

Кинетика и катализ, 2021, T. 62, № 6, стр. 694-705
Каталитическое окисление этилена в растворах катионных комплексов палладия(II) в бинарных и тройных водно-органических растворителях
И. В. Ошанина a, *, А. В. Подтягина b, У. В. Пестунова a, И. Н. Руснак a, О. Н. Темкин a
a МИРЭА – Российский технологический университет (Институт тонких химических технологий
имени М.В. Ломоносова)
119571 Москва, просп. Вернадского, 86, Россия
b ООО “ДНК-Технология”
117587 Москва, Варшавское шоссе, 125Ж, корп. 5, Россия
* E-mail: oshanina.irina@gmail.com
Поступила в редакцию 14.05.2021
После доработки 03.08.2021
Принята к публикации 03.08.2021
Аннотация
Изучено влияние органических растворителей на скорость окисления этилена п-бензохиноном до ацетальдегида в водно-органических растворах катионных комплексов
палладия. Установлено, что повышение скорости реакции наблюдается при увеличении акцепторных
и уменьшении донорных чисел растворителя. Более подробно исследованы процессы окисления
этилена и циклогексена в бинарном (N-метилпирролидон–Н2О) и тройном (ацетонитрил–N-метилпирролидон–Н2О) расторителях. Установлено, что в отличие от системы ацетонитрил–Н2О в бинарном растворителе N-метилпирролидон–Н2О пероксид водорода окисляет этилен до ацетальдегида в присутствии катионных комплексов
Pd(II). Показано, что использование растворителя, приемлемого для технологии окисления
ЦГ (N-метилпирролидона), приводит к снижению скорости и селективности процесса синтеза
циклогексанона.
Графический абстракт
ВВЕДЕНИЕ
Одним из выдающихся успехов гомогенного металлокомплексного катализа за последние 60 лет развития каталитической химии было создание технологий и теории процессов прямого окисления олефинов до карбонильных соединений в водных растворах комплексов палладия(II) (Вакер-окисление) [1, 2]. Важным шагом в повышении активности катализаторов и селективности реакций окисления высших олефинов и циклоолефинов был переход к бинарным водно-органическим растворителям в случае хлоридных (анионных) комплексов палладия(II). Процитируем лишь часть исследований, посвященных использованию бинарных растворителей с различными органическими компонентами в хлоридных каталитических системах Вакер-окисления: DMF [3–8], AN [7–9], NMP [4, 6, 7, 10], DMSO [3, 6, 11], DMA [7, 8, 10], ROH [7, 12–14], сульфолан [4], этиленкарбонат [10]. Сравнение этих каталитических систем, изученных в различных условиях и с разными олефинами, практически невозможно.
Особенно важную роль органические растворители сыграли в создании каталитических систем на основе более активных катионных комплексов палладия для окисления высших и циклоолефинов, выполняя функцию лигандов, стабилизирующих комплексы Pd(0) и Pd(I) [2, 15–24]. Установлено, что в таких системах, например, в системе AN–H2O, циклогексен (ЦГ) окисляется п-бензохиноном (BQ) до циклогексанона (ЦГ-он) с высокой скоростью и селективностью выше 95% [18, 20, 21, 24]. В бинарном растворителе AN–H2O детально изучены кинетика и механизм окисления циклогексена [20] и этилена [23, 25] в растворах катионных комплексов палладия с исследованием кинетических изотопных эффектов [26, 27].
Особая роль катионных комплексов переходных и непереходных металлов в гомогенном катализе обсуждается в статьях [2, 20]. Катионные комплексы в виде синтезированных комплексов Pd(AN)4(BF4)2, ${\text{PdL}}({\text{AN}})_{2}^{{2 + }}$ или образующихся in situ в системе Pd3(OAc)6–HX–растворитель активно использовали для получения сложных органических соединений, включая асимметрический синтез [17, 28–35].
В связи с появлением процесса синтеза циклогексена из бензола фирмы “Асахи” (“Asahi”, Япония) [9, 36, 37] возникает вопрос о разработке технологии прямого окисления ЦГ в циклогексанон, т.е. об использовании в этом конкретном процессе достижений в исследовании реакций окисления олефинов в растворах комплексов палладия. Поскольку окисление ЦГ п-бензохиноном и другими окислителями в водных растворах хлоридных комплексов палладия протекает медленно [38, 39] и для получения хороших результатов требует жестких условий [40], такая технология, естественно, должна разрабатываться в водно-органических растворах катионных комплексов палладия, образующихся in situ из Pd3(OAc)6 и НX.
Оказалось, однако, что система AN–H2O в этом процессе (по результатам исследования фазовых равновесий в системах AN–H2O–ЦГ и AN–H2O–ЦГ–ЦГ-он [21, 41]) создает множество проблем для выделения ЦГ-она из системы – появляются бинарные и тройные азеотропы, образуются двух- и трехфазные системы. Очевидно, что для создания технологии процесса получения циклогексанона окислением циклогексена необходимо подобрать эффективную каталитическую систему с водно-органическим высококипящим растворителем, обеспечивающим одновременно и высокие показатели химического процесса, и возможность организовать процесс выделения ЦГ-она из гомогенного раствора, содержащего комплексы Pd(II), без разрушения катализатора. Поскольку использование легко возгоняемого BQ в промышленном процессе нецелесообразно, необходимо также рассмотреть возможность применения других эффективных окислителей.
На первом этапе решения этой задачи было необходимо изучить кинетические закономерности более простого (модельного) процесса окисления этилена в условиях образования катионных комплексов палладия в различных водно-органических системах для выбора органического компонента бинарного растворителя, соответствующего описанным выше требованиям, и определить оптимальный состав каталитической системы, что и явилось целью настоящего исследования. В конкретные задачи работы входило:
– сравнение активности каталитических систем с различными органическими растворителями в процессе окисления этилена (по начальным скоростям);
– изучение кинетических закономерностей реакции окисления этилена в выбранном бинарном растворителе;
– выяснение возможности применения Н2О2 в качестве окислителя в процессе окисления этилена с использованием выбранного бинарного растворителя;
– оценка эффективности каталитической системы в одном из выбранных растворителей в процессе окисления циклогексена п-бензохиноном.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использованные реагенты
Ацетонитрил (“ч.”, ТУ 2636-092-44493179-04); хлорная кислота (70%, “Acros”, CAS:7601-90-3); толуолсульфокислота; этилен (ГОСТ 25070-87); n-бензохинон (99%, “Acros Organics”, в опытах использовали возогнанный п-бензохинон); перхлорат лития (б/в, “ч.”, ТУ 6-09-3360-73); тример диацетата палладия Pd3(CH3COO)6 (синтезирован в ИОНХ им. Н.С. Курнакова РАН, М = = 224.5 г/моль, Тпл = 205ºС; гидрохинон (ГОСТ 19627-74); эфир диэтиловый (“ч. д. а.”, ТУ 2600-001-45682126-06); кислота уксусная ледяная (“х. ч.”, ГОСТ 61-75); диметилформамид (“х. ч.”, “ЛАБТЕХ”, ГОСТ 20289-74); циклогексен (“Acros”, CAS: 110-83-8); диметилацетамид (“х. ч.”, СТП ТУ КОМП 2-211-10); циклогексанон “Acros”, CAS:108-94-1); адиподинитрил (99%, “Acros Organics”, CAS: 111-69-3); пероксид водорода; N-метилпирролидон (99.5%, “Merck”, CAS: 872-50-4); трет-бутиловый спирт (“РЕАХИМ”, ТУ 6-09-4069-75); нитрил фенилуксусной кислоты (99+%, “Acros Organics”, CAS: 140-29-4).
Методика проведения процесса окисления этилена и циклогексена
Опыты по окислению этилена и циклогексена п-бензохиноном (BQ) проводили в закрытом реакторе смешения, соединенном с газовой бюреткой. Эффективное перемешивание растворов осуществляли при помощи магнитной мешалки.
Методика окисления этилена п-бензохиноном. Перед началом опыта в реактор загружали ВQ ([BQ] = 0.05–0.4 M) и 9 мл бинарного растворителя (вода–органический растворитель), реактор закрывали, продували этиленом. Герметичность установки проверяли при помощи газовой бюретки. Опыты проводили при 30ºС (установка находится в термостатируемом боксе). В отдельной емкости растворяли навеску Pd3(OAc)6 в 1 мл органического растворителя (например, АN) в присутствии кислоты HX. Концентрацию палладия варьировали в диапазоне 0.0005–0.005 М, в качестве HX использовали толуолсульфокислоту ([TCK] = 0.1–0.4 М) или хлорную кислоту ([HClO4] = 0.1–0.4 М). Раствор, содержащий катионные комплексы палладия, выдерживали 20 мин и затем при помощи шприца вводили в реактор. С этого момента начинали отчет времени в опыте. Суммарный объем контактного раствора в реакторе во всех экспериментах равен 10 мл. Количество воды, добавляемой в контактный раствор, определяли, исходя из заданного состава (мольная доля воды – ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$) и содержания воды в хлорной кислоте. Мольную долю воды в опытах варьировали в диапазоне 0.05–0.67. В ряде случаев для поддержания высокого катионного фона или постоянной ионной силы в контактный раствор добавляли LiClO4. Скорость модельной реакции определяли, проводя замеры объема поглощенного газа (этилена) по времени. О кинетической области протекания реакции окисления этилена свидетельствует отсутствие зависимости скорости реакции от объема контактного раствора.
Для изучения влияния природы растворителя на активность каталитической системы использовали следующие вещества: ацетонитрил (AN), адиподинитрил (ADN), бензил цианид (PhCH2CN), диметилацетамид (DMA), диметилформамид (DMF). уксусную кислоту (AAc), трет-бутиловый спирт (t-BuOH), N-метилпирролидон (NMP), диметилсульфоксид (DMSO).
Методика окисления циклогексена n-бензохиноном. Перед началом опыта в термостатируемый реактор загружали навеску ВQ ([BQ] = 0.15–0.4 M) и 9 мл бинарного растворителя (AN–Н2О или NMP–Н2О) или тройного растворителя (AN–NMP–Н2О) и начинали продувать реактор небольшим током азота при перемешивании раствора. При помощи шприца в реактор загружали раствор, содержащий катионные комплексы Pd2+ (методика приготовления описана выше) и навеску ЦГ ([ЦГ] = 0.15–0.3 М). Перед введением в реактор ЦГ выход реактора закрывали, момент ввода циклогексена принимали за начало опыта. Опыты проводили при [Pd(OAc)2]∑ = 0.004–0.05 М и ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$= 0.15–0.67. Концентрации ТСК и хлорной кислоты варьировали от 0.2 до 0.4 М. После завершения опыта продукты и непрореагировавший ЦГ экстрагировали из контактного раствора при помощи диэтилового эфира, экстракт нейтрализовали твердой щелочью (NaOH). Состав раствора анализировали методом ГЖХ на газовом хроматографе ЛХМ-80 (Россия), колонка (длина 3 м, диаметр 3 мм) заполнена насадкой 15% силикона ДС-550 на хроматоне N-AW.
Определение растворимости этилена в бинарных водно-органических растворах, содержащих кислоту
Растворимость этилена измеряли при 30°С и атмосферном давлении 745 мм рт. ст. Опыты проводили с растворами, содержащими 0.15 мольных долей воды, 0.85 мольных долей органического растворителя (AN, NMP, DMF) и 0.2 М толуолсульфокслоты. Объем растворенного газа контролировали волюметрическим методом, последовательно добавляя в установку 3 порции (фиксированный объем) бинарного раствора. Для определения количества растворенного этилена использовали замеры, выполненные после введения второй и третьей порций раствора. Анализ содержания этилена в газовой фазе над раствором выполняли в конце опыта методом ГАХ на газовом хроматографе ЛХМ-80 (Россия), колонка (длина 3 м, диаметр 3 мм), заполнена активированным углем АГ-3.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Влияние природы растворителя на процесс окисления этилена
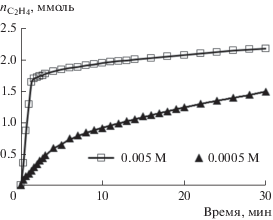
В серии опытов с варьированием органических растворителей концентрация Pd(OAc)2 составляла 0.005 М. В предыдущих работах [23, 24] было показано, что в системе Pd(OAc)2–HClO4–AN–H2O при концентрация Pd(OAc) равной 0.0005 М на кривых зависимостей объема поглощенного этилена от времени можно выделить два квазистационарных участка: быстрый и медленный. На первом быстром участке расходовалось около 50–70% BQ. Два участка кинетических кривых наблюдаются и при окислении циклогексена [20]. Из механизма реакций окисления олефинов в ацетонитрильной системе [20, 25] следует, что появление второго участка связано с образованием в ходе реакций катионных димерных комплексов палладия(I), менее активных в процессе окисления. Для построения кинетических зависимостей начальные скорости реакции (R0) рассчитывали на первом участке кинетических кривых. Увеличение концентрации катализатора с 0.0005 до 0.005 М в настоящей работе привело к значительному повышению скорости реакции. В этом случае на первом участке расходуется почти весь окислитель (рис. 1), поскольку количество поглощенного этилена (ммоль) близко к количеству загруженного BQ (ммоль). Анализ опытов с варьированием мольной доли воды и концентрации кислоты показал, что эти параметры также влияют на время появления 2-го квазистационарного участка.
Рис. 1.
Кинетические зависимости количества поглощенного этилена (${{n}_{{{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{4}}}}}}}$) от времени в системе AN–H2O. Условия реакции: [ТСК] = 0.2 M, [BQ] = 0.2 M (2 ммоль), мольная доля воды ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.15, [Pd(OAc)2]∑ указана на рисунке.
При варьировании органического компонента бинарного растворителя (рис. 2) установлен неизвестный ранее для катализа катионными комплексами палладия факт – вид кинетической кривой и, соответственно, способность катионных комплексов палладия(II) превращаться в ходе процесса в комплексы Pd(I) [20, 25] зависят (в одинаковых условиях) от природы растворителя. Так, в NMP, DMSO и t-BuOH 2-й участок появляется при конверсии BQ в интервале 10–20% (рис. 2б). В PhCH2CN и DMF 1-й участок сохраняется до значений конверсии BQ 80 и 95% соответственно (рис. 2a). Аналогичное наблюдение о появлении двух участков на кинетических кривых в процессе окислительного ацетоксилирования аллилбензола в системе ААс–диоксан было сделано в работе Ш. Стала (S. Stahl) и сотр. [42] с подтверждением образования комплекса Pd(I)–Pd2L2(OAc)2 (L = 4,5-диазафлуорен-9-он).
Рис. 2.
Примеры кинетических зависимостей количества поглощенного этилена (${{n}_{{{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{4}}}}}}}$) от времени в различных растворителях. Условия реакции: [Pd(OAc)2]∑ = 0.005 M, [BQ] = 0.2 M (2 ммоль). Дополнительные условия: все кривые на рис. 2а и кривые 1 и 2 на рис. 2б – [ТСК] = 0.2 M, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.15; кривая 3 (рис. 2б) – [ТСК] = 0.2 M, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.67; кривая 4 (рис. 2б) – [HClO4] = 0.4 M, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.67.

Применение растворителей с разной полярностью, обладающих разными донорными или акцепторными свойствами приводит к значительному изменению скорости реакции окисления этилена. Например, в присутствии таких растворителей, как AAc, DMA, t-BuOH, NMP, DMSO и DMF, не удается достичь высокой скорости окисления этилена (табл. 1, рис. 3а). В этих каталитических системах скорость реакции варьируется в диапазоне от 1.3 до 9.7 ммоль л–1 мин–1.
Таблица 1.
Зависимость начальной скорости окисления этилена от природы органического растворителя*
| Органический растворитель | R0, ммоль л–1 мин–1 | Органический растворитель | R0, ммоль л–1 мин–1 |
|---|---|---|---|
| AAc | 1.3 | DMF | 9.7 |
| DMA | 2.4 | PhCH2CN | 39.3 |
| t-BuOH | 3.1 | ADN | 44.2 |
| NMP | 4.6 | AN | 140.0 |
| DMSO | 7.6 |
Рис. 3.
Влияние растворителей на скорость реакции окисления этилена (сплошная заливка). Условия: [Pd(OAc)2]∑ = 0.005 M, [ТСК] = 0.2 M, [BQ] = 0.2 M (2 ммоль), ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.15–0.16. Пунктиром показаны значения диэлектрической проницаемости ε бинарных растворов.

Использование нитрилов приводит к значительному увеличению скорости реакции окисления этилена. Максимальная скорость ≈140 ммоль л–1 мин–1 получена в ацетонитриле (табл. 1, рис. 3б).
Таким образом, изученные растворители располагаются в следующем ряду по значениям скоростей реакции окисления этилена:
Известно, что влияние растворителя на скорости реакций в металлокомплексном катализе определяется совокупностью ряда факторов [43]. Перечислим основные функции растворителя:
‒ растворитель выступает в роли лиганда и, следовательно, его природа влияет на все характеристики металла-катализатора, включая окислительные потенциалы исходного комплекса и интермедиатов, и кинетику замещения лигандов в координационной сфере комплексов Pd(II), Pd(I) и Pd(0);
‒ растворитель в качестве среды, сольватируя протон (основные свойства), изменяет рКа кислоты НХ (например, ТСК);
‒ растворитель в качестве полярной среды участвует в сольватации всех частиц, участвующих в процессе, особенно в водно-органических средах при наличии ионов и ионных пар [25];
‒ органический компонент растворителя может менять растворимость этилена (${{Р}_{{{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{4}}}}}}}$, константа Генри).
Для такой сложной реакции, механизм которой в системе AN–H2O на первом квазистационарном участке кинетической кривой включает 10 элементарных стадий, а кинетическая модель – 3 константы скорости и 5 констант равновесия [23], непросто установить, какие свойства растворителя оказывают влияние даже на константы скорости медленных стадий.
Из рис. 3 следует, что величина диэлектрической постоянной ε, рассчитанная по аддитивности для бинарных растворителей, не коррелирует со значениями начальных скоростей реакции R0. Все эксперименты этой серии проводили при низких давлениях насыщенного пара бинарных растворителей (30°С) и незначительном изменении в ряду исследованных систем парциального давления этилена (не более чем на 10% от среднего значения), при постоянной мольной доле воды и концентрации кислоты в бинарном растворе и постоянной ионной силе растворов (концентрации комплексов Pd2+ и Н+ в бинарном растворителе с концентрацией воды в интервале 1.5–3.5 моль/л), т.е. при существенном сохранении физико-химических свойств растворов. В связи с этим была сделана попытка оценить в первом приближении, каким образом донорные и акцепторные свойства органических растворителей как лигандов влияют на величины R0.
Найти строгую взаимосвязь между скоростью реакции и донорными (D) или акцепторными (А) числами растворителей [44‒46] нам не удалось. И только использование двух параметров (А и D), при исключении протонных растворителей AAс и t-BuOH, позволило удовлетворительно описать связь свойств 6 растворителей со скоростью реакции окисления этилена уравнением (1) (рис. 4)
Рис. 4.
Сравнение логарифмов расчетных (lnRрасч = = 0.370A ‒ 0.144D) и экспериментальных скоростей реакции окисления этилена.
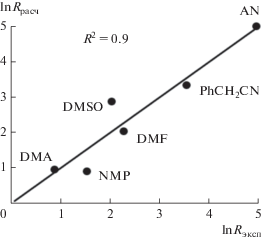
Коэффициент достоверности аппроксимации в этом случае составил 0.90. Для проверки влияния растворимости этилена на вид этой зависимости была измерена растворимость этилена в трех бинарных растворителях (AN, NMP и DMF), содержащих 0.2 моль/л ТСК (табл. 2). Отношение максимального и минимального значений растворимости этилена (системы AN‒H2O и DMF‒H2O) составляет 1.3. Отношение констант Генри (KH = [C2H4]/${{{\text{P}}}_{{\text{C}}}}_{{_{{\text{2}}}{{{\text{H}}}_{{\text{4}}}}}}$) для этих же систем больше – 1.6.
Таблица 2.
Растворимость этилена в бинарных растворителях (${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.15), содержащих ТСК (0.2 М)*
| Система | Концентрация C2H4 в газе, об. % |
Давление пара бинарного растворителя, атм | ${{P}_{{{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}{\text{,}}}}}$ атм | Концентрация C2H4 в растворе, M |
KH = = [C2H4]р-р/${{P}_{{{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}{\text{,}}}}}$ М/атм |
|---|---|---|---|---|---|
| AN–H2O | 90 | 0.137 | 0.76 | 0.057 ± 0.001 | 0.075 |
| NMP–H2O | 90 | 0.0068 | 0.88 | 0.046 ± 0.000 | 0.052 |
| DMF–H2O | 98 | 0.012 | 0.95 | 0.044 ± 0.004 | 0.047 |
Коэффициенты корреляционного уравнения (2) для вышеуказанных трех растворителей изменились несущественно:
Использование значений растворимости этилена в корреляции с двумя параметрами повысило величину коэффициента при D и снизило величину коэффициент при A в уравнении (3), но не изменило вида корреляционного уравнения и не привело к снижению точности описания модели. Коэффициенты достоверности корреляций (2) и (3) близки к 0.99
Значения коэффициентов в уравнении (3) свидетельствуют об одинаковом по степени, но разном по направлению влиянии акцепторных и донорных свойств органического растворителя на активность каталитической системы. Эти факты можно качественно объяснить в рамках ранее предложенного механизма окисления этилена (10 стадий) на основе кинетической модели, полученной в результате использования начальных скоростей реакции [23, 25] в системе АN–H2O. Упрощенную схему механизма можно представить следующей последовательностью стадий:
Сильные акцепторные свойства (А) растворителя, как лиганда, повышают электрофильность Pd(II) и, соответственно, этилена в π-комплексе и его способность к нуклеофильному присоединению молекулы Н2О. Усиление окислительных свойств палладия в случае лигандов с выраженными акцепторными свойствами может ускорять окислительный распад образующегося интермедиата [Pd(II)](CH2CH2OH)+ [23, 25]. Возрастанию электрофильности палладия будут способствовать и низкие донорные свойства растворителя, поддерживая высокие значения окислительных потенциалов палладия(II) в интермедиатах. Эти факторы, вероятно, и объясняют повышенную активность систем на основе нитрилов (см. выше). Присутствие группы ‒C≡N в лиганде может также стабилизировать комплексы Pd(0) и Pd(I) в результате частичного переноса электронов на RCN и, тем самым, препятствовать образованию неактивных форм катализатора.
В дальнейших исследованиях в качестве растворителя использовали NMP. Несмотря на невысокую скорость реакции, NMP представляет особый интерес с технологической точки зрения, так как его можно применять в качестве высококипящего растворителя для сохранения каталитической системы в процессе выделения продуктов реакции. Этот растворитель может на данном этапе (метод ректификации) выступать в качестве эффективного экстрактивного агента [47].
Влияние состава каталитической системы в бинарном растворителе NMP–H2O на показатели процесса окисления этилена
В серии экспериментов в системе NMP–H2O скорость поглощения этилена заметно меняется в ходе опыта. Показано, что увеличение концентрации ацетата палладия приводит к более резкому повышению скорости квазистационарного процесса на первом участке кинетической кривой по сравнению со вторым. При [HСlO4] = 0.2 M, [BQ] = = 0.2 M, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.67, ионной силе 0.5 ([HClO4] + + [LiClO4]) эти зависимости описываются кинетическими уравнениями первого порядка по суммарной концентрации ацетата палладия:
Первый порядок по [Pd(OAc)2]∑ во втором кинетическом уравнении может быть указанием на образование димерных комплексов палладия(I) в результате быстрых необратимых стадий.
Для определения оптимального содержания воды в контактном растворе варьировали ее мольную долю от 0.05 до 0.67. Показано, что при концентрации катализатора 0.005 М максимум скорости наблюдается при ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.15 (рис. 5а). Дальнейшее увеличение мольной доли воды приводит к снижению скорости реакции. Зависимости такого типа наблюдали ранее и в ацетонитриле [24].
Рис. 5.
Зависимость начальной скорости реакции окисления этилена R0 в системе NMP–H2O от: мольной доли воды (а) и [BQ] (б). Условия: [Pd(OAc)2]∑ = 0.005 M, [ТСК] = 0.2 M; [BQ] = 0.2 M (2 ммоль) (a), ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.15 (б).
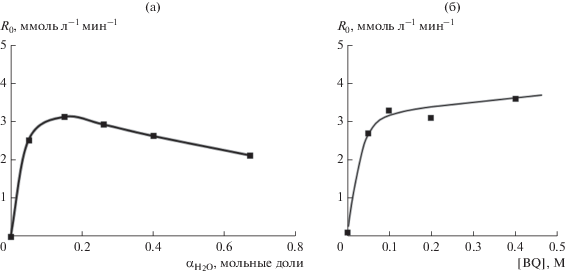
Влияние концентрации ТСК изучали при оптимальном содержании воды в контактном растворе, т.е. при ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.15 и [Pd(OAc)2]∑ = 0.005 M. Повышение концентрации ТСК от 0.05 до 0.4 М привело к росту скорости реакции от 1.6 до 4.8 ммоль л–1 мин–1. Дальнейший рост концентрации кислоты ([ТСК] = 0.4–0.7 М) практически не сказывается на скорости окисления этилена. Изменение концентрации перхлората лития с 0.1 до 0.3 при постоянной концентрации ТСК равной 0.4 М, т.е. изменение ионной силы раствора с 0.4 до 0.7, также не влияет на R0. Интересно отметить, что скорость реакции при использовании ТСК ниже, чем при применении HClO4. Так, повышение концентрации хлорной кислоты от 0.1 до 0.4 М (при изменении ионной силы раствора также с 0.4 до 0.7) и при ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.67 привело к возрастанию начальной скорости реакции от 4.7 до 9.4 ммоль л–1 мин–1. Возможные причины такого эффекта могут быть обусловлены неполной диссоциацией ТСК и участием тозилат-аниона в формировании координационной сферы у части комплексов палладия(II).
Зависимость скорости реакции от концентрации n-бензохинона на рис. 5б описывается кривой с “насыщением”. Увеличение концентрации п-бензохинона с 0.2 до 0.4 M в ацетонитриле в системе Pd(OAc)2–ТСК–AN–H2O также не приводит к изменению скорости реакции окисления этилена. Интересно, что в системе Pd(OAc)2–HClO4–LiClO4–AN–H2O зависимость скорости окисления олефинов от [BQ] имеет экстремум [20, 24]. Например, в реакции окисления этилена при [Pd(OAc)2] = 0.0005 M максимальная скорость достигается при [BQ] ≈ 0.1 М [24]. Можно предположить, что именно использование ТСК вызывает изменение вида кинетической модели процесса окисления олефинов и, соответственно, вида функции R0 = f[BQ], поскольку маловероятно, что увеличение концентрации палладия в 10 раз может сказаться на виде зависимости скорости реакции от концентрации BQ. Сложный характер зависимости R0 = f[BQ] объясняется в рамках предложенного ранее механизма указанной реакции в ацетонитрильной системе [23, 25] образованием комплексов Pd(0) с п-безохиноном.
В результате изучения кинетических закономерностей реакции окисления этилена в бинарной системе NMP–H2O показано, что этот процесс с максимальной скоростью при [Pd(OAc)2]Σ = = 0.005 M следует проводить в каталитической системе при [ТСК] = 0.2–0.4 M, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.15 и [BQ] > 0.1 M. Количество поглощенного в ходе опыта этилена в такой системе практически равно количеству исходного п-бензохинона, что свидетельствует об отсутствии заметного расходования окислителя на образование побочных продуктов. Селективность образования ацетальдегида по этилену составляет около 90% при почти полной конверсии BQ. При этих условиях частота оборотов катализатора равна 0.9 мин–1. В системе AN–H2O селективность образования ацетальдегида составляет 95%, а частота оборотов катализатора – 28 мин–1. Таким образом, замена растворителя (AN на NMP) в процессе окисления этилена приводит в существенному снижению скорости процесса.
Использование пероксида водорода в качестве окислителя этилена
В системе NMP–H2O опыты с использованием в качестве окислителя Н2О2 проводили при следующих условиях: [Pd(OAc)2]∑ = 0.005 M, [ТСК] = 0.2 M, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.67, αNMP = 0.33. Увеличение мольной доли воды в растворе обусловлено применением водного раствора пероксида водорода с концентрацией 30%. Показано, что начальная скорость окисления этилена в опыте с Н2О2 выше, чем в опыте с BQ (табл. 3, рис. 6). Если в исходный контактный раствор загрузить одновременно Н2О2 и гидрохинон, то количество ацетальдегида, образующегося за первые 10 мин, увеличивается в 1.5 и в 1.8 раза по сравнению с опытами только с Н2О2 или с BQ соответственно. Это свидетельствует о том, что и в такой системе пероксид водорода окисляет не только этилен, но и гидрохинон до хинона.
Таблица 3.
Использование H2O2 в процессе окисления этилена в системе NMP–Н2О
| № опыта |
Условия опыта | R0, ммоль л–1 мин–1 |
RII, ммоль л–1 мин–1 |
|---|---|---|---|
| 1 | [BQ] = 0.1 M | 5.9 | 1.8 |
| 2 | [H2O2] = 0.3 M | 11.8 | 0.8 |
| 3 | [H2O2] = 0.3 M, [QH2] = 0.1 M | 9.2 | 0.9 |
Рис. 6.
Влияние пероксида водорода на протекание процесса окисления этилена в системе NMP–H2O: 1 – [QH2] = 0.1 M (1 ммоль), [H2O2] = 0.3 М (3 ммоль); 2 – [BQ] = 0.1 M (1 ммоль); 3 – [H2O2] = 0.3 М (3 ммоль). Условия реакции: [Pd(OAc)2]∑ = 0.005 M, [ТСК] = 0.2 M, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.67.
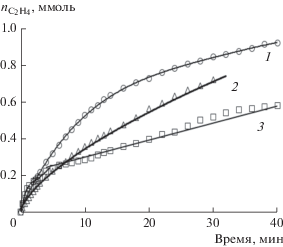
Ранее было показано [24], что в системе AN–H2O при [Pd(OAc)2]Σ = 0.0005 M, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.67, [HClO4] = = 0.2 M и [Н2О2] = 0.3 М не наблюдается поглощения этилена. Процесс окисления этилена начинался только при добавлении 0.2 М раствора BQ (в 1 мл АN). Т.е. BQ является существенно более эффективным окислителем, чем Н2О2, в системе AN–H2O. Опыты с использованием гидрохинона в этой системе показали, что пероксид водорода способен окислять его до п-бензохинона.
Таким образом, в системе NMP–H2O, в отличие от AN–H2O, пероксид водорода можно рассматривать как перспективный стехиометрический окислитель. Изменение поведения окислителей в разных системах, по-видимому, обусловлено изменением свойств комплексов палладия, координированных разными лигандами-растворителями. Например, образование прочного комплекса Pd(0) с NMP может затруднять координацию окислителя с комплексом Pd(0) и, соответственно, процесс окисления.
Отметим, что при использовании ионных жидкостей в качестве компонента бинарного растворителя пероксид водорода также хорошо окисляет этилен [24].
Бинарный органический растворитель в системе АN–NMP–H2O в реакции окисления этилена
Поскольку высокую скорость химической реакции обеспечивает применение ацетонитрила, а для создания эффективной технологии выделения ЦГ-она полезно присутствие в разделяемой смеси N-метилпирролидона в качестве основного высококипящего растворителя и эффективного экстрактивного агента, было интересно изучить процесс в смеси растворителей (АN, NMP и H2O). Варьирование соотношения мольных долей ацетонитрила и NMP проводили при постоянной мольной доле воды.
Увеличение мольной доли AN в тройной системе приводит к повышению скорости реакции окисления этилена от минимальной при использовании системы NMP–H2O (R0 = 4.6 ммоль л–1 мин–1) до максимальной (RI ≈ 140 ммоль л–1 мин–1). Резкое возрастание скорости происходит только когда мольная доля AN становится равной 0.75 (табл. 4). Поэтому, учитывая необходимость присутствия NMP в контактном растворе, его содержание в тройной системе не должно быть выше 0.1 мольной доли. Надо отметить, что в системе c АN основное количество ацетальдегида образуется за период времени, соответствующий первому участку кинетической кривой.
Таблица 4.
Зависимость скорости реакции окисления этилена R0 от мольной доли ацетонитрила (αAN) в тройной системе AN–NMP–H2O*
| αAN | 0 | 0.43 | 0.64 | 0.75 | 0.84 |
|---|---|---|---|---|---|
| R0, ммоль л–1 мин–1 | 4.6 | 10.2 | 17.1 | 33.5 | 140 |
Скорость окисления этилена в контактном растворе с αNMP = 0.1, αAN = 0.75 и ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.15 составляет 33.5 ммоль л–1 мин–1.
Окисление циклогексена в системах NMP–H2O и AN–NMP–H2O п-бензохиноном
Окисление ЦГ в системе AN–H2O ([Pd(OAc)2]∑ = 0.004 M, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.15, [ЦГ] = 0.15 M, [HClO4], = 0.2 M, [BQ] = 0.15 M]) позволяет получить ЦГ-он с селективностью 81.2% при полной конверсии ЦГ. Показано, что повышение мольной доли воды до 0.55 не приводит к возрастанию селективности процесса, а увеличение [HClO4] до 0.4 М и [BQ] до 0.4 M при ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.55 позволяет повысить селективность образования ЦГ-она до 96.6% (при конверсии ЦГ около 100%).
Замена ацетонитрила N-метилпирролидоном ведет к значительному снижению и селективности образования ЦГ-она, и конверсии ЦГ. Так, в системе NMP–H2O ([Pd(OAc)2]∑] = 0.005 M, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.33, [ЦГ] = 0.15 M, [ТСК] = 0.2 M, [BQ] = = 0.2 M) за 30 мин конверсия ЦГ составила 22.7%, а селективность по ЦГ-ону – 50%. Показано, что увеличение содержания воды до 0.67 и концентрации катализатора до 0.05 М повышает конверсию ЦГ, но не влияет на селективность процесса. Максимальная селективность образования ЦГ-она – 52.2% при конверсии ЦГ 90.7% – получена при следующих условиях: [Pd(OAc)2]∑ = 0.05 M, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.67, [ЦГ] = 0.15 M, [ТСК] = 0.2 M.
Использование тройной системы AN–NMP–H2O приводит к улучшению показателей процесса. Варьирование [HClO4] от 0.2 до 0.4 М, ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ от 0.16 до 0.51 и αAN от 0.15 до 0.64 позволило оптимизировать состав контактного раствора. Максимальная селективность образования ЦГ-она – 81.5% при конверсии ЦГ 100% – получена в каталитической системе следующего состава: ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.30, αNMP = = 0.17, αAN = 0.53, [Pd(OAc)2]∑ = 0.005 M, [HClO4] = = 0.4 M и [BQ] = 0.4 M. Дальнейшее увеличение мольной доли AN до 0.64 не улучшает показатели процесса окисления ЦГ.
Идентифицировать побочные продукты в контактном растворе нам не удалось. Метод ГЖХ, к сожалению, не позволяет зафиксировать наличие олигомеров ЦГ, которые, судя по результатам работы [48] по полимеризации ненасыщенных циклических углеводородов в растворах катионных комплексов палладия, могут образоваться и в нашей системе. Вероятно, что в исследованных системах с участием NMP образование олигомеров циклогексена снижает селективность реакции окисления по сравнению с системой AN–H2O. Так, в бинарной системе селективность образования ЦГ составляет ~50%, а в тройной системе ~80%.
ЗАКЛЮЧЕНИЕ
В представленной работе изучено влияние природы водно-органических растворителей на скорость реакции окисления олефинов (этилена, циклогексена) п-бензохиноном. Показано, что использование нитрилов в качестве растворителей позволяет проводить реакцию окисления этилена с высокой скоростью (от 40 до 140 ммоль л–1 мин–1). В присутствии других растворителей скорость реакции значительно ниже.
Впервые установлена корреляция между скоростью реакции окисления этилена и акцепторными и донорными числами апротонных растворителей в форме уравнения lnR = 0.37A ‒ 0.14D. Продемонстрировано, что учет растворимости этилена в бинарных растворителях меняет величину коэффициентов корреляционного уравнения, но не меняет знаков коэффициентов (ln(R/[C2H4]) = 0.163A ‒ 0.169D). Рассмотрены возможные причины появления таких корреляций.
Впервые исследованы кинетические зависимости процесса окисления этилена в системе NMP–H2O. В результате изучения влияния концентраций Pd(OAc)2, воды, кислоты и окислителя на скорость реакции даны рекомендации о составе каталитической системы, обеспечивающем максимальную скорость R0 около 4.6 ммоль л–1 мин–1 при [Pd(OAc)2]∑ = 0.005 М на начальном квазистационарном участке кинетической зависимости. Таким образом, использование NMP, характеризующегося более высоким значением донорного числа (27.3 по сравнению с 14.1 для AN) приводит к значительному снижению скорости реакции окисления этилена. Показано также, что замена HClO4 на толуолсульфокислоту сопровождается изменением вида кинетической зависимости R0 = f[BQ] в процессе окисления п-бензохиноном. Причины этого интересного явления требуют специальных исследований.
Установлено, что, в отличие от ацетонитрильной системы, в случае NMP–H2O пероксид водорода является перспективным стехиометрическим окислителем, окисляя и этилен, и гидрохинон.
Использование в качестве растворителя тройной системы AN–NMP–H2O с αNMP = 0.1, αAN = = 0.75 и ${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.15 при [Pd(OAc)2]∑ = 0.005 M, [ТСК] = 0.2 M, [BQ] = 0.2 M позволяет увеличить скорость реакции окисления этилена по сравнению с системой NMP – H2O до 33.5 ммоль л–1 мин–1.
В случае окисления ЦГ в тройной системе AN–NMP–H2O по сравнению с бинарной системой NMP–H2O наблюдается увеличение конверсии ЦГ (до ~100%) и селективности процесса по ЦГ-ону, которая в тройной системе составляет 81.5% (${{\alpha }_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.30, αNMP = 0.17, αAN = 0.53, [Pd(OAc)2]∑ = 0.005 M, [HClO4] = 0.4 M и [BQ] = = 0.4 M). Такое значение селективности по циклогесанону при проведении реакции в системе AN–NMP–H2O пока не позволяет рассматривать ее в качестве перспективного растворителя для промышленного каталитического процесса получения ЦГ-она. В дальнейшем планируется изучить возможность повышения этого показателя за счет снижения суперэлектрофильности комплексов Pd2+ с помощью лигандов или перехода на другие высококипящие растворители (адиподинитрил, диметилацетамид, диметилсульфоксид и др.).
Полученные в работе результаты являются, по нашему мнению, определенным вкладом в создание теории (и базы данных) многокомпонентных каталитических систем окисления олефинов.
Список литературы
Моисеев И.И. π-Комплексы олефинов в жидкофазном окислении. Москва: Наука, 1970. 270 с.
Темкин О.Н. // Кинетика и катализ. 2020. Т. 61. № 5. С. 595.
Clement W.H., Selwitz C.M. // J. Org. Chem. 1964. V. 29. P. 241.
Fahey D.R., Zuech E.A. // J. Org. Chem. 1974. V. 39. № 22. P. 3276.
Tsuji J., Shimizu I., Yamamoto K. // Tetrahedron Lett. 1976. P. 2975.
Tsuji J. // Synthesis. 1984. P. 369.
Mitsudome T., Umetani T., Nosaka N., Mori K., Mizugaki T., Ebitani K., Kaneda K. // Angew. Chem. Int. Ed. Engl. 2006. V. 45. № 3. P. 48.
Fernandes R.A., Chaudhari D.A. // J. Org. Chem. 2014. V. 79. I. 12. P. 5787.
Kohno M. // Chem. Chem. Ind. (Jpn). 1988. V. 41. № 10. P. 936.
Wang J.-l., He L.-N., Miao C.-X., Li Y.-N. // Green Chem. 2009. V. 11. P. 1317.
Lloyd W.G., Luberoff B.I. // J. Org. Chem. 1969. V. 34. P. 3949.
Григорьев А.А., Моисеев И.И., Клименко М.Я., Липина Ю.Н. // Хим. промышленность. 1972. Т. 48. С. 14.
Nishimura T., Kakiuchi N., Onoue T., Ohe K., Uemura S. // J. Chem. Soc. Perkin Trans. 1. 2000. P. 1915.
Roussel M., Mimoun H. // J. Org. Chem. 1980. V. 45. № 26. P. 5387.
Tsuji J., Minato M. // Tetrahedron Lett. 1987. V. 28. № 32. P. 3683.
Bäckvall J.-E., Hopkins R.B. // Tetrahedron Lett. 1988. V. 29. № 23. P. 2885.
Wenzel T.T. // J. C. S. Chem. Commun. 1989. V. 14. P. 932.
Miller D.G., Wayner D.D.M. // J. Org. Chem. 1990. V. 55. P. 2924.
Scumov M., Balbolov E. // Catal. Lett. 2000. V. 69. P. 103.
Темкин О.Н., Брук Л.Г., Захарова Д.С., Одинцов К.Ю., Кацман Е.А., Петров И.В., Истомина О.Ю. // Кинетика и катализ. 2010. Т. 51. № 5. С. 715.
Захарова Д.С., Семеняко А.Н., Черткова О.А., Фролкова А.В., Кацман Е.А., Брук Л.Г., Темкин О.Н. // Тонкие химические технологии. 2015. Т. 10. № 3. С. 77.
Захарова Д.С., Мартынов И.В., Носова В.М., Темкин О.Н. // Тонкие химические технологии. 2016. Т. 11. № 2. С. 57.
Мартынов И.В., Ефремов Г.Е., Бовырина Е.А., Кацман Е.А., Темкин О.Н. // Кинетика и катализ. 2018. Т. 59. № 4. С. 427.
Ефремов Г.Е., Бовырина Е.А., Подтягина А.В., Ошанина И.В., Темкин О.Н. // Кинетика и катализ. 2019. Т. 60. № 1. С. 54.
Ефремов Г.Е., Бовырина Е.А., Кацман Е.А., Шамсиев Р.С., Темкин О.Н. // Изв. АН. Сер. хим. 2019. № 7. С. 1366.
Захарова Д.С., Черткова О.А., Челкин А.А., Брук Л.Г., Темкин О.Н. // Изв. АН. Сер. хим. 2013. № 3. С. 843.
Мартынов И.В., Ефремов Г.Е., Темкин О.Н. // Изв. АН. Сер. хим. 2017. № 5. С. 922.
Hayashi T., Yamasaki K., Mimura H., Uozumi Y. // JACS. 2004. V. 126. P. 3036.
Michel B.W., Camelio A.M., Cornell C.N., Sigman M.S. // JACS. 2009. V. 131. P. 6076.
Anderson B.J., Keith J.A., Sigman M.S. // J. Amer. Chem. Soc. 2010. V. 132. P. 11872.
SigmanM.S., Werner E.W. // Acc. Chem. Res. 2012. V. 45. P. 874.
Morandi B., Wickens Z.K., Grubbs R.H. // Angew. Chem. Int. Ed. 2013. V. 52. P. 2944.
DeLuca R.J., Edwards J.L., Steffens L.D., Michel B.W., Oiao X., Zhu C., Coek S.P., Sigman M.S. // J. Org. Chem. 2013. V. 78. P. 1682.
Bigi M.A., White M.C. // JACS. 2013. V. 135 (21). P. 7831.
Walker K.L., Dornan L.M., Zare R.N., Waymouth R.M., Muldoon M.J. // JACS. 2017. V. 139 (36). P. 12495.
Fukuoka Y., Nagahara H. // Sci. Technol. Catal. 1995. V. 92. P. 375.
Nagahara H., Ono M., Konishi M., Fukuoka Y. // Appl. Surf. Sci. 1997. V. 121/122. P. 448.
Варгафтик М.Н., Моисеев И.И., Сыркин Я.К. // Докл. АН СССР. 1961. Т. 139. С. 1396.
Kolb M., Bratz E., Dialer K. // J. Mol. Catal. 1977. V. 2. P. 399.
Melgo M.S., Lidner A., Schuchardt U. // Appl. Catal. A: General. 2004. V. 273. P. 217.
Frolkova A., Zakharova D., Frolkova A., Balbenov S. // Fluid Phase Equilibria. 2016. V. 408. P. 10.
Jaworski J.N., Mc Cann S.D., Guzei I.A., Stahl S.S. // Angew. Chem. Int. Ed. 2017. V. 56. P. 3605.
Темкин О.Н. Гомогенный металлокомплексный катализ. Кинетические аспекты // Москва: Академкнига. 2008. 918 с.
Шмид Р., Сапунов В.Н. Неформальная кинетика. Москва: Мир. 1985. 264 с.
Фиалков Ю.А. Растворитель как средство управления химическим процессом // Ленинград: Химия. 1990. 283 с.
Lamb J.D., Nazarenko A.Y., Hansen R.J. // Separation Sci. Technol. 1999. V. 34. № 13. P. 2583.
Frolkova A.V., Mayevskiy M.A., Smirnov A.Y. // J. Chem. Eng. Data. 2019. V. 64. № 5. P. 2888.
Sen A., Lai T.W. // Organometallics. 1982. V. 1. P. 415.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ