рассчитывали по формулам:
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Состав и текстурные характеристики носителей и катализаторов
Данные о массовом соотношении Ce/Mn в смешанные оксидных носителях CeMnOx, а также массовом содержании Ni в катализаторах, приготовленных на их основе, представлены в табл. 1. Установлено, что экспериментально определенные составы носителей и катализаторов близки к расчетным.
Таблица 1.
Результаты элементного анализа
| № | Образец | Состав образцов, мас. % | Соотношение Се/Mn |
||
|---|---|---|---|---|---|
| Mn | Cе | Ni | |||
| 1 | CeMnOx (1 : 1) | 36.9 | 30.2 | – | 0.8 |
| 2 | CeMnOx (9 : 1) | 6.7 | 60.2 | – | 8.9 |
| 3 | CeMnOx (1 : 4) | 76.7 | 21.5 | – | 0.28 |
| 4 | Ni/CeMnOx (1 : 1) | 29.7 | 30.8 | 9.2 | 1.03 |
| 5 | Ni/CeMnOx (9 : 1) | 6.5 | 61.0 | 10.1 | 9.3 |
| 6 | Ni/CeMnOx (1 : 4) | 68.6 | 20.6 | 8.6 | 0.3 |
На рис. 1 приведены рентгенограммы синтезированных носителей и полученных на их основе катализаторов. Для всех образцов наблюдаются интенсивные дифракционные пики, типичные для флюоритной кубической структуры CeO2. По сравнению со стандартной рентгенограммой CeO2 (JCPDS #34–0394) отмечается небольшое смещение в сторону бόльших углов Брэгга. Данный факт может быть связан с образованием твердого раствора CeO2 и MnOx за счет частичного замещения ионов Ce4+ на Mnn+ в кубической структуре флюорита [31–33]. Также следует сказать, что интенсивность и ширина основных дифракционных пиков CeO2 значительно меняются при варьировании содержания марганца вследствие разницы в коэффициентах рассеяния электронов Mn (fMn) и Ce (fCe) [34]. Параметры кристаллической решетки для полученных образцов рассчитаны с помощью программы POWDER CELL 2.5 и представлены в табл. 2. Так, при низком содержании Mn в образце CeMnOx (9 : 1) Mn4+ легко входит в кубическую структуру флюорита CeO2, образуя гомогенный твердый раствор Ce1– xMnxO2 – δ состава Ce0.973Mn0.027O2 (ICSD # 16-5683), о чем свидетельствует отсутствие рефлексов, относящихся к фазам оксида марганца [34]. Параметр решетки а для образца CeMnOx (9 : 1) составляет 5.4101 нм. Образец CeMnOx (1 : 4) с наибольшим содержанием марганца характеризуется наименьшим параметром решетки (а = = 5.4047 нм) в ряду синтезированных носителей и самыми широкими дифракционными пиками. Размеры области когерентного рассеяния (ОКР) кристаллитов фазы Ce1– xMnxO2 – δ, рассчитанные по уравнению Шеррера, закономерно уменьшаются с увеличением содержания Mn и для CeMnOx (9 : 1), CeMnOx (1 : 1) и CeMnOx (1 : 4) составляют 14, 11 и 9 нм соответственно. В дополнение к рефлексам флюоритной структуры CeO2 для образцов CeMnOx (1 : 4) и CeMnOx (1 : 1) наблюдается появление рефлексов Mn2O3 (JCPDS #41-1442) и Mn3O4 (ICSD #68174). Из рентгенограмм на рис. 1а видно, что частицы Mn3O4 лучше окристаллизованы в образце CeMnOx (1 : 1), однако с повышением содержания марганца в CeMnOx (1 : 4) в качестве основной марганецсодержащей фазы кристаллизуется кубический Mn2O3.
Рис. 1.
а – Рентгенограммы для прокаленных носителей CeMnOx: 1 – CeMnOx (9 : 1), 2 – CeMnOx (1 : 1), 3 – CeMnOx (1 : 4); б – рентгенограммы для катализаторов Ni/CeMnOx: 1 – Ni/CeMnOx (9 : 1), 2 – Ni/CeMnOx (1 : 1 ), 3 – Ni/CeMnOx (1 : 4).
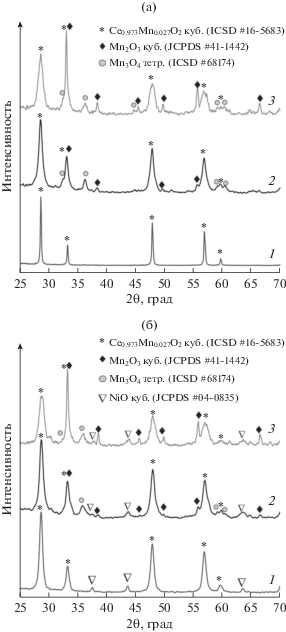
Таблица 2.
Текстурные и структурные характеристики образцов носителей и приготовленных на их основе катализаторов
| № | Образец | Sуд, м2/г | Объем пор, см3/г | Размер пор, нм | а Ce 1–x MnxO2 – δ, Å |
ОКР Ce 1–x MnxO2 – δ |
ОКР Mn2O3 |
ОКР Mn3O4 |
ОКР Mn3O4 |
|---|---|---|---|---|---|---|---|---|---|
| нм | |||||||||
| 1 | CeMnOx (1 : 1) | 20.4 | 0.12 | 12.7 | 5.4061 | 11 | 30 | 16 | − |
| 2 | CeMnOx (9 : 1) | 18.3 | 0.095 | 8.2 | 5.4101 | 14 | − | − | − |
| 3 | CeMnOx (1 : 4) | 17.3 | 0.13 | 27.4 | 5.4047 | 9 | 31 | 12 | − |
| 4 | Ni/CeMnOx (1 : 1) | 15.6 | 0.10 | 9.9 | 5.4027 | 11 | 32 | 9 | 13 |
| 5 | Ni/CeMnOx (9 : 1) | 14.0 | 0.096 | 6.7 | 5.4060 | 14 | − | − | 22 |
| 6 | Ni/CeMnOx (1 : 4) | 15.9 | 0.056 | 20.5 | 5.3996 | 10 | 29 | 12 | 11 |
Низкий предел растворимости Mn в решетке оксида церия, отмечаемый для носителей CeMnOx (1 : 1) и CeMnOx (1 : 4), согласуется с относительно большой разницей между ионными радиусами Ce4+ (0.094 нм) и Mn2+ (0.083 нм), Mn3+ (0.064 нм) или Mn4+ (0.053 нм). По данным [35, 36] замещение Ce4+ катионами марганца меньшего размера приводит к более значительному снижению величины параметра кристаллической решетки CeO2 и уширению дифракционных пиков. Таким образом, сдвиг рефлексов, соответствующих флюоритоподобной фазе CeO2, в сторону более высоких углов Брэгга хорошо коррелирует с включением небольших катионов Mn в решетку CeO2, вызывающим сжатие ее элементарной ячейки в ряду CeMnOx (9 : 1) > CeMnOx (1 : 1) > CeMnOx (1 : 4).
На рис. 1б представлены рентгенограммы прокаленных никельсодержащих катализаторов. С учетом состава носителей можно ожидать образование различных предшественников частиц Ni. Для всех катализаторов Ni/CeMnOx наблюдается формирование фазы NiO, которой соответствуют рефлексы при 2θ = 37°, 43° и 62° (JCPDS #04-0835). Кроме того, наблюдается небольшой сдвиг рефлексов флюоритоподобной фазы в область бóльших углов (от 2θ = 28.4° до 2θ = 28.6°). Это может свидетельствовать, что часть катионов никеля внедряется в приповерхностные слои носителя CeMnOx с образованием совместных структур Ce1 – (x+y)MnxNiyO2 – δ и/или Ce1– yNiyO2 – δ в случае малого содержания оксида марганца, или Ce1 – (x+y)MnxNiyO2 – δ и/или Mn1– yNiyOх для образцов на основе CeMnOx (1 : 1) и CeMnOx (1 : 4). О появлении твердых растворов условного состава Ce1 – (x+y)MnxNiyO2 – δ также говорит уменьшение параметра решетки (табл. 2).
Добавление марганца в CeO2 оказывает влияние на размер кристаллов NiО. Так, ОКР частиц NiО для катализаторов на основе оксидных носителей с разным массовым соотношением Ce/Mn варьируется от 22 до 11 нм (табл. 2). С ростом количества введенного оксида марганца наблюдается снижение размеров ОКР для нанесенного предшественника NiO. Это может быть связано с образованием поверхностных структур между предшественником активного компонента и оксидным носителем.
Согласно данным КР-спектроскопии (рис. 2) для образцов носителей наличие узких полос при ~462 и 459 см–1 соответствует смещению F2g-моды CeO2 при образовании твердого раствора Ce1– xMnxO2 – δ [37]. Оксиды марганца представлены в составе носителей двумя фазами: Mn3O4 и Mn2O3. Группа полос с максимумами при 651–654, 300–312 и 366–400 см–1 свидетельствует о наличии в носителях Mn3O4, максимальное содержание которого обнаружено в CeMnOx (1 : 1) [38]. Полоса при 692 см–1 для CeMnOx (1 : 4) и плечо около 680–690 см–1 для CeMnOx (1 : 1) соответствуют поглощению Mn2O3.
Рис. 2.
Спектры КР носителей CeMnOx и катализаторов Ni/CeMnOx: 1 – CeMnOx (9 : 1), 2 – Ni/CeMnOx (9 : 1), 3 – CeMnOx (1 : 1), 4 – Ni/CeMnOx (1 : 1), 5 – CeMnOx (1 : 4), 6 – Ni/CeMnOx (1 : 4).
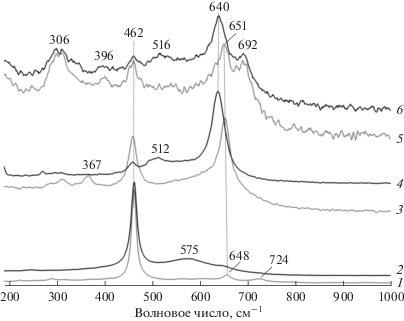
Введение никеля в носитель, обогащенный оксидом церия (образец Ni/CeMnOx (9 : 1)), приводит к появлению в спектре широкой полосы около 575 см–1, которая включает поглощение, обусловленное наличием кислородных вакансий (∼560 см–1) и замещением катионов Ce4+ катионами никеля с образованием твердого раствора Ce1– yNiyO2 – δ (∼600 см–1) [37, 39]. В КР-спектрах образцов Ni/CeMnOx (1 : 1) и Ni/CeMnOx (1 : 4) появление новой полосы при 512–516 см–1 может быть связано с валентными колебаниями Ni–O в образовавшихся частицах NiMn2O4 после пропитки [40]. Вероятно, смещение основной полосы колебаний Mn–O в КР-спектрах никелевых образцов в сравнении со спектрами носителей от 651–654 до 640 см–1 также вызвано образованием смешанного оксида никеля и марганца.
Исследование текстурных характеристик синтезированных носителей и нанесенных никелевых катализаторов проводили методом низкотемпературной адсорбции азота. Основные текстурные характеристики (площадь удельной поверхности, объем пор, размер частиц) приведены в табл. 2. Нанесение никеля на поверхность носителей CeMnOx с разным соотношением Ce/Mn приводит к незначительному уменьшению величины площади удельной поверхности и объема пор, что может свидетельствовать о равномерном распределении никеля на поверхности носителей без блокировки его пор. Следует заметить, что для всех катализаторов значения Sуд близки и составляют от 14.0 до 15.9 м2/г.
Изотермы адсорбции–десорбции азота и кривые распределения пор по размерам по данным десорбционной ветви изотермы для носителей и соответствующих Ni-содержащих катализаторов имеют схожий характер, что говорит о близкой природе их поверхностей. В связи с этим на рис. 3 представлены результаты только для никелевых катализаторов.
Рис. 3.
Изотермы адсорбции–десорбции азота (а) и распределение пор по размерам (б) для синтезированных катализаторов.
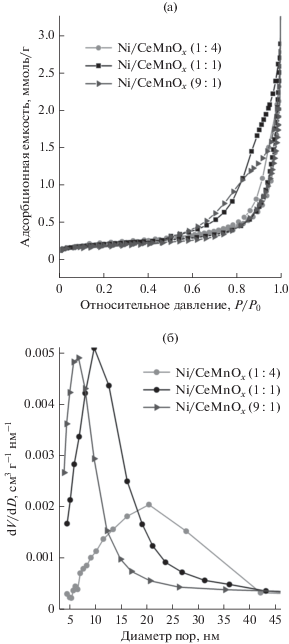
Изотермы адсорбции–десорбции азота для всех образцов характеризуются наличием петли гистерезиса при относительных давлениях 0.45–0.9, что указывает на их мезопористую структуру. Состав двойного оксида CeMnOx оказывает значительное влияние на форму петли гистерезиса [29]. Так, при возрастании содержания Mn в ряду катализаторов Ni/CeMnOx (9 : 1) – Ni/CeMnOx (1 : 1) – Ni/CeMnOx (1 : 4) формирование петли гистерезиса наблюдается при более высоких значениях относительного давления P/P0: 0.45–0.9; 0.5–0.9 и 0.6–0.9 соответственно, что указывает на увеличение вклада крупных мезопор в представленном ряду образцов. Об этом также свидетельствует распределение пор по размерам. Видно, что с ростом доли марганца происходит смещение распределения пор в сторону бóльших размеров. Для образца Ni/CeMnOx (1 : 4) с самым высоким содержанием марганца наблюдается широкое распределение пор с максимумом около 20.5 нм, что связано со структурой носителя, включающего крупные кристаллиты Mn2O3 (ОКР равна 29 нм) и более мелкие кристаллиты Ce1 – xMnxO2 – δ и Mn3O4 (ОКР составляют 10 и 12 нм соответственно). Кристаллизация индивидуальных оксидов марганца Mn2O3 и Mn3O4 с бóльшими размерами ОКР, чем для флюоритоподобной фазы в Ni/CeMnOx (1 : 1), также приводит к увеличению среднего размера мезопор и вкладу пор размером >10 нм по сравнению с Ni/CeMnOx (9 : 1).
Формирование активной поверхности/активных центров катализаторов
Профили ТПВ-H2 для синтезированных оксидных носителей и катализаторов на их основе представлены на рис. 4. Вид профилей ТПВ носителей свидетельствует, что поглощение водорода происходит в несколько стадий в диапазоне температур 160–490°С.
Рис. 4.
Профили ТПВ-H2 носителей CeMnOx и катализаторов Ni/CeMnOx: 1 – CeMnOx (9 : 1), 2 – Ni/CeMnOx (9 : 1), 3 – CeMnOx (1 : 1), 4 – Ni/CeMnOx (1 : 1), 5 – CeMnOx (1 : 4), 6 – Ni/CeMnOx (1 : 4).
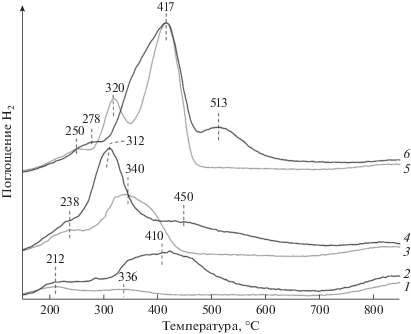
Восстановление СеО2 обычно протекает в области температур 350–650оС с появлением 2-х пиков поглощения Н2, обусловленных восстановлением поверхности наночастиц СеО2 и высокотемпературным восстановлением СеО2 до Се2О3 при Т > 700оС, как было показано ранее в [41]. Введение малого количества оксида марганца (образец CeMnOx (9 : 1)) приводит к сдвигу пиков поглощения Н2 в сторону низких температур, на профиле ТПВ-Н2 обнаружены два пика с максимумами при 212 и 336°С, что связано с образованием твердого раствора состава Ce0.973Mn0.027O2 и, вероятно, присутствием на поверхности рентгеноаморфных наночастиц MnOx. Высокотемпературный пик восстановления CeO2 в Ce2O3 различной интенсивности наблюдается при Т > 700°С для всех образцов синтезированных носителей и катализаторов на их основе.
С увеличением содержания Mnn+ в составе носителя максимумы ТПВ-пиков поглощения Н2 смещаются в область более высоких температур (рис. 4, кривые 1, 3, 5). Важно отметить, что начало пика поглощения Н2 в ТПВ-профилях для всех образцов лежит при одинаковых температурах. Это свидетельствует о присутствии на поверхности всех синтезированных носителей фазы Ce0.973Mn0.027O2, восстановление которой протекает в области температур 160–300°С. Появление в профилях ТПВ-H2 образцов с повышенным содержанием оксида марганца (CeMnOx (1 : 1) и CeMnOx (1 : 4)) широких неразрешенных пиков или пиков с несколькими максимумами в области температур 300–480°С обусловлено восстановлением Mn2O3 → Mn3O4 → MnO [42]. Положение температур максимумов восстановления зависит от размеров наночастиц фаз МnOx. Ступенчатый процесс восстановления Mn2O3 явно выражен для образца CeMnOx (1 : 4) с наибольшим содержанием марганца (пики при 320 и 417°С), что указывает на большое количество хорошо окристаллизованных частиц оксида марганца в составе носителя. Низкотемпературные пики с максимумами при 212–250°С могут быть отнесены к восстановлению поверхностных малых кластеров MnOx [43] либо ионов Mnn+ из твердого раствора Ce1-xMnxO2 – δ. Широкий пик выше 750°С для CeMnOx (9 : 1), а также катализатора на его основе Ni/CeMnOx (9 : 1), связан с восстановлением объемной фазы CeO2 до Ce2O3 (Се4+ до Се3+) [29]. Количество Н2, поглощаемого при восстановлении носителей, увеличивается в ряду CeMnOx (9 : 1) < CeMnOx (1 : 1) < < CeMnOx (1 : 4) и составляет 1.53, 3.01 и 4.25 ммоль/г соответственно (табл. 3).
Таблица 3.
Количество поглощенного Н2 для носителей CeMnOx и катализаторов Ni/CeMnOx
| № | Образец | n(H2)теор*, ммоль/г | n(H2)эксп, ммоль/г | ∆n(H2)**, ммоль/г |
|---|---|---|---|---|
| 1 | CeMnOx (9 : 1) | − | 1.53 | 2.28 |
| 2 | Ni/CeMnOx (9 : 1) | 1.73 | 3.81 | |
| 3 | CeMnOx (1 : 1) | − | 3.01 | 1.58 |
| 4 | Ni/CeMnOx (1 : 1) | 1.30 | 4.59 | |
| 5 | CeMnOx (1 : 4) | − | 4.25 | 0.98 |
| 6 | Ni/CeMnOx (1 : 4) | 1.38 | 5.23 |
Широкий пик в температурной области 300–550°С в профиле ТПВ катализатора Ni/CeMnOx (9 : 1) (рис. 4, кривая 2) обусловлен восстановлением наночастиц NiO [44], а также ионов марганца. Поглощение Н2 в области 250–350°С можно объяснить восстановлением кислорода, адсорбированного с участием кислородных вакансий, появление которых вызвано внедрением Ni2 + в приповерхностные слои CeO2 [45] и/или CeMnOx.
Согласно данным [46] восстановление ионов Ni2+ из структуры твердого раствора Ni0.1Ce0.9O2 −y протекает при более высоких температурах – 400–700°С. Снижение температуры поглощения Н2 для исследуемого катализатора Ni/CeMnOx (9 : 1) может быть связано с тем, что в этом случае катионы Ni2+ внедряются в приповерхностную область твердого раствора состава Ce0.973Mn0.027O2, а не индивидуального CeO2, как это было показано авторами [46]. Образование твердого раствора Ce0.973Mn0.027O2 в образце Ni/CeMnOx (9 : 1) подтверждено результатами РФА.
Количество поглощенного водорода после введения предшественника активного компонента на CeMnOx (9 : 1) составляет 2.28 ммоль/г, что превышает количество Н2, необходимого для восстановления Ni2+. Это может быть обусловлено частичным восстановлением поверхности носителя, а также образованием высокоокисленных состояний типа Ni3+ в составе твердого раствора [47]. Таким образом, пропитка носителя CeMnOx (9 : 1) раствором активного компонента приводит к появлению на его поверхности твердых растворов Ce1 – (x+y)MnxNiyO2 – δ различного состава, что подтверждается снижением температуры поглощения водорода на профиле ТПВ-Н2.
Для никельсодержащего катализатора Ni/CeMnOx (1 : 1) профиль ТПВ-H2 характеризуется наличием низкотемпературного плеча при 238°С, что, как и для Ni/CeMnOx (9 : 1), свидетельствует об образовании общего твердого раствора CeMnNiOx. Интенсивный пик поглощения водорода при 312°С соответствует совместному восстановлению Nin+ и Mnn+, скорее всего, из общего твердого раствора, поскольку восстановление MnOx в составе носителя CeMnOx (1 : 1) протекает в области более высоких температур (кривые 3, 4 на рис. 3). Плечо при 450°С связано с восстановлением наночастиц (НЧ) NiO, слабо взаимодействующих с носителем, а плечо при ~550°С можно отнести к восстановлению НЧ NiO, сильно взаимодействующих с поверхностью носителя [16]. Количество поглощенного водорода для Ni/CeMnOx (1 : 1) составляет 1.58 ммоль/г, что превышает количество Н2, необходимое для восстановления Ni2+. Из этого можно заключить, что на стадиях пропитки и последующей термообработки образуются Ni3+-центры.
Восстановление частиц NiO в катализаторе Ni/CeMnOx (1 : 4) в режиме ТПВ-Н2 происходит совместно с восстановлением Mn2O3 и Mn3O4 до MnO в диапазоне температур 300–460°С. Пик поглощения Н2 с максимумом при 513°С связан с восстановлением дисперсных частиц NiO, сильно взаимодействующих с поверхностью носителя. Для этого катализатора количество поглощенного Н2 (0.98 ммоль/г) за счет восстановления NiO ниже теоретического (в расчете на Ni2+). Увеличение доли трудно восстанавливаемых Ni-содержащих центров вызвано их стабилизацией оксидом марганца с образованием общих соединений [48].
Обработка катализаторов в токе восстановительной среды при 750°С в течение часа приводит к формированию частиц Ni0, размер которых зависит от состава носителя, природы и размеров частиц фаз-предшественников (NiO, CeMnNiOx, MnNiOx). Размеры ОКР Ni0 по данным РФА приведены в табл. 4 и составляют от 19 до 29 нм, при этом частицы меньшего размера наблюдаются для Ni/CeMnOx (1 : 1) и Ni/CeMnOx (1 : 4). Для высокотемпературных каталитических процессов большое значение имеет не только первичный размер частиц активного компонента, но и их устойчивость к спеканию в процессе эксплуатации. Так, под воздействием реакционной среды и высоких температур дисперсность исходных частиц Ni0 со временем снижается [49, 50], что может быть вызвано миграцией мелких кристаллитов с последующей их коалесценцией или миграцией атомов к более крупным кристаллитам. С этой точки зрения на стабильность работы катализатора влияет не только размер Ni0 в предвосстановленных катализаторах, но и прочность связи частиц активного компонента с носителем. Результаты исследования влияния состава носителя на устойчивость частиц Ni0 к спеканию методом РФА in situ показали, что размер ОКР Ni0 на носителях CeMnOx (1 : 4) и CeMnOx (9 : 1) почти не изменяется при нагреве образцов до 800оС в вакууме (табл. 4) и составляет 23 и 30 нм соответственно. Для образца Ni/CeMnOx (1 : 1) размер ОКР Ni0 увеличивается с 19 до 26 нм. Таким образом, носитель CeMnOx (1 : 4) обеспечивает максимальную устойчивость частиц активного компонента при высоких температурах в изученной серии катализаторов. Вероятно, предшественниками частиц Ni0 в образце Ni/CeMnOx (1 : 4) являются частицы NiO, прочно связанные с поверхностью носителя за счет образования твердых растворов MnNiOх, что согласуется с результатами ТПВ-Н2.
Каталитические свойства образцов в реакции УКМ
На рис. 5 и в табл. 5 представлены результаты исследования стабильности работы катализаторов Ni/CeMnOx в реакции УКМ при 650°С в течение 24 ч. Наибольшие значения конверсии X(СО2) и X(СН4) наблюдаются для катализатора Ni/CeMnOx (1 : 4), составляя 72 и 53% соответственно. Величины конверсии реагентов в присутствии Ni/CeMnOx (9 : 1) и Ni/CeMnOx (1 : 1) близки между собой (рис. 5а). Для этих двух катализаторов отмечается падение X(СО2) во времени. Выход Н2 снижается в исследуемой серии катализаторов в той же последовательности, что и конверсия метана (рис. 5а и 5б): Ni/CeMnOx (1 : 4) > > Ni/CeMnOx (1 : 1) > Ni/CeMnOx (9 : 1), и повышается с уменьшением размеров частиц Ni0 (табл. 4, ОКР800). Соотношение H2/CO при 650°С (рис. 5б и табл. 5) в течение 24 ч для всех образцов было ниже единицы – 0.74 для Ni/CeMnOx (1 : 1) и 0.71 для Ni/CeMnOx (1 : 4) и Ni/CeMnOx (9 : 1). Повышенное содержание CO в продуктах реакции может быть связано с реализацией побочных реакций, таких как обратная конверсия водяного пара (СО2 + Н2 → СО + Н2О), окисление поверхностного углерода в обратной реакции Будуара (С + СО2 → 2СО). Накопление углерода на поверхности катализаторов в процессе УКМ при температуре > 600°С в основном обусловлено разложением метана (СН4 → С + 2Н2). Следует отметить, что образующийся в этом процессе углерод более реакционноспособен, чем тот, что формируется в результате реакции Будуара, поэтому он может легко окисляться в присутствии CO2 до СО [51]. Кроме того, снижение H2/CO может быть вызвано участием Н2 в реакции с поверхностным MnCO3 с выделением СО и Н2О [52].
Рис. 5.
Результаты тестирования синтезированных катализаторов на стабильность в течение 24 ч при 650°С: а – конверсия метана (закрытые символы) и конверсия СО2 (открытые символы); б – мольное соотношение Н2/СО (открытые символы) и выход Н2 (закрытые символы).
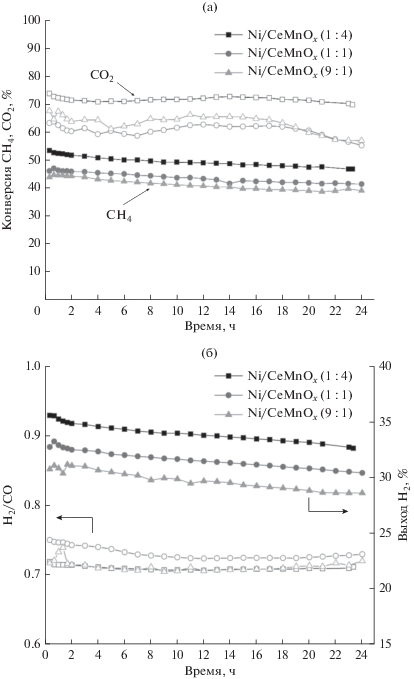
Таблица 5.
Каталитические свойства синтезированных Ni-содержащих материалов в реакции углекислотной конверсии метана
| Катализатор | Х(CH4)/Х(CO2) после 2 ч, % |
Х(CH4)/Х(CO2) после 24 ч, % |
Соотношение H2/CO после 2 ч |
Соотношение H2/CO после 24 ч |
Потеря массы, %* |
|---|---|---|---|---|---|
| Ni/CeMnOx (1 : 1) | 46/60 | 42/55 | 0.74 | 0.73 | 29.1 |
| Ni/CeMnOx (9 : 1) | 44/64 | 39/57 | 0.71 | 0.72 | 24.8 |
| Ni/CeMnOx (1 : 4) | 52/73 | 47/70 | 0.71 | 0.71 | 5.6 |
Анализ данных о конверсии реагентов и соотношении H2/CO показывает, что наиболее стабильным среди изученных образцов является катализатор Ni/CeMnOx (1 : 4).
Для оценки накопления углеродсодержащих продуктов на поверхности катализаторов после проведения экспериментов на стабильность были выполнены исследования методом ТГА (рис. 6). ТГ-кривые для всех катализаторов показывают небольшой прирост массы от 1.4% для Ni/CeMnOx (9 : 1) до 5.2% для Ni/CeMnOx (1 : 4) в диапазоне 200–450°С. Увеличение массы отработанных катализаторов может быть связано с окислением наночастиц Ni0 и частиц MnO [53] (рис. 7). При температуре выше 400–450°С происходит окисление продуктов углеотложения, накопленных в ходе реакции в основном в результате реакции разложения метана. По ТГ-кривым потеря веса для образцов Ni/CeMnOx (1 : 1) и Ni/CeMnOx (9 : 1), составляет 29.1 и 24.8% соответственно. Незначительная потеря массы (5.6%) за счет выгорания углерода наблюдается для Ni/CeMnOx (1 : 4). Разное содержание углерода на поверхности отработанных катализаторов подтверждается различной интенсивностью рефлекса при 2θ = 26.18° (С, графит #3-401) по данным РФА (рис. 7).
Рис. 6.
Результаты ТГ-анализа образцов катализаторов Ni/CeMnOx после испытания на стабильность при 650°С: 1 – Ni/CeMnOx (1 : 1), 2 – Ni/CeMnOx (9 : 1), 3 – Ni/CeMnOx (1 : 4).
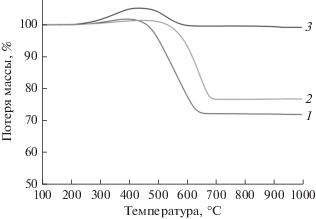
Рис. 7.
Рентгенограммы катализаторов Ni/CeMnOx после испытания на стабильность при 650°С: 1 – Ni/CeMnOx (1 : 1), 2 – Ni/CeMnOx (9 : 1), 3 – Ni/CeMnOx (1 : 4).
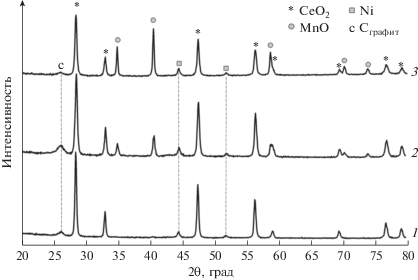
Малое количество углерода в образце Ni/CeMnOx (1 : 4) обусловлено стабилизацией частиц активного компонента оксидом марганца MnO, который взаимодействует с СО2 в процессе УКМ с образованием максимального количества поверхностных адсорбированных карбонатных центров, способных вступать в реакцию с углеродом [52]. Таким образом, MnO, обладая умеренной основностью, снабжает поверхность НЧ Ni0 адсорбированным CO2. Активные карбонатные центры способствуют быстрому удалению углеродсодержащих форм, образующихся на поверхности катализатора, прежде чем они переходят в инертный углерод.
Размер ОКР Ni0 для образцов после катализа (табл. 4) и составляет 13, 17 и 23 нм для Ni/CeMnOx (1 : 1), Ni/CeMnOx (9 : 1) и Ni/CeMnOx (1 : 4) соответственно. Следует обратить внимание, что только для катализатора Ni/CeMnOx (1 : 4) размер ОКР Ni0 не меняется в условиях теста на стабильность. Наблюдаемое уменьшение размера частиц никеля для Ni/CeMnOx (1 : 1) и Ni/CeMnOx (9 : 1) по сравнению с предвосстановлеными образцами (табл. 4, ОКР25 и ОКР800), а также значительное количество продуктов углеотложения для этих образцов, указывают на редиспергирование наночастиц активного компонента под действием реакционной смеси в результате формирования углеродных нитей. Поскольку для никельсодержащих катализаторов характерно образование нитевидного или вермикулярного углерода [47, 54, 55], можно предположить, что в процессе УКМ более мелкие частицы металлического никеля выносятся углеродными нитями без инкапсуляции, так как активность всех катализаторов в течение 24 ч меняется незначительно.
Взаимодействие катионов никеля с оксидами церия и марганца, а также твердого раствора Ce0.973Mn0.027O2 приводит к изменению взаимодействия наночастиц активного компонента – Ni – с носителем. Часть катионов никеля внедряется в приповерхностные слои носителей с образованием твердых растворов Ce1 – (x+y)MnxNiyO2 – δ или Mn1– yNiyOх, что способствует снижению размеров ОКР NiO от 22 до 11–13 нм. С увеличением содержания MnOx растет доля частиц оксида никеля, прочно связанных с поверхностью носителя и восстанавливающихся при более высоких температурах (>500оС). Кроме того, количество катионов активного компонента в составе твердого раствора Mn1 –yNiyOх повышается с ростом содержания оксида марганца в составе образца Ni/CeMnOx (1 : 4), в результате чего наблюдается самое низкое поглощение Н2 при восстановлении катионов никеля на поверхности этого катализатора (табл. 3). Формирование частиц активного компонента в контакте с оксидными фазами Mn1– yNiyOх и оксида марганца способствует стабилизации наночастиц активного компонента (что подтверждается результатами РФА, полученными для образцов после их прокаливания в вакууме), а также благоприятствует активации СО2 и снижению содержания углеродистых отложений.
Изменение каталитических свойств синтезированных Ni-катализаторов, нанесенных на CeMnOx с разным соотношением Ce/Mn, обусловлено отличиями в составе активной поверхности катализаторов (размеры кристаллитов, окружение наночастиц Ni и прочность их взаимодействия с носителем), что приводит к различной способности поверхности к активации CH4 и СО2. Результаты тестов на стабильность в процессе УКМ показывают, что соотношение Ce/Mn в составе образцов не определяет их активность, так как, в отличие от ранее изученных систем [29], в полученных в настоящей работе катализаторах не образуется непрерывный ряд твердых растворов Ce1– xMnxO2 – δ, а распределение активного компонента зависит от природы и соотношения фаз индивидуальных или двойных оксидов в носителе. Так, наиболее стабильным и активным среди исследованных в настоящей работе образцов является Ni/CeMnOx (1 : 4), для которого значения конверсии СH4 и СО2 в начале каталитического теста составляют 52 и 73% соответственно, и за 24 ч работы эти показатели снижаются на 4.1 и 9.6 отн. %. Для двух других катализаторов с меньшим содержанием марганца, Ni/CeMnOx (1 : 1) и Ni/CeMnOx (9 : 1), конверсия обоих реагентов в течение 24 ч снижается пропорционально на 8.7–8.3 и 11 отн. % соответственно. Максимальная каталитическая активность и стабильность Ni/CeMnOx (1 : 4) обусловлена наличием более развитой границы раздела MnOx и присутствием на поверхности высокодисперсных частиц Ni и MnNiOx.
Часть поверхности НЧ Ni0 может быть декорирована частицами восстановленного оксида марганца MnOx, особенно в случае образца Ni/CeMnOx (1 : 4). Такое декорирование препятствует выносу Ni-наночастиц нитевидными продуктами углеотложения и обеспечивает близкий контакт между активным компонентом и носителем. Частичное покрытие металла оксидом обычно наблюдается при использовании в качестве промоторов или носителей оксидов переходных металлов, таких как MnOx, La2O3 и CeO2, частично восстанавливающихся при высоких температурах [56, 57]. Одним из эффектов декорирования Ni-частиц с помощью MnOx является контроль размера доменов на поверхности никелевых частиц, доступных для активации молекул реагентов [57]. Так, высокая скорость активации молекул CO2 на оснóвных центрах восстановленных наночастиц MnOх и высокая скорость диссоциации CH4 на дисперсных Ni-наночастицах делают образец Ni/CeMnOx (1 : 4) наиболее активным.
Таким образом, по результатам исследования свойств Ni-содержащих нанесенных катализаторов, приготовленных на основе носителей CeO2–MnOx, было выявлено оптимальное соотношение Сe/Mn = 1 : 4 в составе носителя, которое приводит к увеличению активности катализаторов Ni/CeMnOx без значительного снижения стабильности в реакции углекислотной конверсии метана.
ЗАКЛЮЧЕНИЕ
По итогам проведенной работы предложены каталитические материалы на основе Ni-наночастиц, нанесенных на бинарные оксиды CeО2–MnOx с различным соотношением Сe/Mn. Установлено, что увеличение содержания Mn в серии образцов Ni/CeMnOx (9 : 1), Ni/CeMnOx (1 : 1) и Ni/CeMnOx (1 : 4) приводит к уменьшению размеров ОКР Ni-содержащего оксидного предшественника за счет частичного внедрения катионов никеля в приповерхностные слои носителей с образованием совместных структур Ce1 – (x+y)MnxNiyO2 – δ и/или Mn1– yNiyOх. Как следствие на поверхности предвосстановленного катализатора Ni/CeMnOx (1 : 4) формируются частицы металлического никеля, стабилизированные оксидом марганца(II), устойчивые к спеканию.
Каталитические испытания в реакции углекислотной конверсии метана при 650°C в течение 24 ч выявили высокую активность и стабильность образца Ni/CeMnOx (1 : 4), обусловленные свойствами межфазной границы Ni/MnO, на которой формирование поверхностных карбонатных частиц способствует быстрому удалению углерода с поверхности катализатора. Катализатор Ni/CeMnOx (1 : 4) показал высокую устойчивость к спеканию Ni-частиц, что было подтверждено методом РФА in situ (ОКР25Ni0 = 22 нм и ОКР800Ni0 = 23 нм). Также для этого образца было характерно малое содержание продуктов углеотложения после проведения каталитического эксперимента: потеря массы в режиме ТГА составила ~5.6 мас. %.
Список литературы
Kuckshinrichs W., Leitner W., Linssen J., Zapp P., Bongartz R., Schreibera A., Müller T.E. // Energy Environ. Sci. 2012. V. 5. P. 7281.
Yabe T., Sekine Y. // Process. Technol. 2018. V. 181. P. 187.
Nikoo M.K., Amin N.A.S. // Fuel Processing Technology. 2011. V. 92. P. 678.
Aramouni N.A.K., Touma J.G., Tarboush B.A., Zeaiter J., Ahmad M.N. // Renewable and Sustainable Energy Reviews. 2018. V. 82. P. 2570.
Lavoie J.M. // Front. Chem. 2014. V. 2. P. 1.
Pakhare D., Spivey J. // Chem. Soc. Rev. 2014. V. 43. P. 7813.
Muraleedharan Nair M., Kaliaguine S. // New J. Chem. 2016. V. 40. P. 4049.
Tsipouriari V.A., Verykios X.E. // J. Catal. 1999. V. 187. P. 85.
Sokolov S., Kondratenko E.V., Pohl M.M., Barkschat A., Rodemerck U. // Appl. Catal. B: Environ. 2012. V. 113–114. P. 19.
Singha R.K., Yadav A., Agrawal A., Shukla A., Adak S. // Appl. Catal. B: Environ. 2016. V. 191. P. 165.
Amin M.H. // Catalysts. 2020. V. 10. Article number 51.
Cheng H., Feng S., Tao W., Lu X., Yao W., Li G. // Int. J. Hydrog. Energy. 2014. V. 39. P. 12604.
Hou Z., Chen P., Fang H., Zheng X., Yashima T. // Int. J. Hydrogen Energy. 2006. V. 31. P. 555.
Yu M., Zhu Y.-A., Lu Y., Tong G., Zhu K., Zhou X. // Appl. Catal. B: Environ. 2015. V. 165. P. 43.
Theofanidisa S.A., Galvita V.V., Sabbe M., Poelman H., Detavernier C., Marin G.B. // Appl. Catal. B: Environ. 2017. V. 209. P. 405.
Gao X., Tan Z., Hidajat K., Kawi S. // Catal. Today. 2017. V. 281. P. 250.
Luisetto I., Tuti S., Di Bartolomeo E. // Int. J. Hydrogen Energ. 2012. V. 37. P. 15992.
Argyle M.D., Bartholomew C.H. // Catalysts. 2015. V. 5. P. 145.
Papadopoulou C., Matralis H., Verykios X. // Catalysis for Alternative Energy Generation. 2012. P. 57.
Seo H.O. // Catalysts. 2018. V. 8. Article number 110.
Roh H-S., Potdar H., Jun K-W. // Catal. Today. 2004. V. 93. P. 39.
Tatiparthi V., Pintar S.A. // Appl. Catal. A: General. 2020. V. 599. P. 117603.
Aghamohammadi S., Haghighi M., Maleki M., Rahemi N. // Mol. Catal. 2017. V. 431. P. 39.
Zhang S., Muratsugu S., Ishiguro N., Tada M. // ACS Catal. 2013. V. 3. P. 1855.
Yan X., Hu T., Liu P., Li S., Zhao B., Zhang Q., Jiao W., Chen S., Wang P., Lu J., Fan L., Deng X., Pan Y.-X. // Appl. Catal. B: Environ. 2019. V. 246. P. 221.
Wang N., Qian W., Chu W., Wei F. // Catal. Sci. Technol. 2016. V. 6. P. 3594.
Laosiripojana N., Assabumrungrat S. // Appl. Catal. B: Environ. 2005. V. 60. P. 107.
Chang K., Zhang H., Cheng M., Lu Q. // ACS Catal. 2020. V. 10. P. 613.
Mousavi S.M., Meshkani F., Rezaei M. // Int. J. Hydrogen Energ. 2017. V. 42. P. 24776.
Littlewood P., Xie X., Bernicke M., Thomas A., Schomacker R. // Catal. Today. 2015. V. 242. P. 111.
Jampaiah D., Venkataswamy P., Tur K.M., Ippolito S.J., Bhargava S.K., Reddy B.M. // Anorg. Allg. Chem. 2015. P. 1141.
Machida M., Uto M., Kurogi D., Kijima T. // Chem. Mater. 2000. V. 12. P. 3158.
Mousavi S.M., Niaei A., Illan Gomez M.J., Salari D., Panahi P.N., Abaladejo-Fuentes V. // Mater. Chem. Phys. 2014. V. 143. P. 921.
He H., Junheng L., Ping S., Song Y., Bingxia L. // RSC Advances. 2017. V. 7. P. 7406.
Huang C., Liu J., Sun P., Liang X., Tang C., Wu H. // Energy Sources, Part A: Recovery, Utilization, and Environmental Effects. 2019. V. 1. P. 11.
Lin X., Li S., He H., Wu Z., Wu J., Chen L., Fu M. // Appl. Catal. B: Environ. 2018. V. 223. P. 91.
Матус Е.В., Шляхтина А.С., Сухова О.Б., Исмагилов И.З., Ушаков В.А., Яшник С.А., Никитин А.П., Bharali P., Керженцев М.А., Исмагилов З.Р. // Кинетика и катализ. 2019. Т. 60. № 2. С. 245.
Buciuman F., Patcas F., Craciun R., Zahn D.R.T. // Phys. Chem. Chem. Phys. 1999. V. 1. P. 185.
Atzori L., Cutrufello M.G., Meloni D., Cannas C., Gazzoli D., Monaci R., Sini M.F., Rombi E. // Catal. Today. 2018. V. 299. P. 183.
Karmakar S., Behera D. // Ceram. Int. 2019. V. 45. P. 13052.
Grabchenko M.V., Mamontov G.V., Zaikovskii V.I., La Parola V., Liotta L.F., Vodyankina O.V. // Appl. Catal. B: Environ. 2020. V. 260. P. 118148(1).
Andreoli S., Deorsola F.A., Pirone R. // Catal. Today. 2015. V. 253. P. 199.
Tang X., Li Y., Huang X., Xu Y., Zhu H., Wang J., Shen W. // Appl. Catal. B: Environ. 2006. V. 62. P. 265.
Yu M., Zhu Y.-A., Lu Y., Tong G., Zhua K., Zhou X. // Appl. Catal. B: Environ. 2015. V. 165. P. 43.
Nematollahi B., Rezaei M., Lay E.N. // Int. J. Hydrogen Energ. 2015. V. 40. P. 8539.
Barrio L., Kubacka A., Zhou G., Estrella M., Martınez-Arias A., Hanson J.C., Fernandez-Garcia M., Rodriguez J.A. // J. Phys. Chem. C. 2010. V. 114. P. 12689.
Grabchenko M., Pantaleo G., Puleo F., Vodyankina O., Liotta L.F. // Int. J. Hydrogen Energ. 2021. In press.
Lu B., Zhuang J., Du J., Gu F., Xu G., Zhong Z., Liu Q., Su F. // Catalysts. 2019. V. 9. P. 282.
Tamagawa H., Oyama K., Yamaguchi T., Tanaka H., Tsuiki H., Ueno A. // J. Chern. Soc., Faraday Trans. 1987. V. 83(10). P. 3189.
Bai X., Wang S., Sun T., Wang S. // Reac. Kinet. Mech. Catal. 2014. V. 122. P. 437.
Wang S., Lu G., Millar G.J. // Energy Fuels. 1996. V. 10. P. 896.
Krylov O.V., Mamedov A.Kh., Mirzabekova S.R. // Catal. Today. 1998. V. 42. P. 211.
Pike J., Hanson J., Zhang L., Chan S.-W. // Chem. Mater. 2007. V. 19. P. 5609.
Yao L., Zhu J., Peng X., Tong D., Hu C. // Int. J. Hydrogen Energ. 2013. V. 38. P. 7268.
Touahra F., Sehailia M., Halliche D., Bachari K., Saadi A., Cherifi O. // Int. J. Hydrogen Energ. 2016. V. 41. P. 21140.
Lu G.Q., Wang S. // CHEMTECH. 1999. P. 37.
Seok S.-Ho, Han S.H., Lee J.S. // Appl. Catal. A: General. 2001. V. 215. P. 31.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ